

Abstract
We previously showed that newborn piglets who develop pulmonary hypertension during exposure to chronic hypoxia have diminished pulmonary vascular nitric oxide (NO) production and evidence of endothelial NO synthase (eNOS) uncoupling (Fike CD, Dikalova A, Kaplowitz MR, Cunningham G, Summar M, Aschner JL. Am J Respir Cell Mol Biol 53: 255–264, 2015). Tetrahydrobiopterin (BH4) is a cofactor that promotes eNOS coupling. Current clinical strategies typically invoke initiating treatment after the diagnosis of pulmonary hypertension, rather than prophylactically. The major purpose of this study was to determine whether starting treatment with an oral BH4 compound, sapropterin dihydrochloride (sapropterin), after the onset of pulmonary hypertension would recouple eNOS in the pulmonary vasculature and ameliorate disease progression in chronically hypoxic piglets. Normoxic (control) and hypoxic piglets were studied. Some hypoxic piglets received oral sapropterin starting on day 3 of hypoxia and continued throughout an additional 7 days of hypoxic exposure. Catheters were placed for hemodynamic measurements, and pulmonary arteries were dissected to assess eNOS dimer-to-monomer ratios (a measure of eNOS coupling), NO production, and superoxide (O2·−) generation. Although higher than in normoxic controls, pulmonary vascular resistance was lower in sapropterin-treated hypoxic piglets than in untreated hypoxic piglets. Consistent with eNOS recoupling, eNOS dimer-to-monomer ratios and NO production were greater and O2·− generation was less in pulmonary arteries from sapropterin-treated than untreated hypoxic animals. When started after disease onset, oral sapropterin treatment inhibits chronic hypoxia-induced pulmonary hypertension at least in part by recoupling eNOS in the pulmonary vasculature of newborn piglets. Rescue treatment with sapropterin may be an effective strategy to inhibit further development of pulmonary hypertension in newborn infants suffering from chronic cardiopulmonary conditions associated with episodes of prolonged hypoxia.
Keywords: nitric oxide signaling, sapropterin dihydrochloride, superoxide, pulmonary resistance arteries
infants suffering from chronic cardiopulmonary diseases associated with intermittent or prolonged periods of hypoxia often develop pulmonary hypertension (45, 50). The lack of effective treatment strategies for infants with hypoxia-induced pulmonary hypertension results in a chronic and progressive condition that too often culminates in right-sided heart failure and death (1, 30). There is evidence that impaired nitric oxide (NO) signaling plays a pivotal role in the pathogenesis of many forms of pulmonary hypertension, including chronic progressive hypoxia-induced pulmonary hypertension (16, 35, 52). Potential targets for novel, mechanistically based therapies can be identified by delineating the sites in the NO signaling pathway that are impaired in chronic cardiopulmonary conditions.
Endothelial NO synthase (eNOS) uncoupling has been shown to contribute to impaired NO signaling in a variety of cardiopulmonary disorders, including pulmonary hypertension (18, 24, 32, 52). In the homodimeric or coupled state, electrons are transferred from the eNOS reductase domain to the oxygenase domain, and NO is produced. When eNOS becomes uncoupled, electrons are diverted to molecular oxygen producing superoxide (O2·−) instead of NO (51, 52).
Tetrahydrobiopterin (BH4) is a cofactor that regulates eNOS activity in part by stabilizing the eNOS dimer and promoting eNOS coupling (24, 55, 56). Notably, the eNOS dimer is destabilized, and eNOS becomes uncoupled when BH4 levels are depleted. During conditions of oxidative stress, reactive oxygen species (ROS), including O2·−, plays a prominent role in depleting cellular BH4 via oxidation of BH4 to dihydrobiopterin (BH2). Augmenting BH4 levels provides a reasonable strategy to improve NO signaling and ameliorate the development of vascular diseases in situations where O2·− is elevated and eNOS is uncoupled (36).
Our laboratory has previously shown that newborn piglets develop pulmonary hypertension when exposed to chronic hypoxia for either 3 or 10 days (19, 20). Our laboratory has also shown that eNOS becomes uncoupled during both in vitro and in vivo exposure to chronic hypoxia, with concomitant increases in O2·− generation and decreases in both eNOS dimer formation and NO production (12, 17, 18). The major purpose of this study was to determine whether treatment with an oral BH4 compound, sapropterin dihydrochloride, recouples eNOS in the pulmonary vasculature and ameliorates the development of pulmonary hypertension in newborn piglets exposed in vivo to chronic hypoxia. Initiating treatments after the diagnosis of pulmonary hypertension, rather than prophylactically, better approximates current clinical strategies. Therefore, we designed the study to determine whether a “rescue” treatment strategy, i.e., starting BH4 after the onset of pulmonary hypertension, inhibits the progressive development of pulmonary hypertension that we have shown to occur when exposure to in vivo hypoxia is extended from 3 to 10 days (19, 20).
METHODS
Animal care and in vivo hypoxia model.
Use of animals was approved by the Institutional Animal Care and Use Committees of Vanderbilt University Medical Center and the University of Utah Health Sciences Center and were in adherence to the National Institutes of Health guidelines for the use of experimental animals. Both animal resource facilities are fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Use. For the hypoxic group, piglets were obtained from the vendor on day of life 2 and raised in a normobaric hypoxic environment until day of life 11–12 (9–10 days of hypoxia). Oxygen content was regulated at 10–12% O2. CO2 was absorbed with soda lime and was maintained at 3–6 Torr. Our laboratory has previously found no differences in pulmonary vascular resistance (PVR) between piglets raised by us in a room air environment and piglets raised by a vendor (19, 20). Therefore, most of the normoxic (control) animals were studied on the day of arrival from the vendor at day of life 12 (comparable postnatal age for the hypoxic piglets on the day of study).
Animals: chronic BH4 supplementation.
Some hypoxic piglets were given oral BH4 starting on the 3rd day of hypoxic exposure and continued for an additional 6–7 days of hypoxia. Piglets were treated with a synthetic form of BH4, sapropterin dihydrochloride (sapropterin). Piglets received either a lower (10 mg·kg−1·day−1 on the 3rd day of hypoxic exposure followed by 20 mg·kg−1·day−1 on subsequent days of hypoxia) or higher (20 mg·kg−1·day−1 on the 3rd day of hypoxic exposure followed by 40 mg·kg−1·day−1 on subsequent days of hypoxia) dosing strategy. For both dosing strategies, the sapropterin was given orally by syringe once a day in the morning. These doses and intervals of sapropterin were chosen based on pharmacokinetic studies in human infants with phenylketonuria (15).
Animals: in vivo hemodynamics.
For these measurements, piglets were preanesthetized with ketamine (15 mg/kg im) and acepromazine (2 mg/kg im). As previously described (19), with all animals breathing room air, a tracheostomy, venous and arterial catheters, and thermistor were placed using intravenous pentobarbital for sedation. Blood samples were drawn and spun, and the plasma was collected and frozen at −80°C for later determination of nitrite/nitrate (NOx) levels. Pulmonary arterial pressure, systemic blood pressure, left ventricular (LV) end-diastolic pressure, and cardiac output were measured. Cardiac output was measured by a thermodilution technique using a thermistor in the aortic arch and the LV catheter as an injection port. Most of the animals were breathing spontaneously in room air during the hemodynamic measurements. Some animals became apneic when sedated for placement of the catheters and required ventilation with room air using a piston-type ventilator at a tidal volume of 10–15 ml/kg, end-expiratory pressure of 2 Torr, and a respiratory rate of 20–25 breaths/min during the hemodynamic measurements. An arterial blood gas was drawn at the time of the hemodynamic measurements. At the completion of the hemodynamic measurements, the animals were given additional anesthesia and heparin (1,000 IU/kg iv) and exsanguinated.
Right ventricular mass assessment.
The heart was removed, and the right ventricular (RV) free wall and LV wall and septum (LV+S) were separated and weighed. Fulton's index, the ratio of the RV to LV+S weights, was calculated and used to assess RV mass.
Pulmonary artery isolation.
Small pulmonary arteries (≤300 μm) were dissected from lungs of all groups of piglets. Some pulmonary arteries were used for cannulated artery studies. Other pulmonary arteries were used to assess levels of NO production and O2·− generation, or were frozen in liquid nitrogen and stored at −80°C until used to assess BH4/BH2 levels or for protein analysis by Western blot technique.
Cannulated artery studies.
For the cannulated artery studies, 80- to 200-μm-diameter pulmonary arteries were isolated, cannulated, and pressurized using our laboratory's previously published methods (18). Each artery was equilibrated for 30–60 min to establish basal tone. Normoxic (control) and hypoxic arteries were equilibrated, respectively, at 15 or 25 cmH2O, transmural pressures that represent their respective in vivo pressures (19, 20). Our laboratory previously found that these different transmural pressures had no effect on pulmonary arterial responses in pulmonary arteries from normoxic or hypoxic piglets (21). Contraction to the thromboxane mimetic, U-46619, was used to test all arteries for viability. To check for a functional endothelium, we tested responses of normoxic arteries to acetylcholine (ACh) (10−6 M). For hypoxic arteries, responses to A-23187 were tested to check for a functional endothelium, because our laboratory previously found that hypoxic arteries constricted to ACh but dilated to the calcium ionophore A-23187 (21).
We then performed studies to determine whether responses to the endothelium-dependent agonist ACh (10−9 to 10−5 M), or the endothelium-independent NO donor S-nitroso-N-acetyl-penicillamine (SNAP; 10−9 to 10−5 M) were altered by chronic in vivo hypoxia or by BH4 treatment during chronic in vivo hypoxia. While vessel diameter was continuously monitored, cumulative doses of either ACh or SNAP were added to the reservoir. Responses to ACh were assessed with the vessels at basal tone, while responses to SNAP were measured after the vessel tone was elevated (40–50%) by addition of either endothelin (10−10 to 10−9 M) or U-46619 (10−9 to 10−8 M) to the reservoir.
NO measurements by electron spin resonance.
Small pulmonary arteries (≤300 μm diameter) were incubated for 60 min in 1.5 ml of Krebs/HEPES buffer containing 200 μmol/l iron diethyldithiocarbamate (Fe[DETC]2) and 10 μmol/l A-23187 at 37°C. Electron spin resonance (ESR) was used as described previously to detect the NO-Fe[DETC]2 complex (14). Values were normalized to the vessel protein content, which was determined by the Bradford assay.
O2·− measurements by ESR.
A modification of previously described methods was used to measure O2·− in small pulmonary arteries (≤300 μm diameter) (11). Vessels were incubated for 40 min at 37°C in 1 ml of Krebs/HEPES buffer (pH = 7.4) containing 10 μmol/l diethylenetriaminepentaacetic acid and 10 mmol/l 1-hydroxy-3-ethoxycarbonyl-2,2,5,5-tetramethyl-pyrrolidine. Some vessels were incubated for 15 min in the presence of NG-nitro-l-arginine methyl ester (l-NAME; 10−4 M). Vessels were frozen in liquid nitrogen, and ESR spectra were recorded with a Bruker EMX ESR spectrometer and a super-high Q microwave cavity. The following ESR instrument settings were used: field sweep, 50 G; microwave frequency, 9.78 GHz; microwave power, 20 mW; modulation amplitude, 5 G; conversion time, 327.68 ms; time constant, 5,242.88 ms; 512-point resolution and receiver gain, 1 × 104. The amplitude of the signal was measured, and the concentration of 3-carboxymethoxyl (CM)-radical was determined by calibration with standard concentrations of CM-nitroxide. The portion of the signal due to O2·− was determined by preincubating duplicate samples with polyethylene glycol superoxide dismutase (PEG-SOD; 100 U/ml) for 3 h in Krebs/HEPES buffer. PEG-SOD pretreatment inhibited 65–75% of CM-nitroxide formation. The formation of CM-nitroxide was normalized to the protein content, which was determined by the Bradford assay.
BH4 measurements by differential oxidation and HPLC.
As previously described, HPLC analysis and a differential oxidation method were used to measure oxidized (BH2) and reduced BH4 content in small pulmonary arteries (≤300 μm diameter) (13). The difference between total and alkaline-stable oxidized biopterin was used to determine the amount of BH4. A C-18 column was used with 5% methanol/95% water as a solvent at a flow rate of 1.0 ml/min. The fluorescence detector was set at 350 nm for excitation and 450 nm for emission.
Immunoblot analysis of total eNOS, eNOS dimers and monomers, and NOX1.
Frozen samples of small pulmonary arteries (≤300 μm diameter) were used for immunoblot analysis of eNOS (eNOS 1:2,000, eNOS antibody from BD-Transduction Laboratory, San Diego, CA) and NOX1 (NOX1 1:500, NOX1 antibody from Santa Cruz Biotechnology, Dallas, TX) using previously published methods (10, 16). Specifically, frozen samples of small pulmonary arteries were crushed under liquid nitrogen, transferred to a regular Triton (1%) lysis buffer containing protease inhibitors, and then sonicated on ice. Protein concentrations of the vessel homogenates were determined by the Bio-Rad protein assay. Aliquots of the protein concentrations were solubilized in equal volumes of denaturing, reducing sample buffer [Novex/0.25 M Tris·HCl, 5% (wt/vol) SDS, 2.5% (vol/vol) 2-mercaptoethanol, 10% glycerol, 0.05% bromphenol blue, pH 6.8] heated to 80°C for 15 min, and centrifuged in a microfuge. Equal volumes and, therefore, equal protein amounts of the supernatants were applied to Tris-glycine precast polyacrylamide gels (10% for eNOS and 4–20% for NOX1, Novex). Electrophoresis was carried out at 125 V for 90–120 min at room temperature. The proteins were transferred from the gel to a polyvinylidene difluoride (PVDF), membrane at 100 V for 60 min in 25 mM Tris, 192 mM glycine, and 20% methanol (pH 8.3) with an ice pack.
The immunoblot methods used for measurement of eNOS dimers/monomers have been previously described (34) and differed from those used for total eNOS. For determination of eNOS dimers/monomers (eNOS antibody 1:1,000), 10–20 mg of freshly dissected small pulmonary arteries were minced and then mechanically dounced in a regular Triton (1%) lysis buffer containing protease inhibitors, phosphatase inhibitors, and HEPES, and then agitated at 4°C for 10 min. Aliquots of the protein concentration were solubilized in the denaturing, reducing sample buffer and immediately centrifuged in the microfuge. Equal volumes of the supernatants were applied to Tris-glycine precast gels (10%, Novex). Electrophoresis was carried out at 90 V for 2 h at 4°C. The protein were transferred from the gel to a PVDF membrane at 35 V overnight at 4°C.
The membranes for total eNOS, NOX1, and eNOS monomers/dimers were incubated overnight at 4°C in PBS containing either 5% nonfat dried milk or 5% bovine serum albumin and 0.1% Tween 20 to block nonspecific protein binding. The membranes were developed using enhanced chemiluminescence reagents (ECL, Amersham), and the chemiluminescent signal was captured on X-ray film (ECL Hyperfilm, Kodak). The membranes were reprobed for β-actin (1:100,000, Sigma-Aldrich, St. Louis, MO). The bands for each protein were quantified using densitometry.
Plasma NOx measurements.
Using a modification of previously described methods, plasma NOx concentrations were measured by chemiluminescence analysis (2). Plasma samples were deproteinized, diluted 1:3 with ethanol, and injected (20 μl) into the reaction chamber of a chemiluminescent NO analyzer (model 170B NOA, Sievers). The reaction chamber contained vanadium (III) chloride in 1 M HCl heated to 90°C to reduce nitrite and nitrate to NO gas. The NO gas was carried into the analyzer using a constant flow of N2 gas via a gas bubble trap containing 1 M NaOH to remove HCl vapor. A standard cure was generated by adding known amounts of NaNO3 to distilled water and assaying, as described for the plasma samples.
Statistical analysis.
Data are presented as means ± SE. Data were compared by unpaired t-test or one-way ANOVA, with Fisher's protected least significant difference post hoc comparison test as appropriate. P values <0.05 were considered significant (43).
RESULTS
Consistent with our laboratory's previous findings (2, 17, 19), pulmonary arterial pressure was higher in the untreated group of chronically hypoxic piglets than in the comparable age normoxic control group (Fig. 1A). Pulmonary arterial pressures measured in both low- and high-dose groups of sapropterin-treated hypoxic animals were also higher than those measured in the control animals (Fig. 1A). There was no difference between pulmonary arterial pressures measured in the untreated and low-dose sapropterin-treated groups of chronically hypoxic piglets. However, pulmonary arterial pressure was significantly lower in the high-dose sapropterin-treated group of hypoxic animals than in both untreated and low-dose sapropterin-treated groups of hypoxic animals (Fig. 1A). LV end-diastolic pressure and aortic pressure measurements were similar in all groups of piglets (Table 1). Cardiac output was higher in the high-dose sapropterin-treated group of hypoxic animals than in all other groups and did not differ significantly between any of the other groups (Table 1). Measurements of arterial pH, arterial Po2, and arterial Pco2 were similar for all four groups of piglets (Table 1). Body weights measured on the day of study were lower in all three groups of hypoxic piglets than in the control group (Table 1).
Fig. 1.
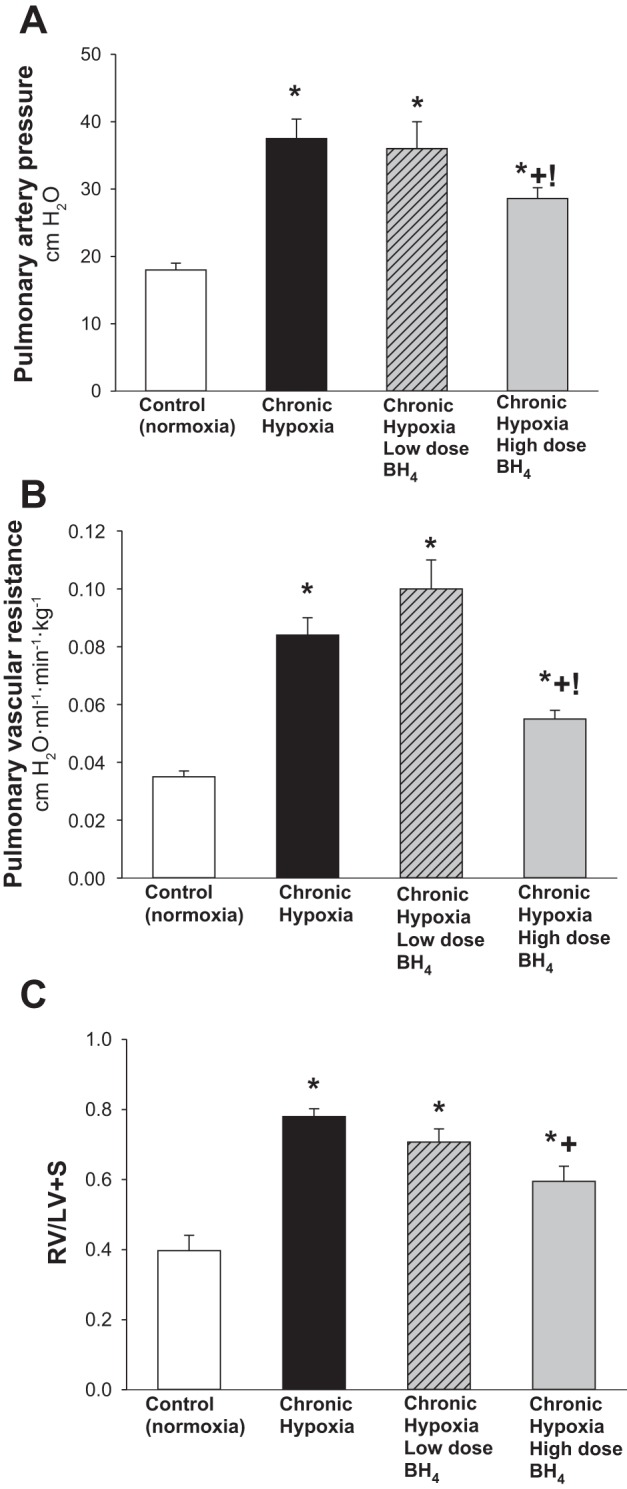
A: pulmonary artery pressure in control (normoxic) and chronically hypoxic piglets. High-dose (n = 12), but not low-dose (n = 4), treatment with oral BH4 (sapropterin dihydrochloride) reduced pulmonary artery pressure in hypoxic piglets to values less than those in untreated (n = 8) hypoxic piglets. Pulmonary artery pressure was higher in all groups of hypoxic piglets than values in normoxic controls (n = 10). Moreover, pulmonary artery pressure was less in hypoxic piglets treated with the high- vs. low-dose oral BH4. B: pulmonary vascular resistance (PVR) in control (normoxic) and chronically hypoxic piglets. High-dose (n = 12), but not low-dose (n = 4), treatment with oral BH4 reduced PVR in hypoxic piglets to values less than those in untreated (n = 8) hypoxic piglets. PVR was higher in all groups of hypoxic piglets than values in normoxic controls (n = 10). Moreover, PVR was less in hypoxic piglets treated with the high- vs. low-dose oral BH4. C: Fulton index (RV/LV+S) measurements of right ventricular mass in control (normoxic) and chronically hypoxic piglets. All three groups of chronically hypoxic piglets developed increased right heart mass compared with normoxia controls (normoxic controls, n = 10; untreated chronic hypoxia, n = 8; low-dose BH4-treated chronic hypoxia, n = 4; high-dose BH4-treated chronic hypoxia, n = 12). The high-dose, but not the low-dose, oral BH4-treatment reduced right ventricular mass in chronically hypoxic piglets to values below those measured in untreated chronically hypoxic piglets. Values are means ± SE. Different from *control (normoxic), +untreated chronic hypoxic, and !low-dose BH4-treated chronic hypoxic: P < 0.05; ANOVA with post hoc comparison test.
Table 1.
Data for normoxic (control), chronic hypoxic, and oral BH4-treated chronic hypoxic piglets
| Treatment Group | N | Weight, kg | LVEDP, cmH2O | Cardiac Output, ml·min−1·kg−1 | Aortic Pressure, cmH2O | pH | Pao2, Torr | Paco2, Torr |
|---|---|---|---|---|---|---|---|---|
| Controls(normoxic) | 10 | 3.65 ± 0.23 | 4.3 ± 0.4 | 383 ± 26 | 82 ± 3 | 7.33 ± 0.02 | 91 ± 3 | 35 ± 1 |
| Chronic hypoxic | 8 | 2.5 ± 0.16* | 5.3 ± 0.3 | 349 ± 18 | 77 ± 3 | 7.39 ± 0.02 | 93 ± 8 | 34 ± 1 |
| Low-dose BH4-treated chronic hypoxic | 4 | 2.61 ± 0.16* | 5.3 ± 1 | 305 ± 12 | 78 ± 6 | 7.36 ± 0.01 | 89 ± 3 | 34 ± 2 |
| High-dose BH4-treated chronic hypoxic | 12 | 2.65 ± 0.09* | 4.8 ± 0.3 | 428 ± 15*+! | 76 ± 2 | 7.35 ± 0.01 | 95 ± 4 | 34 ± 1 |
Values are means ± SE; N, no. of animals.
LVEDP, left ventricular end-diastolic pressure; Pao2, arterial Po2; Paco2, arterial Pco2. Different from
normoxic controls,
chronic hypoxia, and
low-dose oral BH4 (sapropterin dihydrochloride)-treated chronic hypoxic (P < 0.05; ANOVA with post hoc comparison test).
As shown in Fig. 1B, findings for PVR paralleled those for pulmonary arterial pressures (Fig. 1A). Specifically, high-dose, but not low-dose, treatment with sapropterin reduced PVR in hypoxic piglets to values less than those in untreated hypoxic piglets, although higher than values in normoxic controls. Four of the normoxic control piglets and three of the high-dose sapropterin-treated hypoxic piglets required mechanical ventilation during the hemodynamic measurements. However, there was no difference in PVR between unventilated (spontaneously breathing) and ventilated animals of either group (PVR was 0.035 ± 0.003 vs. 0.039 ± 0.004 cmH2O·ml−1·min−1·kg−1, respectively, for unventilated and ventilated normoxic control piglets; PVR was 0.056 ± 0.004 vs. 0.047 ± 0.005 cmH2O·ml−1·min−1·kg−1, respectively, for unventilated and ventilated high-dose sapropterin-treated hypoxic piglets). Moreover, PVR was lower in hypoxic piglets treated with the high vs. the low sapropterin dosage strategy (Fig. 1B). The impact on RV mass also differed between the high-dose and low-dose sapropterin-treatment strategies, with the high, but not the low, sapropterin-treated hypoxic group demonstrating a reduced RV mass compared with that observed in untreated hypoxic animals (Fig. 1C). The differential impact of sapropterin treatment on pulmonary arterial pressure, PVR, and RV mass correlated with the plasma NOx levels that were measured on the day of study (Fig. 2). Specifically, higher plasma NOx levels were found in the high- vs. the low-dose treated animals.
Fig. 2.
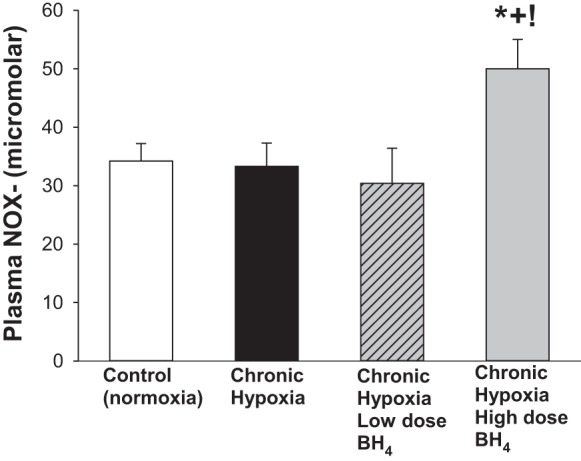
Plasma NOx levels in control (normoxic) and chronically hypoxic piglets. Plasma NOx levels were greater in chronically hypoxic piglets receiving high-dose oral BH4 (sapropterin dihydrochloride) treatment (n = 7) than in normoxic control piglets (n = 9), untreated chronically hypoxic piglets (n = 9), or in piglets receiving low-dose oral BH4 treatment (n = 4). Values are means ± SE. Different from *control (normoxic), +untreated chronic hypoxic, and !low-dose BH4-treated chronic hypoxic: P < 0.05; ANOVA with post hoc comparison test.
Consistent with our laboratory's previous findings, functional responses to the endothelium-dependent dilator, ACh, and the endothelium-independent NO donor, SNAP, were diminished in small pulmonary arteries from the untreated group of hypoxic animals compared with responses to these agents in pulmonary arteries from the comparable-age normoxic control group (16, 18) (Fig. 3). Responses to ACh did not differ between pulmonary arteries from the untreated and low-dose sapropterin-treated groups of hypoxic animals (Fig. 3A). However, pulmonary arteries from the high-dose sapropterin-treated group of hypoxic animals exhibited responses to ACh that were different from both the untreated and low-dose sapropterin-treated groups of hypoxic animals, although also different from responses in the normoxic control group (Fig. 3A). Likewise, responses to SNAP differed between the high-dose sapropterin-treated hypoxic animals and the other two groups of hypoxic animals (Fig. 3B). Pulmonary arteries from the high-dose sapropterin-treated group of piglets exhibited responses to SNAP that were similar to those in pulmonary arteries from control animals (Fig. 3B).
Fig. 3.
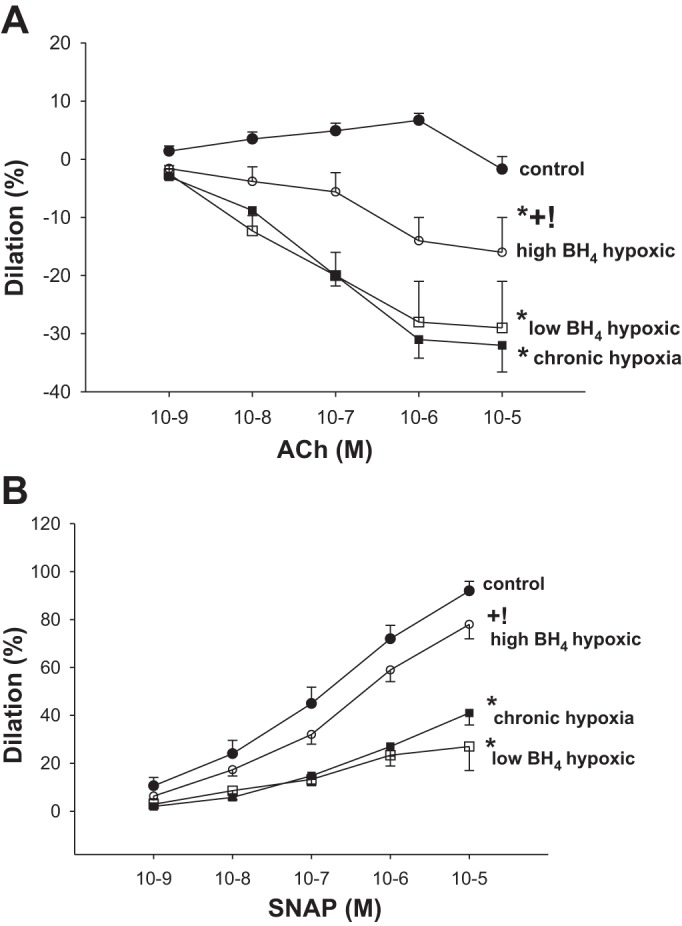
A: responses to the endothelium-dependent vasodilator, ACh, in small pulmonary arteries from control (normoxic) and chronically hypoxic piglets. Small pulmonary arteries from normoxic control piglets dilated to ACh (n = 8); ACh elicited a similar degree of constriction in small pulmonary arteries from the untreated group of hypoxic piglets (n = 4) and the low-dose oral BH4 (sapropterin dihydrochloride)-treated group of hypoxic piglets (n = 4). ACh elicited less constriction in small pulmonary arteries from the high-dose oral BH4-treated group of hypoxic piglets (n = 11) than measured in small pulmonary arteries from the other two groups of hypoxic piglets. B: responses to the NO donor, SNAP, in small pulmonary arteries from control (normoxic) and chronically hypoxic piglets. Responses to SNAP were similar for small pulmonary arteries from normoxic control piglets (n = 10) and high-dose oral BH4-treated chronically hypoxic piglets (n = 11). Responses to SNAP were less for small pulmonary arteries from both the untreated group of hypoxic piglets (n = 6) and the low-dose oral BH4-treated group of hypoxic piglets (n = 4) than for the other two groups of piglets. Values are means ± SE. Different from *control (normoxic), +untreated chronic hypoxic, and !low-dose BH4-treated chronic hypoxic: P < 0.05; ANOVA with post hoc comparison test.
BH4 levels were lower (Fig. 4A) and BH2 levels were higher (Fig. 4B) such that BH4-to-BH2 ratios (BH4/BH2) were markedly reduced (Fig. 4C) in small pulmonary arteries from all groups of hypoxic animals compared with values in small pulmonary arteries from control piglets. Combined BH4+BH2 levels also differed between small pulmonary arteries of control piglets and those from both untreated and low-dose sapropterin-treated groups of hypoxic animals (Fig. 4D). In contrast, combined BH4+BH2 values were similar in control piglets and high-dose sapropterin-treated hypoxic animals (Fig. 4D). Not surprisingly, BH4 levels were greater in both the high- and low-dose groups of sapropterin-treated hypoxic piglets than in the untreated group of hypoxic piglets (Fig. 4A). Moreover, BH2 levels were less in both high- and low-dose groups of sapropterin-treated hypoxic piglets than in the untreated group of hypoxic piglets (Fig. 4B). Furthermore, when comparing values between treatment groups, BH4 levels were greater and BH2 levels were less in the high-dose than the low-dose group of sapropterin-treated hypoxic piglets. Notably, for small pulmonary arteries from the high-dose but not the low-dose group of sapropterin-treated hypoxic piglets, BH4/BH2 (Fig. 4C) and combined BH4+BH2 levels (Fig. 4D) differed from respective values measured in the untreated group of hypoxic piglets.
Fig. 4.
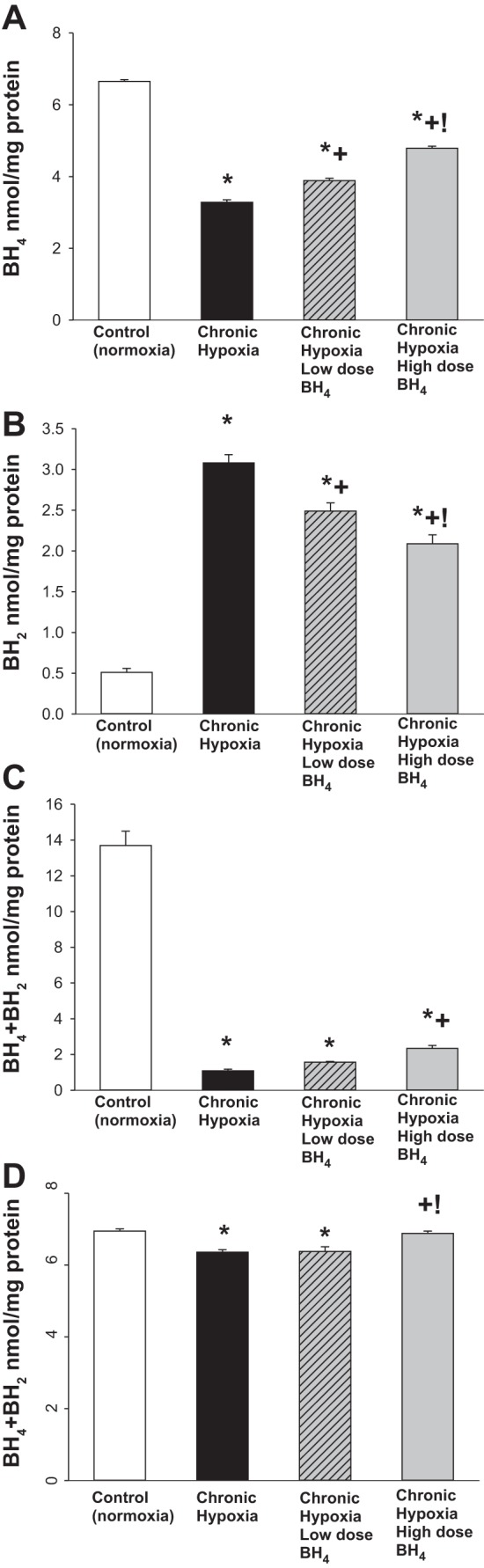
A: BH4 levels in small pulmonary arteries from control (normoxic) and chronically hypoxic piglets. BH4 levels were greater in small pulmonary arteries from normoxic control piglets (n = 6) than in all groups of hypoxic piglets. BH4 levels were greater in small pulmonary arteries from chronically hypoxic piglets receiving either low-dose (n = 4) or high-dose (n = 6) oral BH4 treatment than in small pulmonary arteries from untreated chronically hypoxic piglets (n = 6). BH4 levels were greater for pulmonary arteries from high-dose compared with low-dose oral BH4-treated chronically hypoxic piglets. B: BH2 levels in small pulmonary arteries from control (normoxic) and chronically hypoxic piglets. BH2 levels were less in small pulmonary arteries from normoxic control piglets (n = 6) than in all groups of hypoxic piglets. BH2 levels were less in small pulmonary arteries from chronically hypoxic piglets receiving either low-dose (n = 4) or high-dose (n = 6) oral BH4 treatment than in small pulmonary arteries from untreated chronically hypoxic piglets (n = 6). BH2 levels were less for pulmonary arteries from high-dose compared with low-dose oral BH4-treated chronically hypoxic piglets. C: BH4-to-BH2 ratios (BH4/BH2) in small pulmonary arteries from control (normoxia) and chronically hypoxic piglets. BH4/BH2 were greater in small pulmonary arteries from normoxic control piglets (n = 6) than in all groups of hypoxic piglets. BH4/BH2 were greater in small pulmonary arteries from the high-dose oral BH4-treated group of chronic hypoxic piglets (n = 6) than in the untreated group of chronic hypoxic piglets (n = 6). D: BH4+BH2 levels in small pulmonary arteries from control (normoxia) and chronically hypoxic piglets. BH4+BH2 ratios were greater in small pulmonary arteries from normoxic control piglets (n = 6) than in the untreated (n = 6) or low-dose oral BH4-treated (n = 4) groups of chronic hypoxic piglets. Values are means ± SE. Different from *control (normoxic), +untreated chronic hypoxic, and !low-dose BH4-treated chronic hypoxic: P < 0.05; ANOVA with post hoc comparison test.
NO production was higher and O2·− generation was lower in small pulmonary arteries from control piglets than all groups of hypoxic piglets (Fig. 5). Likewise, eNOS dimer-to-monomer ratios (Fig. 6A) were less in pulmonary arteries from all groups of hypoxic piglets compared with their respective values in pulmonary arteries from normoxic control piglets. There was no difference in total eNOS expression between pulmonary arteries from any of the piglet groups (Fig. 6B). Compared with pulmonary arteries from the untreated group of hypoxic piglets, pulmonary arteries from both the high- and low-dose groups of sapropterin-treated hypoxic piglets demonstrated greater NO production (Fig. 5A), less O2·− generation (Fig. 5B), and higher values for eNOS dimer-to-monomer ratios (Fig. 6A). Ex vivo treatment with l-NAME reduced O2·− generation for pulmonary arteries dissected from all groups of hypoxic piglets (Fig. 5B), but the impact was greatest for the arteries dissected from the untreated (32 ± 1% reduction) group of hypoxic animals. Of interest, the impact of ex vivo l-NAME treatment on O2·− generation was less for pulmonary arteries from the high-dose (16 ± 1% reduction) than for the low-dose (25 ± 2% reduction) sapropterin-treated group of hypoxic animals. In addition, O2·− generation was less (Fig. 5B) and eNOS dimer-to-monomer ratios (Fig. 6A) were greater with high-dose than with low-dose sapropterin treatment.
Fig. 5.
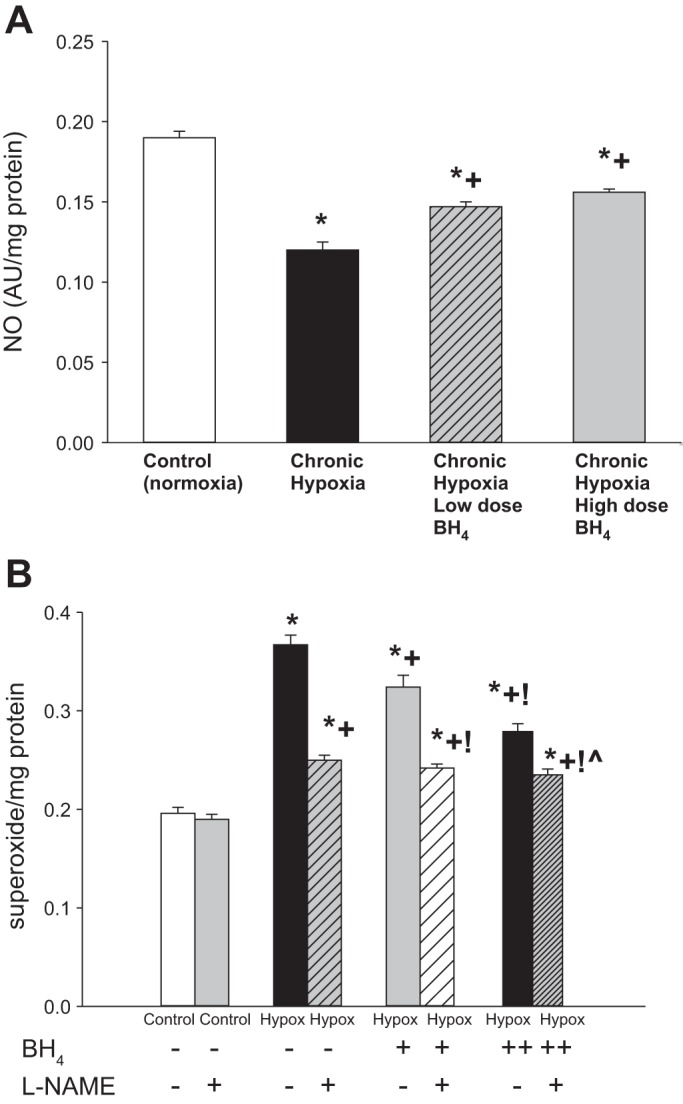
A: nitric oxide (NO) production by small pulmonary arteries from control (normoxic) and chronically hypoxic piglets. NO production was greater for small pulmonary arteries from normoxic control piglets (n = 7) than all groups of hypoxic piglets. Small pulmonary arteries from both low- (n = 4) and high-dose (n = 6) oral BH4 (sapropterin dihydrochloride)-treated groups of piglets had greater NO production than untreated (n = 7) hypoxic animals. B: superoxide production by small pulmonary arteries from control (normoxic) and chronically hypoxic piglets. Superoxide production was less for small pulmonary arteries from normoxic control piglets (n = 6) than all groups of hypoxic piglets. Small pulmonary arteries from both low- (n = 4) and high-dose (n = 6) oral BH4-treated groups of piglets had less superoxide production than untreated (n = 6) hypoxic animals. Superoxide production was less for pulmonary arteries from high-dose compared with low-dose oral BH4-treated chronically hypoxic piglets. Superoxide production was unchanged by ex vivo l-NAME (10−4 M) treatment in small pulmonary arteries from control (normoxic) piglets (n = 6). Ex vivo treatment with l-NAME reduced superoxide generation in small pulmonary arteries from all three groups of hypoxic piglets. Values are means ± SE. Different from *control (normoxic), +untreated chronic hypoxic, !low-dose BH4-treated chronic hypoxic, and ĥigh-dose BH4-treated chronic hypoxic: P < 0.05; ANOVA with post hoc comparison test.
Fig. 6.
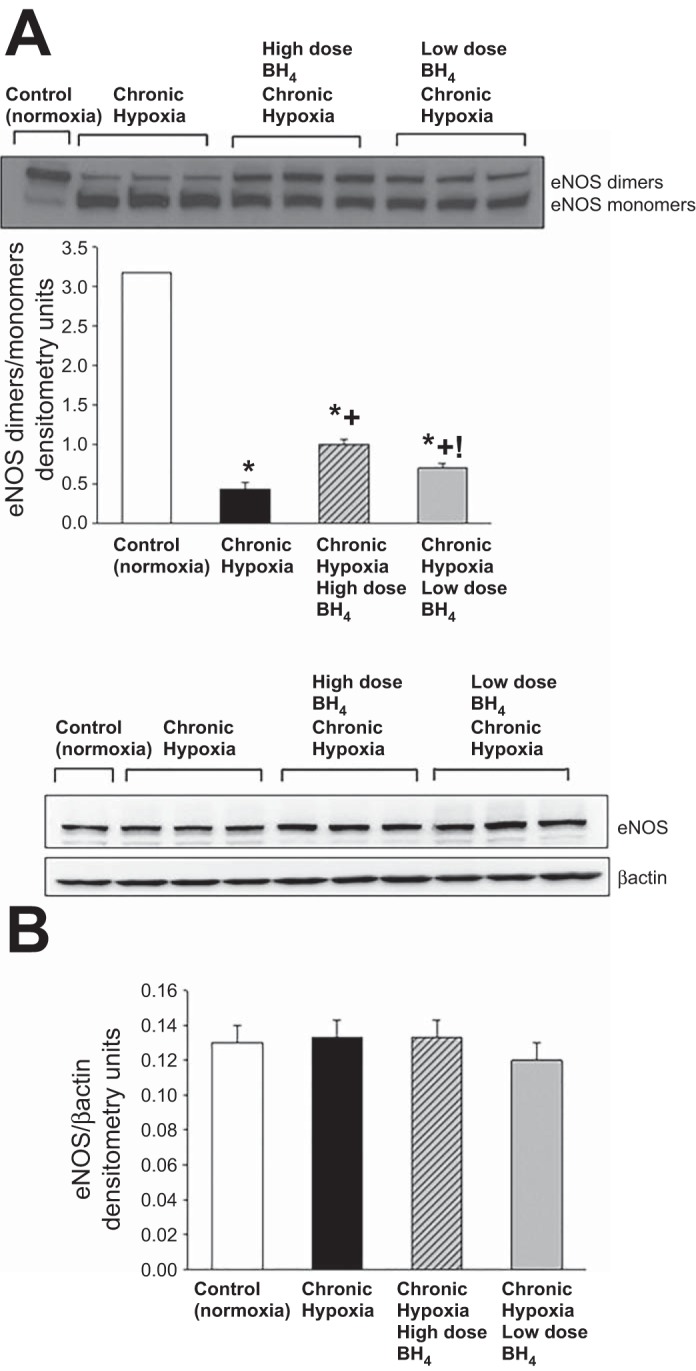
A: representative Western blot for eNOS dimer-to-monomer ratios with corresponding densitometry shows that small pulmonary arteries from both low- and high-dose oral BH4 (sapropterin dihydrochloride)-treated groups of chronic hypoxic piglets had greater eNOS dimer-to-monomer ratios than untreated hypoxic animals. In addition, eNOS dimer-to-monomer ratios were greater for pulmonary arteries from high-dose compared with low-dose oral BH4-treated chronically hypoxic piglets. B: representative Western blot for eNOS expression with corresponding densitometry shows that eNOS expression was similar in small pulmonary arteries from all groups of piglets. Values are means ± SE. Different from *control (normoxic), +untreated chronic hypoxic, and !high-dose BH4-treated chronic hypoxic: P < 0.05; ANOVA with post hoc comparison test.
Similar to previous findings, expression of NOX1, another enzymatic source of O2·−, was greater in small pulmonary arteries from the untreated group of hypoxic piglets than in small pulmonary arteries from normoxic control animals (Fig. 7; Ref. 10). Notably, NOX1 expression was less in small pulmonary arteries from both low- and high-dose sapropterin-treated groups of hypoxic animals compared with values in small pulmonary arteries from the untreated group of hypoxic piglets (Fig. 7). Moreover, the reduction in NOX1 expression was much greater for the high-dose than for the low-dose sapropterin-treated group of hypoxic piglets (Fig. 7).
Fig. 7.
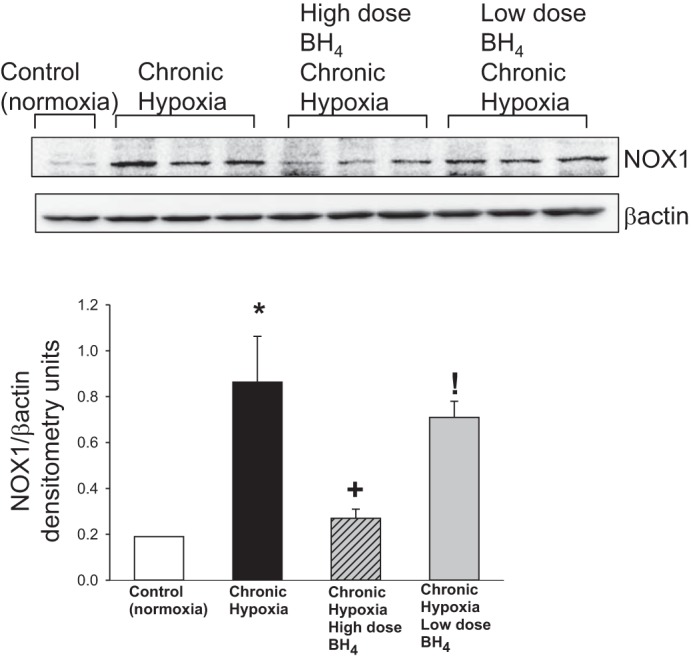
Representative Western blot for NOX1 with corresponding densitometry shows that small pulmonary arteries from both low- and high-dose oral BH4 (sapropterin dihydrochloride)-treated groups of chronic hypoxic piglets had lower NOX1 expression than untreated hypoxic animals. In addition, NOX1 expression was less for pulmonary arteries from high-dose compared with low-dose oral BH4-treated chronically hypoxic piglets. Different from *control (normoxic), +untreated chronic hypoxic, and !high-dose BH4-treated chronic hypoxic: P < 0.05; ANOVA with post hoc comparison test.
DISCUSSION
An important finding in this study is that, when used as a “rescue” strategy, oral treatment with the BH4 compound, sapropterin dihydrochloride, can ameliorate chronic hypoxia-induced pulmonary hypertension in newborn piglets. Underlying this finding, we are the first to report that small pulmonary arteries, the vessels most relevant to the increased PVR, of oral BH4-treated newborn piglets produce more NO, generate less O2·−, and exhibit a greater amount of eNOS dimer-to-monomer formation compared with the small pulmonary arteries of newborn piglets that were untreated throughout exposure to chronic hypoxia. Taken together, these latter findings indicate that mechanistically, recoupling of eNOS underlies the improved NO production found in sapropterin-treated piglets.
It is potentially of translational interest that the ability to ameliorate pulmonary hypertension, as exhibited by reductions in pulmonary arterial pressure, PVR, and RV mass, was only found in piglets receiving the high-dose sapropterin treatment strategy. Our findings indicate a dose-dependent ability of oral sapropterin treatment to recouple eNOS, reduce O2·− generation, and inhibit the progression of pulmonary hypertension that occurs in newborn piglets when hypoxic exposure is extended from 3 to 10 days. It is possible that the lower dose would be effective if given over a longer period of time, but our data suggest that dose is important and potentially point to a dose threshold for the efficacy of sapropterin treatment in a neonatal animal model of pulmonary hypertension.
Likewise, the impact of sapropterin dihydrochloride treatment on plasma NOx levels in hypoxic piglets was dose dependent. Indeed, the therapeutic effectiveness of sapropterin treatment correlated with the finding that plasma NOx levels were increased only in the hypoxic piglets that received the high-dose, and not the low-dose, treatment strategy. It should be noted that plasma NOx levels were similar in normoxic control piglets and the untreated group of chronic hypoxic piglets, despite marked differences in pulmonary arterial NO production between these two groups of animals. Altogether, these findings indicate that plasma NOx levels may not consistently reflect tissue NOx levels, limiting the use of plasma NOx levels as a dependable biomarker.
We also found a dose-dependent effect of sapropterin treatment on expression of NOX1, another enzymatic source of O2·− generation. Consistent with our laboratory's previous findings, NOX1 expression was increased in pulmonary arteries of piglets who did not receive sapropterin treatment during exposure to chronic hypoxia (10). ROS, including O2·−, have been shown to play a role in the pathogenesis of pulmonary hypertension by both us (10) and other investigators (5, 23). It follows that the greater reduction in NOX1 expression would contribute to the larger decrease in O2·− generation and help explain why the high-dose, but not the low-dose, treatment with sapropterin had a physiological effect on the development of pulmonary hypertension. It also merits comment that other investigators have provided evidence that BH4 has an antioxidant effect independent of its role as a cofactor that recouples NOS (42). Thus it is possible that an antioxidant effect of sapropterin treatment, unrelated to either NOX1 expression or eNOS recoupling, contributed in a dose-dependent fashion to the ability of this drug to inhibit chronic hypoxia-induced pulmonary hypertension.
The mechanism for the reduction in NOX1 expression found in pulmonary arteries of sapropterin-treated hypoxic animals is not known and will require future investigation. However, in a positive feedback fashion, NOX1 expression has been shown to be upregulated by ROS, including O2·− (41). Hence, sapropterin may act as a O2·− reducing agent with both eNOS-dependent and -independent effects to alter NOX1 expression in a dose-dependent manner.
Along these lines, we also found a dose-dependent effect of sapropterin treatment on BH4 and BH2 levels measured in small pulmonary arteries of chronically hypoxic piglets. BH4 is important to eNOS function because it modulates binding of the substrate, arginine (38, 53), and plays a role in dimer stabilization (34). The oxidation products of BH4, including BH2, have a negative impact on eNOS coupling by competing with BH4 for binding to eNOS (8, 9). Our findings support the notion that a reduction in BH4 levels, accompanied by an increase in BH2 levels, contributes to the eNOS uncoupling that develops in small pulmonary arteries of newborn piglets exposed to chronic hypoxia. In fact, it is possible that O2·− derived from NOX1 contributes to the oxidative loss of BH4 in pulmonary arteries of chronically hypoxic piglets. Moreover, our findings are consistent with the idea that the eNOS recoupling found in small pulmonary arteries of both groups of sapropterin-treated chronically hypoxic piglets can be attributed, at least in part, to the elevations in BH4 and reductions in BH2 levels that occurred with both low- and high-dose treatment strategies. Of interest, BH4/BH2 may have a greater influence on eNOS coupling than absolute levels of either BH4 or BH2 (8, 9, 54). It is possible that the failure of low-dose sapropterin treatment to ameliorate pulmonary hypertension could be due to the finding that, despite changes in absolute levels of both BH4 and BH2, BH4/BH2 were not sufficiently altered.
BH4 therapy has been used in other animal models of vascular disease. Oral BH4 was shown to be protective in a number of adult mice and rat models of systemic hypertension (27, 36, 48). Genetic manipulations that increase lung BH4 levels have been shown to inhibit chronic hypoxia-induced pulmonary hypertension in adult mice (31). Restoring pulmonary vascular BH4 levels by treating adult mice with folic acid has also been shown to inhibit the development of chronic hypoxia-induced pulmonary hypertension (6). Of particular interest, oral BH4 therapy used in a rescue treatment strategy was recently shown to inhibit the development of pulmonary hypertension in chronically hypoxic adult rats (22). Neither cardiac output nor left ventricular end-diastolic pressure were measured in the adult rats so that the impact of BH4 on PVR could not be assessed. Nonetheless, similar to our findings in newborn piglets, the effect of BH4 on pulmonary arterial pressures and RV mass in chronically hypoxic adult rats was dose dependent, with no impact when a low-dose strategy similar to ours was used. Of note, the higher dosage strategy tested and shown to be efficacious in adult rats was 100 mg·kg−1·day−1. Our higher dosage strategy, 40 mg·kg−1·day−1, was more comparable to doses used to treat newborn infants with phenylalanine hydroxylase deficiency (49). We add to the literature by showing that pulmonary hypertension was ameliorated in a newborn animal model using less than one-half the amount of BH4 given to adult animals.
We also found that cardiac output was greater in piglets treated with our higher dose strategy of sapropterin than in all other groups. Consistent with this finding, BH4 has been shown to improve cardiac function in a dose-dependent fashion in a mouse model of pressure-overload-induced hypertrophy and heart failure (44). Indeed, in addition to vascular diseases, there is ongoing interest in the potential use of BH4 to treat a variety of cardiac pathologies (4). The mechanisms underlying the positive impact of BH4 on cardiac output in our study is not currently clear and merits future investigation.
Another new finding in our study is that oral treatment with sapropterin improves pulmonary vascular responses to NO-dependent agents in a dose-dependent fashion. It is likely that the responses to the NO-dependent agents were improved due, at least in part, to the dose-dependent reductions in O2·− generation in pulmonary arteries from sapropterin-treated piglets. This is because poor functional responses to both endogenous and exogenous NO can be explained by interactions between O2·− and NO (25) that reduce the amount of NO available to activate the downstream signaling target, soluble guanylate cyclase. Specifically, the reduced constrictor response to the endothelium-dependent agonist, ACh, found in the small pulmonary arteries from high-dose sapropterin-treated piglets could be due to greater bioavailability of endogenously released NO. Likewise, increased bioavailability of exogenous NO could explain the enhanced dilator response to the endothelium-independent agent and NO donor, SNAP, found in pulmonary arteries of the high- but not the low-dose sapropterin-treated hypoxic piglets. Furthermore, our findings are consistent with the possibility that the low-dose BH4 treatment did not reduce O2·− generation enough to effectively improve NO bioavailability and thereby improve responses to SNAP, whereas the high-dose treatment did.
The improved response to exogenous NO in pulmonary arteries from high-dose sapropterin-treated piglets has important potential therapeutic implications. It is well known that infants with chronic progressive pulmonary hypertension have poor and erratic pulmonary dilator responses to inhaled NO therapy (3, 39). It is also known that term and preterm infants suffering from acute hypoxemic respiratory failure and pulmonary hypertension, a condition commonly referred to as persistent pulmonary hypertension of the newborn (PPHN) do not always respond adequately to inhaled NO therapy. It is possible that sapropterin treatment could be a strategy to reduce O2·− generation and improve in vivo responses to inhaled NO in both of these clinical scenarios. This possibility merits future exploration, including study in other newborn animal models of pulmonary hypertension. A limitation of our findings is that the use of chronic hypoxia may not accurately reflect the pathogenesis of all forms of neonatal pulmonary hypertension.
Interest in the therapeutic use of BH4 in humans, particularly for adults with cardiovascular diseases, has been increasing (4, 28, 33). Not all studies have shown positive outcomes with BH4 treatment (4, 28). Nonetheless, a number of studies performed in adults with a variety of systemic vascular diseases, including coronary artery disease (40), hypercholesterolemia (7), systemic hypertension (46), and diabetic vasculopathy (26), have demonstrated therapeutic effectiveness with BH4 supplementation. In addition, there is one report in adults with pulmonary hypertension in which sapropterin dihydrochloride was shown to be safe and associated with clinical improvement, as assessed by 6-min walk tests (47). We are not aware of studies evaluating the use of BH4 in any pediatric population with vascular disease. Yet it merits comment that treatment with BH4 has been shown to be safe, effective, and well tolerated in infants and children with phenylketonuria, a metabolic disorder due to deficiency of the enzyme phenylalanine hydroxylase (29, 37, 49).
In summary, via a mechanism involving recoupling of eNOS, use of a clinically relevant “rescue” treatment strategy with oral sapropterin dihydrochloride ameliorates the development of pulmonary hypertension in chronically hypoxic newborn piglets. Sapropterin treatment also improves pulmonary arterial vasodilator responses to the NO donor, SNAP. These findings could lead to advances in the treatment of pulmonary hypertension for a number of infants, particularly if our findings are validated in other animal models of neonatal pulmonary hypertension. It is notable that inhaled NO is a safe and oftentimes effective therapy that was Food and Drug Administration approved in 1999 for use for up to 14 days in term and near-term infants with PPHN. However, there are infants with pulmonary hypertension who need therapeutic approaches additional or alternative to inhaled NO. Infants who respond inadequately to inhaled NO could benefit from a treatment to augment NO responsiveness. Moreover, due to the difficulties incumbent with administering inhaled therapies, use of inhaled NO is problematic for infants requiring protracted treatments to reverse or prevent the progressive development of pulmonary vascular disease, such as infants with bronchopulmonary dysplasia. These latter infants would benefit from an effective therapy that can be easily administered long term, particularly in the outpatient setting. The safety profile of long-term use of oral sapropterin dihydrochloride is already established in human infants and children with metabolic disorders (29, 37, 49). Thus our findings provide the impetus to further explore oral sapropterin dihydrochloride supplementation as a treatment for infants suffering from a number of forms of pulmonary hypertension, especially those with chronic cardiopulmonary conditions associated with prolonged hypoxia.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grants RO1-HL-097566 (C. D. Fike) and R56-HL-097566 (C. D. Fike).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
A.E.D., J.L.A., M.S., and C.D.F. conception and design of research; A.E.D., M.R.K., and C.D.F. performed experiments; A.E.D., M.R.K., and C.D.F. analyzed data; A.E.D., J.L.A., M.S., and C.D.F. interpreted results of experiments; A.E.D., J.L.A., and C.D.F. edited and revised manuscript; A.E.D., J.L.A., M.R.K., M.S., and C.D.F. approved final version of manuscript; C.D.F. prepared figures; C.D.F. drafted manuscript.
ACKNOWLEDGMENTS
We thank BioMarin Pharmaceutical for the generous gift of the oral BH4 compound, sapropterin dihydrochloride.
REFERENCES
- 1.Abman SH. Monitoring cardiovascular function in infants with chronic lung disease of prematurity. Arch Dis Child 87: F15–F18, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ananthakrishnan M, Barr FE, Summar ML, Smith HA, Kaplowitz M, Cunningham G, Magarik J, Zhang Y, Fike CD. l-Citrulline ameliorates chronic hypoxia-induced pulmonary hypertension in newborn piglets. Am J Physiol Lung Cell Mol Physiol 297: L506–L511, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Banks BA, Seri I, Ischiropoulos H, Merrill J, Rychik J, Ballard RA. Changes in oxygenation with inhaled nitric oxide in severe bronchopulmonary dysplasia. Pediatrics 103: 610–618, 1999. [DOI] [PubMed] [Google Scholar]
- 4.Bendall JK, Douglas G, McNeill E, Channon KM, Crabtree MJ. Tetrahydrobiopterin in cardiovascular health and disease. Antioxid Redox Signal 20: 3040–3077, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brennan LA, Steinhorn RH, Wedgwood S, Mata-Greenwood E, Roark EA, Russell JA, Black SM. Increased superoxide generation is associated with pulmonary hypertension in fetal lambs: a role for NADPH oxidase. Circ Res 92: 683–691, 2003. [DOI] [PubMed] [Google Scholar]
- 6.Chalupsky K, Kracun D, Kanchev I, Bartram K, Gorlach A. Folic acid promotes recycling of tetrahydrobiopterin and protects against hypoxia-induced pulmonary hypertension by recoupling endothelial nitric oxide synthase. Antioxid Redox Signal 23: 1076–1091, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Consentino F, Hurlimann D, Gatti CD, Chenevard R, Blau N, Alp NJ, Channon KM, Eto M, Lerch P, Enseleit F, Ruschitzka F, Volpe M, Luscher TF, Noll G. Chronic treatment with tetrahydrobiopterin reverses endothelial dysfunction and oxidative stress in hypercholesterolaemia. Heart 94: 487–492, 2008. [DOI] [PubMed] [Google Scholar]
- 8.Crabtree MJ, Smith CL, Lam G, Goligorsky MS, Gross SS. Ratio of 5,6,7,8-tetrahydrobiopterin to 7,8-dihydrobiopterin in endothelial cells determines glucose-elicited changes in NO vs. superoxide production by eNOS. Am J Physiol Heart Circ Physiol 294: H1530–H1540, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Crabtree MJ, Tatham AL, Al-Wakeel Y, Warrick N, Hale AB, Cai S, Channon KM, Alp NJ. Quantitative regulation of intracellular endothelial nitric oxide synthase (eNOS) coupling by both tetrahydrobiopterin-eNOS stoichiometry and biopterin redox status: insights from cells with tet-regulated GTP cyclohydrolase I expression. J Biol Chem 284: 1136–1144, 2009. [DOI] [PubMed] [Google Scholar]
- 10.Dennis KE, Aschner JL, MIlatovic D, Schmidt JW, Aschner M, Kaplowitz MR, Zhang Y, Fike CD. NADPH oxidases and reactive oxygen species at different stages of chronic hypoxia-induced pulmonary hypertension in newborn piglets. Am J Physiol Lung Cell Mol Physiol 297: L596–L607, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dikalova A, Clempus R, Lassegue B, Cheng G, McCoy J, Dikalov S, Martin AS, Lyle A, Weber DS, Weiss D, Taylor WR, Schmidt HHW, Owens GK, Lambeth JD, Griendling KK. Nox 1 overexpression potentiates angiotensin II-induced hypertension and vascular smooth muscle hypertrophy in transgenic mice. Circulation 112: 2668–2676, 2005. [DOI] [PubMed] [Google Scholar]
- 12.Dikalova A, Fagiana A, Aschner JL, Aschner M, Summar M, Fike CD. Sodium-coupled neutral amino acid transporter 1 (SNAT 1) modulates l-citrulline transport and nitric oxide (NO) signaling in piglet pulmonary arterial endothelial cells. PLoS One 9: e85730, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dikalova A, Gongora MC, Harrison DG, Lambeth JD, Dikalov S, Griendling KK. Upregulation of Nox1 in vascular smooth muscle leads to impaired endothelium-dependent relaxation via eNOS uncoupling. Am J Physiol Heart Circ Physiol 299: H673–H679, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dikalova AE, Bikineyeva AT, Budzyn K, Nazarewicz RR, McCann L, Lewis W, Harrison DG, Dikalov SI. Therapeutic targeting of mitochondrial superoxide in hypertension. Circ Res 107: 106–116, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feillet F, Clarke L, Meli C, Lipson M, Morris AA, Harmatz P, Mould DR, Green B, Dorenbaum A, Giovannini M, Foehr E. Pharmacokinetics of sapropterin in patients with phenylketonuria. Clin Pharmacokinet 47: 817–825, 2008. [DOI] [PubMed] [Google Scholar]
- 16.Fike CD, Aschner JL, Zhang Y, Kaplowitz MR. Impaired NO signaling in small pulmonary arteries of chronically hypoxic newborn pigs. Am J Physiol Lung Cell Mol Physiol 286: L1244–L1254, 2004. [DOI] [PubMed] [Google Scholar]
- 17.Fike CD, Dikalova A, Kaplowitz MR, Cunningham G, Summar M, Aschner JL. Rescue treatment with l-citrulline inhibits hypoxia-induced pulmonary hypertension in newborn pigs. Am J Respir Cell Mol Biol 53: 255–264, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fike CD, Dikalova A, Slaughter JC, Kaplowitz MR, Zhang Y, Aschner JL. Reactive oxygen species reducing strategies improve pulmonary arterial responses to nitric oxide in piglets with chronic hypoxia-induced pulmonary hypertension. Antioxid Redox Signal 18: 1727–1738, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fike CD, Kaplowitz MR. Chronic hypoxia alters nitric oxide-dependent pulmonary vascular responses in lungs of newborn pigs. J Appl Physiol 81: 2078–2087, 1996. [DOI] [PubMed] [Google Scholar]
- 20.Fike CD, Kaplowitz MR. Effect of chronic hypoxia on pulmonary vascular pressures in isolated lungs of newborn pigs. J Appl Physiol 77: 2853–2862, 1994. [DOI] [PubMed] [Google Scholar]
- 21.Fike CD, Pfister SL, Kaplowitz MR, Madden JA. Cyclooxygenase contracting factors and altered pulmonary vascular responses in chronically hypoxic newborn piglets. J Appl Physiol 92: 67–74, 2002. [DOI] [PubMed] [Google Scholar]
- 22.Francis BN, Hale A, Channon KM, Wilkins MR, Zhao L. Effects of tetrahydrobiopterin oral treatment in hypoxia-induced pulmonary hypertension in rat. Pulm Circ 4: 462–470, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fresquet F, Pourageaud F, Leblais V, Brandes RP, Savineau JP, Marthan R, Muller B. Role of reactive oxygen species and gp91 phox in endothelial dysfunction of pulmonary arteries induced by chronic hypoxia. Br J Pharmacol 148: 714–723, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gielis JF, Lin JY, Wingler K, Van Schil PE, Schmidt HH, Moens AL. Pathogenetic role of eNOS uncoupling in cardiopulmonary disorders. Free Radic Biol Med 50: 765–776, 2011. [DOI] [PubMed] [Google Scholar]
- 25.Gryglewski RJ, Palmer RMJ, Moncada S. Superoxide anion is involved in the breakdown of endothelium-derived vascular relaxing factor. Nature 320: 454–456, 1986. [DOI] [PubMed] [Google Scholar]
- 26.Heitzer T, Krohn K, Albers S, Meinertz T. Tetrahydrobiopterin improves endothelium-dependent vasodilation by increasing nitric oxide activity inpatients with type II diabetes. Diabetologia 43: 1435–1438, 2000. [DOI] [PubMed] [Google Scholar]
- 27.Kase H, Hashikabe Y, Uchida K, Nakanishi N, Hattori Y. Supplementation with tetrahydrobiopterin prevents the cardiovascular effects of angiotensin II-induced oxidative and nitrosative stress. J Hypertens 23: 1375–1382, 2005. [DOI] [PubMed] [Google Scholar]
- 28.Katusic ZS, d'Uscio LV, Nath KA. Vascular protection by tetrahydrobiopterin: progress and therapeutic prospects. Trends Pharmacol Sci 30: 48–54, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Keil S, Anjema K, van Spronsen FJ, Lambruschini N, Burlina A, Belanger-Quintana A, Couce ML, Feillet F, Cerone R, Lotz-Havla AS, Muntau AC, Bosch AM, Meli CA, Billette de Villemeur T, Kern I, Riva E, Giovannini M, Damaj L, Leuzzi V, Blau N. Long-term follow-up and outcome of phenylketonuria patients on sapropterin: a retrospective study. Pediatrics 131: e1881–e1888, 2013. [DOI] [PubMed] [Google Scholar]
- 30.Khemani E, McElhinney DB, Rhein L, Andrade O, Lacro RV, Thomas KC, Mullen MP. Pulmonary artery hypertension in formerly premature infants with bronchopulmonary dysplasia: clinical features and outcomes in the surfactant era. Pediatrics 120: 1260–1269, 2007. [DOI] [PubMed] [Google Scholar]
- 31.Khoo JP, Zhao L, Alp NJ, Bendall JK, Nicoli T, Rockett K, Wilkins MR, Channon KM. Pivotal role for endothelial tetrahydrobiopterin in pulmonary hypertension. Circulation 111: 2126–2133, 2005. [DOI] [PubMed] [Google Scholar]
- 32.Kietadisorn R, Juni RP, Moens AL. Tackling endothelial dysfunction by modulating NOS uncoupling: new insights into its pathogenesis and therapeutic possibilities. Am J Physiol Endocrinol Metab 302: E481–E495, 2012. [DOI] [PubMed] [Google Scholar]
- 33.Kim HK, Ha SH, Han J. Potential therapeutic applications of tetrahydrobiopterin: from inherited hyperphenylalaninemia to mitochondrial diseases. Ann NY Acad Sci 1201: 177–182, 2010. [DOI] [PubMed] [Google Scholar]
- 34.Klatt P, Schmidt K, Lehner D, Glatter O, Bachinger HP, Mayer B. Structural analysis of porcine brain nitric oxide synthase reveals a role for tetrahydrobiopterin and l-arginine in the formation of an SDS-resistant dimer. EMBO J 14: 3687–3695, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Klinger JR, Abman SH, Gladwin MT. Nitric oxide deficiency and endothelial dysfunction in pulmonary arterial hypertension. Am J Respir Crit Care Med 188: 639–646, 2013. [DOI] [PubMed] [Google Scholar]
- 36.Landmesser U, Dikalov S, Price SR, McCann L, Fukai T, Holland SM, Mitch WE, Harrison DG. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J Clin Invest 111: 1201–1209, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leuret O, Barth M, Kuster A, Eyer D, Parseau Ld Odent S, Gilgert-Dussardier B, Feillet F, Labarthe F. Efficacy and safety of BH4 before the age of 4 years inpatients with mild phenylketonuria. J Inherit Metab Dis 35: 975–981, 2012. [DOI] [PubMed] [Google Scholar]
- 38.Liu Q, Gross SS. Binding sites of nitric oxide synthases. Methods Enzymol 268: 311–324, 1996. [DOI] [PubMed] [Google Scholar]
- 39.Lonnqvist PA, Jonsson B, Winberg P, Frostell CG. Inhaled nitric oxide in infants with developing or established chronic lung disease. Acta Paediatr 84: 1188–1192, 1995. [DOI] [PubMed] [Google Scholar]
- 40.Maier W, Cosentino F, Lutolf RB, Fleisch M, Seiler C, Hess OM, Meier B, Luscher TF. Tetrahydrobiopterin improves endothelial function in patients with coronary artery disease. J Cardiovasc Pharmacol 35: 173–178, 2000. [DOI] [PubMed] [Google Scholar]
- 41.Manea A, Tanase LI, Raicu M, Simionescu M. Transcriptional regulation of NADOPH oxidase isoforms, Nox 1 and Nox 4, by nuclear factor-kappaB in human aortic smooth muscle cells. Biochem Biophys Res Commun 396: 901–907, 2010. [DOI] [PubMed] [Google Scholar]
- 42.Mayahi L, Heales S, Owen D, Casas JP, Harris J, MacAllister RJ, Hingorani AD. (6R)-5,6,7,8-tetrahydro-l-biopterin and its stereosiomer prevent ischemia reperfusion injury in human forearm. Arterioscler Thromb Vasc Biol 27: 1334–1339, 2007. [DOI] [PubMed] [Google Scholar]
- 43.Meier U. A note on the power of Fisher's least significant difference procedure. Pharm Stat 5: 253–263, 2006. [DOI] [PubMed] [Google Scholar]
- 44.Moens AL, Ketner EA, Takimoto E, Schmidt TS, O'Neill CA, Wolin MS, Alp NJ, Channon KM, Kass DA. Bi-modal dose-dependent cardiac response to tetrahydrobiopterin in pressure-overlaid induced hypertrophy and heart failure. J Mol Cell Cardiol 51: 564–569, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mourani PM, Abman SH. Pulmonary vascular disease in bronchopulmonary dysplasia: pulmonary hypertension and beyond. Curr Opin Pediatr 25: 329–337, 2013. [DOI] [PubMed] [Google Scholar]
- 46.Porkert M, Sher S, Reddy U, Cheema F, Niessner C, Kolm P, Jones DP, Hooper C, Taylor WR, Harrison D, Quyyumi AA. Tetrahydrobiopterin: a novel antihypertensive therapy. J Hum Hypertens 22: 401–407, 2008. [DOI] [PubMed] [Google Scholar]
- 47.Robbins IM, Hemnes AR, Gibbs JS, Christman BW, Howard L, Meehan S, Cabrita I, Gonzalez R, Oyler T, Zhao L, Du RH, Mendes LA, Wilkins MR. Safety of sapropterin dihydrochloride (6r-bh4) in patients with pulmonary hypertension. Exp Lung Res 37: 26–34, 2011. [DOI] [PubMed] [Google Scholar]
- 48.Shinozaki K, Nishio Y, Okamura T, Yoshida Y, Maegawa H, Kojima H, Masada M, Toda N, Kikkawa R, Kashiwagi A. Oral administration of tetrahydrobiopterin prevents endothelial dysfunction and vascular oxidative stress in the aortas of insulin-resistant rats. Circ Res 87: 566–573, 2000. [DOI] [PubMed] [Google Scholar]
- 49.Shintaku H, Ohura T. Sapropterin is safe and effective in patients less than 4 years old with BH4 responsive phenylalanine hydroxylase deficiency. J Pediatr 265: 1241–1244, 2014. [DOI] [PubMed] [Google Scholar]
- 50.Stenmark K, Abman S. Lung vascular development: implications for the pathogenesis of bronchopulmonary dysplasia. Annu Rev Physiol 67: 623–661, 2005. [DOI] [PubMed] [Google Scholar]
- 51.Stuehr D, Pou S, Rossen GM. Oxygen reduction by nitric oxide synthases. J Biol Chem 276: 14533–14536, 2001. [DOI] [PubMed] [Google Scholar]
- 52.Tabima DM, Frizezell S, Gladwin MT. Reactive oxygen and nitrogen species in pulmonary hypertension. Free Radic Biol Med 52: 1970–1986, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vasquez-Vivar J, Kalyanaraman B, Martasek P, Hogg N, Masters BS, Karoui H, Tordo P, Pritchard KA Jr. Superoxide generation by endothelial nitric oxide synthase: the influence of cofactors. Proc Natl Acad Sci U S A 95: 9220–9225, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vasquez-Vivar J, Martasak P, Whitsett J, Joseph J, Kalyanaraman B. The ratio between tetrahydrobiopterin and oxidized tetrahydrobiopterin analogues controls superoxide release form endothelial nitric oxide synthase: an EPR spin trapping study. Biochem J 362: 733–739, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wever RM, van Dam T, van Rijn HJ, de Groot F, Rabelink TJ. Tetrahydrobiopterin regulates superoxide and nitric oxide generation by recombinant endothelial nitric oxide synthase. Biochem Biophys Res Commun 237: 340–344, 1997. [DOI] [PubMed] [Google Scholar]
- 56.Xia Y, Tsai A, Berka V, Zweier JL. Superoxide generation from endothelial nitric-oxide synthase. J Biol Chem 273: 25804–25808, 1998. [DOI] [PubMed] [Google Scholar]