

Abstract
This study evaluated the pulmonary pathophysiology of pigs with transgenic CFTR “gut-corrected” cystic fibrosis (CF). Four sows produced 18 piglets of which 11 were stillborn, with only 2 animals surviving beyond 2 wk. Failure to survive beyond the neonatal period by five piglets was judged to result from metabolic dysfunction related to genetic manipulation for CFTR gut expression or due to cloning artifact. Plasma analysis showed very low plasma proteins, highly elevated liver enzymes, and severe acidosis. All surviving offspring received furosemide for systemic edema. Physiologic evaluation was performed with lung tissues from the two surviving pigs. Both acetylcholine and forskolin induced mucous liquid secretion that was significantly lower in bronchi of pigs with CF than those without CF. The percent of nonvolatile solids in mucus secreted from CF bronchi was elevated following acetylcholine or forskolin treatment. Mucociliary transport in excised tracheas was reduced in the CF tracheas relative to non-CF tracheas. The diameter of tracheas in pigs with CF was less than that of pigs without CF despite their greater body weight. Despite exhibiting severe metabolic dysfunction during the neonatal period, this CF animal model appears to express important characteristics of human CF pulmonary disease.
Keywords: pig, airway, CFTR, cystic fibrosis, pathophysiology
cystic fibrosis is a lethal, autosomal recessive genetic disease in humans that is caused by loss of function mutations in the gene that codes for the cystic fibrosis (CF) transmembrane conductance regulator (CFTR) (13, 21, 23). The CFTR normally functions in a variety of tissues as a channel that conducts Cl− ions (20) and HCO3− ions (19, 24). All CF-related pathology is linked in some way to loss of anion transport by this protein. Multiple organ systems are affected in CF disease. The gastrointestinal manifestations of CF include meconium ileus in newborns, distal intestinal obstruction syndrome in older patients, pancreatic insufficiency, and focal biliary cirrhosis (6). With appropriate and timely medical treatment, these gastrointestinal complications can be successfully managed for decades. Reproduction is affected in CF disease in that men are sterile due to atrophy of the vas deferens, and women have reduced fertility due to thickened cervical mucus (6). Mortality in most patients with CF is caused by chronic pulmonary disease that includes mucus plugging of airways, colonization of the lung with opportunistic bacteria, development of bronchiectasis, and progressive loss of lung function (6). Historically, the lack of appropriate animal models that faithfully reproduce the unique pattern of human CF pulmonary pathology has hindered our understanding of CF disease pathogenesis and the development of effective treatments for CF lung disease.
The first genetically engineered animal model of CF was the CFTR gene-knockout mouse (25). Unlike human patients with CF, all mice with CF develop severe intestinal obstruction at weaning that results in significant mortality (25). Addition of polyethylene glycol to the drinking water significantly ameliorates mortality due to intestinal obstruction in the CF mouse model (8). Zhou et al. (32) found that introduction of a functional murine CFTR gene sequence into the CF mouse genome linked to an intestinal fatty acid binding protein promoter that resulted in expression of functional CFTR only in intestinal tissues was successful at reducing mortality due to intestinal obstruction. Unfortunately, CFTR-knockout mice eventually proved to be of limited usefulness for developing therapeutic treatments for CF because they do not develop the characteristic lung disease observed in human patients with CF (26) that is responsible for mortality in the great majority of humans with CF.
More recently, genetic models of CF in higher mammals have been developed. CFTR gene-knockout models of both domestic pigs (22) and ferrets (29) express the progressive mucus obstruction of airways that appears to mirror that in human CF disease. However, both pig and ferret models also exhibit severe, life-threatening meconium ileus at birth (18, 29). In CFTR gene-knockout pigs, severe meconium ileus developed in all newborns within the first few days of life (22). Some newborn pigs with CF with meconium ileus can be rescued by invasive gastrointestinal surgery to correct the obstruction; however, this intervention is labor-intensive and frequently unsuccessful. To improve the survival rate of neonatal pigs with CF, Stoltz and colleagues (27) generated pigs with CF using the CFTR “gut-correction” approach that was developed for mice with CF. In their initial publication, Stoltz et al. (27) reported that the pigs with CFTR gut-corrected CF exhibited a less severe gastrointestinal disease phenotype and survived to develop significant lung disease within weeks of birth. Additionally, the bioelectric properties of airway epithelia from these pigs with CFTR gut-corrected CF mirrored the responses of human CF airways by failing to respond to the CFTR-activating agonists forskolin and 3-isobutyl-1-methylxanthine (IBMX), indicating very low or absent CFTR expression. In older pigs with gut-corrected CF, the authors noted liver changes ranging from focal biliary cirrhosis to multilobular cirrhosis (27).
Based on the promising report by Stoltz et al. (27), we chose this CFTR gut-corrected CF pig model to evaluate epithelial Cl− and HCO3− secretion, mucus liquid secretion, and mucociliary transport in CF airways, processes whose corruption is likely responsible for CF lung pathogenesis in humans. Although we found evidence that anion secretion, mucous liquid secretion, and mucociliary transport were abnormal in the airways of pigs with CFTR gut-corrected CF, our studies were unexpectedly confounded by very high neonatal mortality, severe systemic edema, and cardiovascular and hepatic abnormalities in newborn pigs. These complications were not described by Stoltz et al. in their initial communication (27) and were not disclosed to us by Exemplar Genetics, the commercial vendor for this model. Herein, we document our experiences and findings with this model of CF disease.
MATERIALS AND METHODS
Animals.
All procedures involving animals were reviewed and approved by the University of South Alabama Institutional Animal Care and Use Committee. Wild-type mature Yorkshire gilts implanted with cloned “gut-corrected” CF embryos (CFTR−/−; TgFABP > pCFTR), generated as previously described by Stoltz et al. (27), were purchased from Exemplar Genetics (Sioux Center, IA) and transported to our research facility ∼2 wk before term. These animals were housed according to the Guide for the Care and Use of Laboratory Animals (8th ed., Washington, DC: National Academies Press) and the Guide for the Care and Use of Agricultural Animals in Research and Teaching (3rd ed., Champaign, IL: Federation of Animal Science Societies). All animals were fed commercial pig chow (Harlan) and a variety of fruits and vegetables twice daily. Routine husbandry included twice-daily cleaning, enrichment, and interaction with caregivers. The gilts were observed via web camera every 3 h, 24 h/day as they approached 115 days of gestation. Wild-type Yorkshire pigs (approximately 5–8 wk old and 20–25 lb body wt) were obtained from the Auburn University Swine Research and Education Center (Auburn, AL) and served as non-CF control animals. At the time of the animals' death, non-CF pigs weighed in the range of 20–35 lb.
At or near term, delivery of the newborns with CF was induced following an established protocol after 115 days of gestation (12), and the young were born via assisted vaginal delivery or by terminal Cesarean delivery under isoflurane anesthesia. None of the four sows used in this study exhibited normal parturition. Eighteen piglets were produced by the four sows (Table 1). Eleven of the piglets with CF were dead at delivery and seven were born alive. Despite intensive rescue efforts including intubation, positive-pressure ventilation, holding in Thermocare isolets, intravenous catheterization, and nasogastric tube feeding, only two of the seven piglets with CF that were born alive survived beyond 13 days (Table 1).
Table 1.
Neonatal survival statistics for pigs with gut-corrected CF*
| Litter | Stillborn | Viable | Disposition of Viable Piglets |
|---|---|---|---|
| 1 | 4 | 1 | Piglet survived to 61 days (16.8 kg) and was killed for experimental end point |
| 2 | 1 | 3 | Piglet 1 survived to 4 days of age and was killed because it had congenital anomalies (large head and tongue, small eyes) and failure to thrive |
| Piglet 2 survived to 13 days and was humanely killed due to inability to successfully treat edema and subsequent right heart failure | |||
| Piglet 3 survived to 55 days (17.2 kg) and was humanely killed after development of dyspnea and edema that could not be clinically managed | |||
| 3 | 2 | 3 | Piglet 1 was never responsive and died within 1 h of birth despite efforts to revive it |
| Piglet 2 was never responsive and died within 2 h of birth despite efforts to revive it | |||
| Piglet 3 was never responsive but stabilized with intensive care, including ventilation; the piglet died 3 h after birth and was observed to be edematous | |||
| 4 | 4 | 0 |
Percent survival beyond 13 days, 11.1%.
To procure tissues for study from the two surviving pigs with CF and the pigs without CF, the animals were first sedated with intramuscular Telazol (tilemine HCl and zolazepam HCl) and ketamine, then killed by pentobarbital overdose administered through an ear vein. A thoracotomy was performed, and the heart, trachea, and lungs were removed.
Measurement of mucociliary transport.
Mucociliary transport was measured using a technique previously described (2, 4). Tracheas were removed intact by sectioning the tracheas just below the larynx and just above the first bronchial branch, which in swine is to the right cranial lobe above the carina. The tracheas were each placed in room-temperature Krebs solution that was constantly bubbled with 95% O2/5% CO2 gas and gradually warmed to 37°C over ∼1 h. The posterior trachealis muscle was then resected to the length of the tracheas. The tracheas were then tied onto cannulas, which were mounted in a specially designed rack with the slot formed by the muscle resection facing uppermost [see online supplement in Ref. 2]. The rack held the tissues horizontal in a bath of warm (37°C) Krebs solution that was constantly bubbled with 95% O2/5% CO2 gas. The bath solution was of sufficient depth to hydrate the tracheal adventitia without contaminating the native airway surface liquid in the tracheal lumen. In this configuration, the ventral mucosal surface was easily viewed through a removable, warmed, tempered glass chamber lid and the slot created by the trachealis muscle resection. The tracheas were allowed to stabilize for 60 min, after which airway liquid that accumulated at the cranial end was removed and discarded. To measure mucociliary transport, a few small flakes of dried India ink were placed on the tracheal mucosa at the caudal end, and the movement of the ink flakes was visualized with an analog video camera and recorded on videotape. A scale was placed on the rack, alongside the tracheas, to track the position of the ink particles. Particle velocities were determined later by offline analysis of the videotape images displayed on a conventional video monitor.
Collection and analysis of mucous liquid.
Mucous liquid was generated from intrapulmonary bronchi that were dissected intact from the lung parenchyma. Side branches of the bronchi were ligated with suture, and the airways were then immersed in room-temperature Krebs solution that was constantly aerated with 95% O2/5% CO2 gas. The Krebs bath solution was gradually warmed to 37°C over 1 h. The bronchi were then removed from the Krebs bath, and all accessible luminal fluid was aspirated from the airway lumens. The bronchi were then tied onto polyethylene cannulas and returned to the Krebs bath. At this time, either acetylcholine (ACh) or forskolin was added to bath to induce mucous liquid secretion. Bronchi that were not treated with either agonist served as controls. Following a 2-h exposure to the agonists, the bronchi were removed from the bath and the cannulas, the bronchi were resected, and all accessible luminal mucous liquid was removed and weighed to determine its mass and volume (assuming that 1 mg = 1 μl). The rate of mucous liquid secretion was determined from the volume collected from each airway and normalized to the time of agonist exposure and the airway mucosal surface area. The airway mucosal surface area of each bronchus was calculated from the airway dimensions as previously described (30). The percent solids of the mucous liquid was determined by drying the mucus samples overnight in an 80°C oven and expressing the mucus sample dry weight as a percentage of its wet weight.
Solutions and chemicals.
Krebs, HCO3−-buffered, physiological salt solution contained 112 mM NaCl, 25 mM NaHCO3, 11.6 mM glucose, 4.7 mM KCl, 2.5 mM CaCl2, 2.4 mM MgSO4, and 1.2 mM KH2PO4. Krebs solution was bubbled with 95% O2/5% CO2 gas to maintain the solution pH at ∼7.4. DMSO was used as the vehicle for forskolin. Telazol was purchased from Patterson Veterinary Supply (Devens, MA). Ketamine was purchased from Cardinal Health (Dublin, OH). Pentobarbital sodium was obtained from Vortech Pharmaceuticals (Dearborn MI). All other chemicals and drugs were purchased from Sigma-Aldrich (St. Louis, MO).
RESULTS
Pre- and postmortem observations of pigs with CF.
Four litters of piglets with CF were produced (Table 1). Neonatal mortality at birth was very high. From the total of 18 piglets produced, 11 were born dead and 7 were alive at birth. Of the piglets that were born alive, 3 died within 3 h, 1 died at 4 days, and 1 died at 13 days. Two piglets survived to ∼2 mo of age (55 and 60 days), when they were both killed for physiological evaluation. Thus, the neonatal survival rate of the piglets with CF beyond 2 wk was 11.1%. Piglets that died before 13 days exhibited systemic edema, labored breathing, pneumonia, jaundice, and meconium ileus. Four piglets were intubated and ventilated. Although transient meconium ileus was observed in all piglets (Fig. 1), it did not appear to affect neonatal viability. All seven live-born piglets passed feces and were not obstructed. Live-born piglets received iron and colostrum replacement at birth. All live-born animals were hypoglycemic and required oral or intravenous dextrose to maintain serum glucose during the first 24 h. Plasma chemical analysis was performed on one piglet with CF that died 3 h postpartum (Table 2). That animal was profoundly hypoproteinemic with highly elevated aspartate aminotransferase and creatine kinase, and low gamma-glutamyltransferase levels. A large anion gap and low HCO3− concentration was also evident. This pattern is indicative of acidosis and compromised liver function. Hypoproteinemia was judged to be the likely cause of the systemic edema that affected all the pigs with CF. Evidence of extracellular volume expansion is apparent in Fig. 2, where the septal spaces in the lung of a pig with CF are widened and prominent compared with the lung of a normal pig. High glucose levels (Table 2) were likely due to intravenous dextrose administration during resuscitation and stress-induced hyperglycemia. All pigs with CF had noticeably enlarged hearts (Fig. 3). The underlying cause for cardiomegaly was likely to have been a consequence of the expanded extracellular volume and development of pulmonary hypertension. Klebsiella pneumoniae, Enterococcus faecalis, Streptococcus suis, and coagulase-negative Staphylococcus were identified in pleural fluid from deceased piglets. Positive Dinese stain indicated the presence of mycoplasma in the lung of one deceased piglet.
Fig. 1.

Newborn pig with “gut-corrected” cystic fibrosis (CF). Note the string-of-pearls appearance of the meconium, which is commonly observed in human newborns with CF disease. Several of the newborn pigs with CF exhibited this clinical sign, but none appeared to have life-threatening obstructive meconium ileus.
Table 2.
Plasma analysis of a pig with CF*
| Test | Result | Reference Values† |
|---|---|---|
| Total protein | 1.29 g/dl | 5.8–8.3 g/dl |
| Albumin | 0.4 g/dl | 2.3–4.0 g/dl |
| Globulin | 0.9 g/dl | 3.9–6.0 g/dl |
| Albumin/Globulin ratio | 0.4 | |
| Sorbitol dehydrogenase | 34.8 U/l | 0.5–4.9 U/l |
| Aspartate amino transferase | 137 U/l | 15–55 U/l |
| Gamma-glutamyltransferase | 18 U/l | 31–52 U/l |
| Bilirubin | 0.11 mg/dl | 0–0.5 mg/dl |
| Creatine kinase | 1,988 U/l | 66–489 U/l |
| Blood urea nitrogen | 11.3 mg/dl | 8.2–25 mg/dl |
| Creatinine | 1.3 mg/dl | 0.8–2.3 mg/dl |
| Calcium | 14.9 mg/dl | 9.3–11.5 mg/dl |
| Phosphorus | 24.0 mg/dl | 5.5–9.3 mg/dl |
| Magnesium | 3.7 mg/dl | 2.3–3.5 mg/dl |
| Glucose | 259 mg/dl | 66–116 mg/dl |
| Bicarbonate | 13.8 mM/l | 18–27 mM/l |
| Sodium | 146 mM/l | 139–153 mM/l |
| Potassium | 8.3 mM/l | 4.4–6.5 mM/l |
| Chloride | 98 mM/l | 97–106 mM/l |
| Anion gap (Na+ + K+) − (Cl− + HCO3−) | 42.5 mM/l | |
| Osmolality (calculated) | 309 mOsm/kg |
Pig died 3 h postpartum, blood collected postmortem.
Latimer et al. (14)
Fig. 2.

Evidence of expanded extracellular space in the lung of a pig with gut-corrected CF. A: lung of a pig with gut-corrected CF. The septal spaces between the lung lobules are widened and prominent. B: lung of a normal pig without CF. No evidence of overt edema or enlarged septal space is apparent.
Fig. 3.

Cardiomegaly in a neonatal pig with gut-corrected CF. A: a pig with gut-corrected CF. Note the large size of the heart, which spans the width of the thoracic cavity. B: the heart of a normal pig without CF. The heart in this 10-kg pig is of normal proportions.
The two piglets that survived to a mean of 58 days were able to maintain normal O2 saturation within 2 h of birth without ventilation. Systemic edema, a persistent issue with surviving pigs, was controlled with furosemide. Both pigs were given daily Bactrim (STI Pharma), nystatin, cimetidine, iron-containing multivitamin, and Zegerid (Salix Pharmaceuticals) by mouth. They were also given PancreVed (Vedco) mixed with milk-replacer every other feeding, and their diets were supplemented with Ensure (Abbott), yogurt, and fresh fruit. At the time of death, those animals weighed 16.8 and 17.2 kg, respectively.
Before CF pig 1 died it showed no signs of dyspnea at rest. Following thoracotomy, the lungs of the animal appeared unusually white in contrast to the typical pinkish coloration observed in lungs of normal, healthy pigs. This was judged to be a consequence of the very low hematocrit. Gross appearance of the lungs was otherwise unremarkable with no obvious consolidation. The larynx was unusually narrow in appearance, as if it had been pinched in from the sides. The diameter of the trachea was notably small relative to the size of the animal. In cross-section, it was apparent that the ends of the cartilage rings that met the smooth muscle at the posterior aspect of the trachea curled inward toward the lumen forming a ridge of cartilage that ran along its full length. Together with the unusually narrow trachea, airflow through the upper airways was likely restricted, thereby contributing to the dyspnea that was observed in the animals during exertion. Large bronchi were rigid and somewhat difficult to dissect. The distal ends of the bronchi were fragile and easily damaged during dissection. CF pig 2 exhibited pronounced labored breathing that was exacerbated by mild exercise. It was judged at the time the animal was killed that it may have been in respiratory distress, but labored breathing was probably due to the structural narrowing of the airways. Similar to that of CF pig 1, the trachea was extremely narrow in this animal. As in CF pig 1, cartilage malformation was apparent in the trachea, where inwardly curled cartilage formed a ridge inside the tracheal lumen. The trachealis muscle had a pronounced zigzag appearance the length of the trachea.
The gross appearance of the lungs from CF pig 2 was also normal except for the very white appearance, again the likely consequence of anemia. The bronchi generally appeared normal although bronchiectasis was noted in some bronchi in an upper lobe. One mainstem bronchus was “floppy,” indicative of abnormal cartilage. Compared with the upper and middle lobes, bronchi in the lower lobes appeared to have extensive branching that made dissection more difficult. In some airways, it was very difficult to separate the pulmonary vessels from the bronchi. This appeared to be due to increased vascularization. Indeed, bronchial arteries and veins were prominent and visibly enlarged, relative to normal pigs, which suggests chronic inflammation and/or infection in the cartilaginous airways. The heart in this animal was very large (115 g) for its size (17.3 kg) (Fig. 3).
Pathophysiology.
Dissection and cannulation of intrapulmonary bronchi for measurement of mucous liquid secretion required lung tissues from animals that were at least 3–4 wk of age. Because of the very high mortality in the newborn pigs with CF, suitable airway tissues for physiological experiments could be obtained only from the two oldest surviving animals. Consequently, we could not generate enough CF tissues to allow full statistical analyses to be performed for all treatments. For treatments in which CF pig measurements consisted of only one or two samples, comparisons are made with non-CF tissues, where 95% confidence intervals were determined.
The rates of mucous liquid secretion by cannulated bronchi from pigs with and without CF are shown in Fig. 4. For these experiments, multiple bronchi were excised from both of the pigs with CF. Aspiration of mucous liquid from CF bronchi prior to cannulation, as well as collection of mucus from the bronchial lumens at the completion of liquid secretion experiments, was difficult, especially following exposure to ACh. Mucus appeared to be adhered to the airway surfaces in these animals. Under basal unstimulated conditions, bronchi from pigs with CF secreted significantly less mucous liquid than non-CF bronchi (non-CF, 1.50 ± 0.13 μl·cm2·h−1, n = 19; CF, 0.80 ± 0.13 μl·cm2·h−1, n = 5; P < 0.05). When stimulated with 10 μM ACh, CF bronchi secreted significantly less mucous liquid than non-CF bronchi (non-CF, 7.48 ± 0.13 μl·cm2·h−1, n = 6; CF, 2.33 ± 1.19 μl·cm2·h−1, n = 4; P < 0.05). When stimulated with forskolin, non-CF bronchi secreted 4.63 ± 0.40 μl·cm2·h−1 (n = 16), whereas CF bronchi secreted 0.91 ± 0.18 μl·cm2·h−1 (n = 4), which was significantly lower (P < 0.05).
Fig. 4.
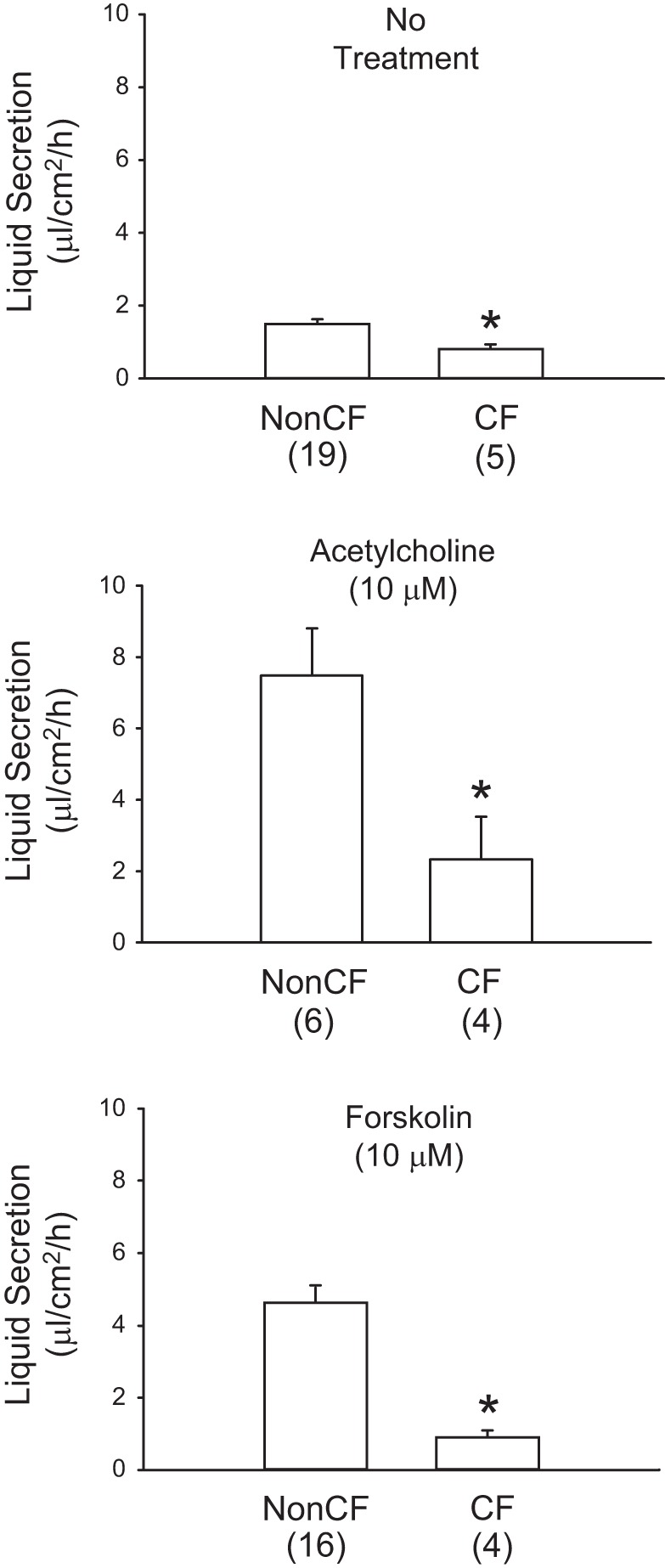
Mucous liquid secretion responses of gut-corrected CF and non-CF porcine bronchi. Bronchi, dissected as intact tubes from pig lungs, were cannulated and immersed in Krebs solution at 37°C. Bronchi were treated with the indicated secretogogue. Secreted mucous liquid was collected from the airway lumens after 2 h and weighed to determine the volume. Secretion rates were calculated and normalized to airway surface area and time. Bars represent means ± SE. *Significant difference (P < 0.05) from non-CF airways. Number of individual airways in each group is shown in parentheses.
The percentage of nonvolatile solids in the bronchial mucous liquid of pigs with and without CF are shown in Fig. 5. Untreated non-CF bronchi secreted mucous liquid with 7.48 ± 0.41% solids (n = 17). Non-CF bronchi that were treated with 10 μM ACh secreted mucous liquid with 3.21 ± 0.22% solids (n = 5), whereas non-CF bronchi that were treated with 10 μM forskolin secreted mucous liquid with 4.41 ± 1.5% solids (n = 15). Although the percent of solids measured in the mucus from bronchi of pigs with CF represent only single measurements for each of the three groups (no treatment, 10.41% solids; 10 μM ACh treatment, 9.38% solids; and 10 μM forskolin treatment, 6.02% solids), these single values each fell well outside the 95% confidence intervals for the three treatment groups of non-CF bronchi. Thus, it is likely that the bronchial mucus from these gut-corrected pigs with CF contain a higher percentage of nonvolatile solids than mucus from non-CF pig bronchi.
Fig. 5.
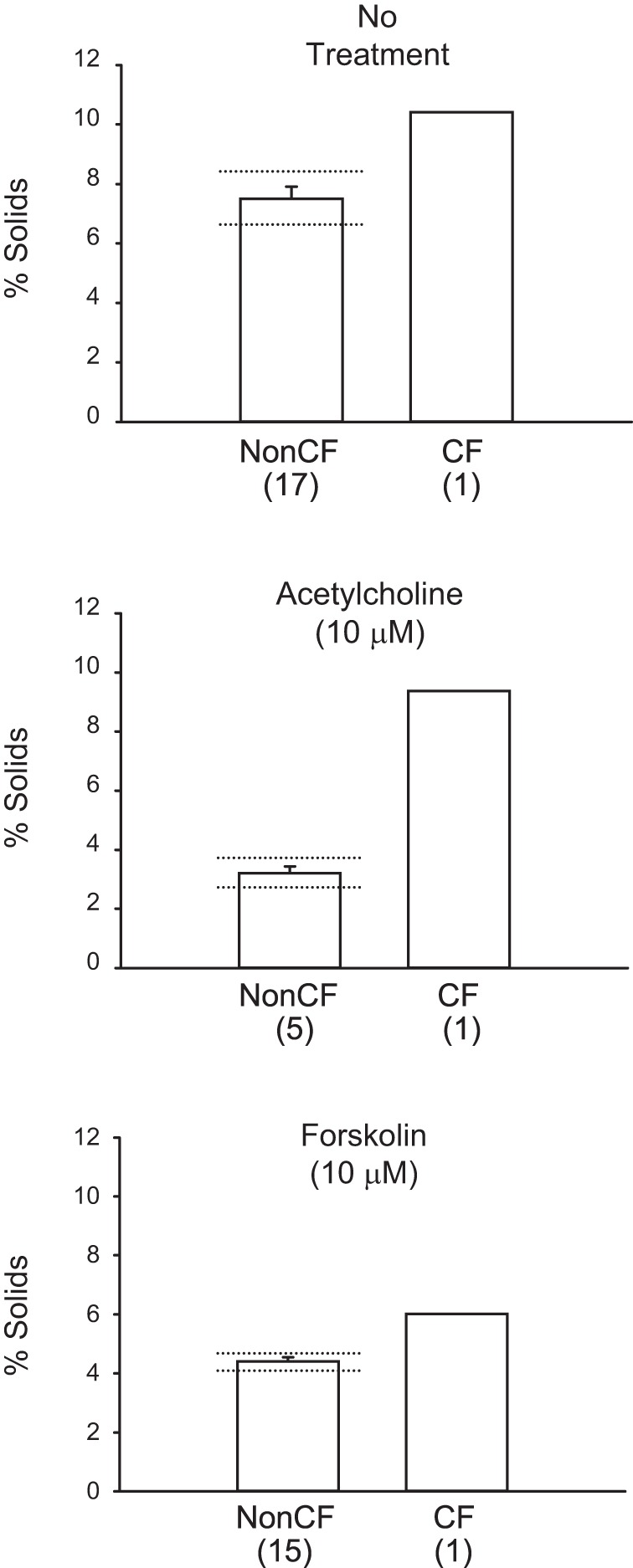
Percent of nonvolatile solids content of mucous liquid secreted by gut-corrected CF and non-CF porcine bronchi. Percent solids are shown for each of the indicated treatments. Number of individual airways are shown in parentheses. Means ± SE are shown for non-CF bronchi, whereas only single samples were obtained for bronchi from pigs with CF in each treatment group. Dotted horizontal lines (above and below bars) indicate the 95% confidence intervals for non-CF percent solids in each group. All three single values for mucous liquid percent solids from the CF bronchi fell well outside the 95% confidence intervals for non-CF bronchi responses.
Baseline and ACh-induced rates in mucociliary transport for non-CF tracheas (n = 13) and CF tracheas (n = 2) are shown in Fig. 6. For the three 20-min pre-ACh periods, the rates of mucociliary transport were low and comparable between the non-CF and CF tracheas. In non-CF tracheas, pre-ACh mucociliary transport rates were 1.71 ± 0.56, 1.19 ± 0.31, and 1.54 ± 0.41 mm/min for the 0–20, 20–40, and 40–60 min control periods, respectively. For the two CF tracheas, pre-ACh mucociliary transport rates were 4.42 and 2.87, 0.30 and 0.06, and 1.05 and 0.06 mm/min, respectively, for the same time periods. Following ACh addition (100 μM) to the bath solution, the mucociliary transport rates were substantially and significantly (P < 0.05) increased in the non-CF tracheas (Fig. 6). Mucociliary transport rates after ACh for non-CF tracheas were 13.83 ± 0.97, 15.29 ± 1.49, and 15.10 mm/min, respectively for the 70–90, 90–110, and 110–130 min periods, respectively. The mucociliary transport rates for the two CF tracheas after ACh were 2.16 and 10.02, 2.62 and 11.37, and 2.47 and 11.15 mm/min, respectively, for the same time periods. Mucociliary transport rates for both of the two CF tracheas were outside of the lower 95% confidence limits for the non-CF tracheal responses for each of the three post-ACh time intervals (12.00 mm/min for 70–90 min, 12.48 mm/min for 90–110 min, and 13.54 mm/min for 110–130 min).
Fig. 6.
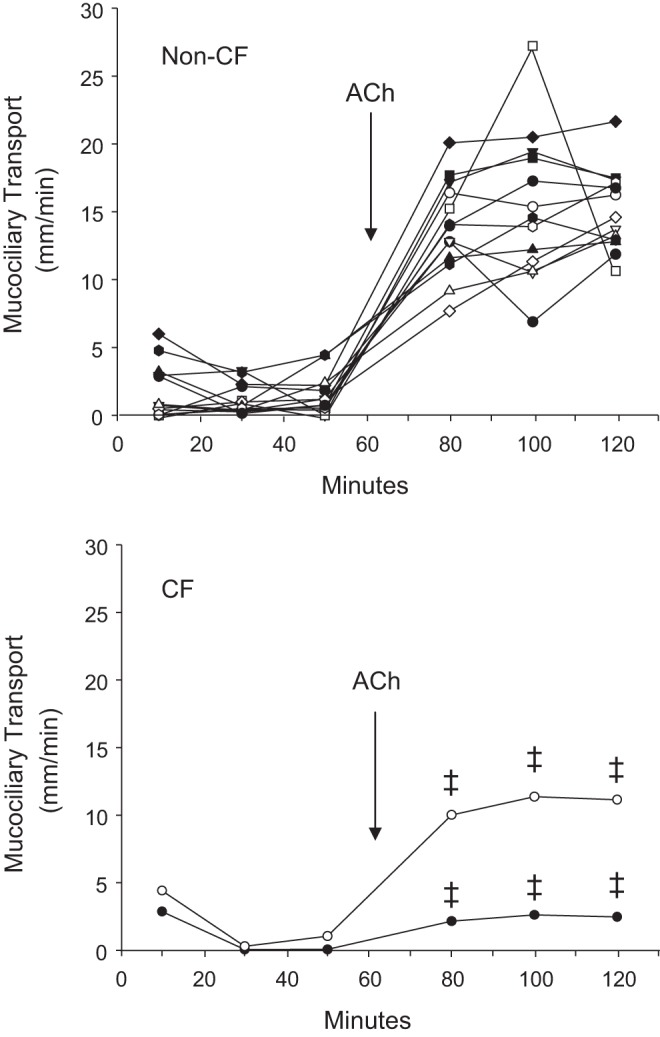
Effect of acetylcholine on mucociliary transport by gut-corrected CF and non-CF pig tracheas. Basal rates of mucociliary transport in non-CF tracheas were measured in individual tissues over three consecutive 20-min periods. Acetylcholine (ACh, 100 μM) was then added to the bath and the mucociliary transport was again measured over three consecutive 20-min periods. Dotted lines show the 95% confidence intervals for non-CF percent solids in each group. The mucociliary transport responses to ACh for both CF tracheas in the bottom graph were outside of the 95% confidence intervals of the responses of non-CF tracheas (‡) for all three ACh exposure periods. Each line represents responses of a different trachea.
Adams et al. (1) previously noted that the tracheas of transgenic CF pigs were narrow relative to those of wild-type pigs. Figure 7 shows a plot of trachea diameters against body weight for non-CF pigs and the two CF pigs that were killed at 55 and 61 days. As expected, linear regression analysis of the non-CF pig tracheas indicates that there is a significant (P < 0.05) positive correlation of tracheal diameter with body weight. The diameters of the tracheas of the two pigs with CF clearly lie outside the lower 95% confidence interval for the regression line for the non-CF pigs (Fig. 7). Indeed, the diameters of the two CF pig tracheas were smaller than the diameters of any of the non-CF pig tracheas despite the substantially greater body weights of the CF animals.
Fig. 7.
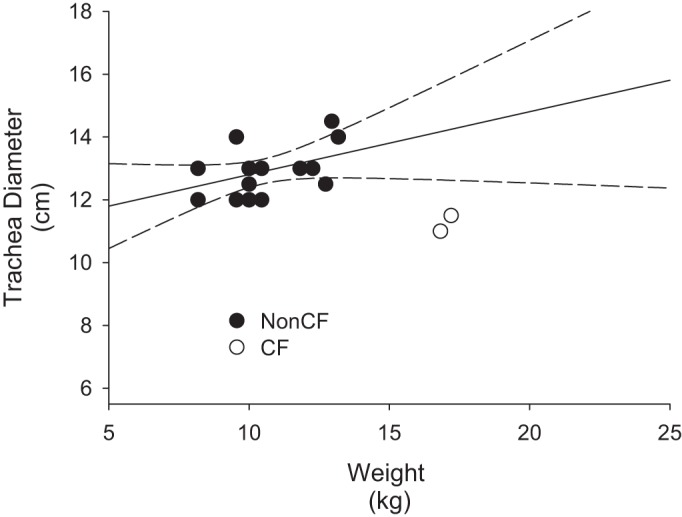
Trachea diameter differences between gut-corrected CF and non-CF pigs. Filled symbols show the relationship between body weight and tracheal diameter for non-CF pigs. Linear line fit to the data points indicates that a significant (P < 0.01) positive correlation exists between body weight and tracheal diameter among non-CF pigs. The two curved dashed lines show the 95% confidence intervals for this relationship in non-CF pigs. Open symbols represent the trachea diameter and body weight relationship for the two CF pigs that survived infancy. Values for both pigs with CF lie outside the 95% confidence intervals for the non-CF pigs. Note that both CF tracheas have diameters that are less than any of the non-CF tracheas despite the substantially greater body mass of the pigs with CF.
DISCUSSION
This study documents our experiences with the gut-corrected CF pig developed by researchers at the University of Iowa (27). We found that this model expressed numerous characteristics of CF pathology that have been documented in human and porcine CF disease. Unfortunately, we also found that this model was plagued by unexpectedly high rates of fetal and neonatal mortality that were unlikely to have been caused by simple loss of CFTR function. Although we were able to obtain important and useful information about CF pathophysiology from the two surviving gut-corrected CF pigs, these complications in this commercial model were unexpected and confounding. Apart from the complication of meconium ileus (which was not a significant morbidity issue in the present study for the gut-corrected CF pigs), the very high neonatal mortality rates that we document here have not been reported in previous papers in which CF porcine models (including the gut-corrected model) were used, nor in the literature provided by the vendor.
In experimental mammals, knockout of the CFTR gene results in relatively high incidence of meconium ileus. In pigs with CF, severe meconium ileus occurs in all newborns and is usually lethal without surgical intervention (18). Neonatal mortality due to meconium ileus in mice is close to 100% after weaning (25) and, in ferrets with CF, it is about 75% (28). Neonatal mortality in rats with CF is only about 30% (31). Administration of polyethylene glycol laxative solution substantially improves survival in both rats (31) and mice (8). By comparison, human newborns with CF have a relatively low incidence of meconium ileus. A retrospective review of newborn screening and clinical records for people with CF born in Victoria, Australia, between 1989 and 2008, found the incidence of meconium ileus to be about 15% (16). The promise of the gut-corrected porcine model of CF used by us in the present study was to avoid the need for invasive surgery to correct the meconium ileus in all offspring of pigs with CF. Although that objective was achieved with the gut-corrected CF pigs, the extremely high fetal and neonatal mortality, apparently associated with the gut correction maneuver in this animal model, abrogated any advantage gained from the reduced severity of meconium ileus.
The most confounding and unexpected feature of the gut-corrected CF pig model was the hepatic disease. All offspring exhibited severe systemic edema from birth. Plasma analysis revealed severe hypoproteinemia, and absolute concentrations of both plasma albumin and globulin were far below normal reference values. Although wild-type neonatal piglets are moderately hyproteinemic at birth, this is reversed within the first 3 days of life (7) and is not as striking as the hypoproteinemia observed in the mutant piglets. The low concentration of plasma proteins was certainly the cause of the severe systemic extracellular edema and the likely cause of the enlarged hearts. In addition, plasma levels of sorbitol dehydrogenase, aspartate amino transferase, and creatine kinase were greatly elevated compared with reference values. Plasma phosphorus concentration was also substantially elevated. These findings are consistent with liver dysfunction. Severe acidosis, evidenced by the low HCO3− level and very large anion gap, was also a likely contributor to this animal's death. Despite these complications, two pigs survived for nearly 2 mo in relatively good health, although they required frequent furosemide administration to control systemic edema. We did not perform plasma analyses in these two surviving animals, so we cannot judge whether the severity of their hepatic complications improved over time. However, no overt jaundice was observed in the two surviving pigs with CF prior to death. Although transient elevation of hepatic enzymes can be observed in up to 50% of human infants with CF, it often normalizes by 2–3 yr of age (5). Hepatic synthetic failure is unusual in human patients with CF (5). In the initial CFTR gene-knockout model of CF in pigs, only mild to moderate hepatic lesions were noted in newborns (22).
We observed that bronchi isolated from the lungs of the two surviving CF pigs secreted liquid at significantly lower rates than non-CF pig bronchi in the presence of ACh or forskolin, or in the absence of a secretogogue (Fig. 4). This finding is consistent with CFTR playing an important role in the active transepithelial secretion of Cl−, HCO3−, and, thereby, liquid in non-CF animals. These responses importantly indicate that CFTR plays a significant role in cholinergic as well as forskolin-sensitive pathways in bronchial airways. Our previous studies with normal pig bronchi, which were pretreated with pharmacological inhibitors of Cl− and HCO3− secretion to mimic the CF condition in vitro, blocked both ACh- and forskolin-induced liquid secretion by approximately the same proportions we observed in the present study with CF pig bronchi (2). These responses are also consistent with the higher percent of solids we observed in the airway liquid collected from the pigs with CF under these three conditions (Fig. 5). That is, a lower rate in the volume of secreted liquid due to the absence of the CFTR would be predicted to increase the percentage of nonvolatile solids in airway mucous liquid if the rate of biomolecule secretion was largely CFTR-independent. This reduced water-to-solids ratio in airway liquid results in a thicker, more viscous mucus, which would likely be poorly cleared from CF airways by mucociliary transport and cough. These results mirror those obtained in previous studies with non-CF pig bronchi in which inhibition of active Cl− and HCO3− secretion with specific anion transport inhibitors significantly reduced the rate of liquid secretion (15, 30) and significantly increased the percent of solids in the secreted liquid (15). Thus, the thick mucus produced by CF airways is inherent to the loss of anion and liquid secretion by airways. Nonetheless, it should be appreciated that goblet and mucous cell metaplasia, which accompanies chronic infection in airways of individuals with CF, exacerbates this problem by further increasing the capacity of the airways to secrete mucous biomolecules.
We found that ACh-induced mucociliary transport rates are lower in tracheas of pigs with CF than in non-CF tracheas. The transport rates in the tracheas of the two pigs with CF were both outside the 95% confidence intervals for rates measured in non-CF pig tracheas in all three post-ACh periods. These results also mirror our previous in vitro studies with excised, cannulated tracheas from normal domestic pigs in which the CF condition was mimicked with bumetanide and dimethylamiloride (DMA) pretreatment, which respectively inhibited transepithelial Cl− and HCO3− secretion (4). In 1997, our laboratory reported that airway submucosal glands secreted a low-viscosity fluid when stimulated with ACh (11); but in the presence of bumetanide, to inhibit Cl− secretion, and DMA to inhibit HCO3− secretion, the gland ducts became filled and distended with a thick mucous gel that formed long, slowly moving strands as it exited the gland ducts (4, 10). We proposed at that time that these mucus strands reduced mucus transport by joining together with the surface mucus sheet, thus tethering the mucus blanket to the gland ducts from which they emerged (4). Hoegger and coworkers (9), using the simple CFTR gene-knockout pig model of CF, recently confirmed our hypothesis that impaired mucociliary transport in CF airways is indeed due to mucus strands that are tethered to submucosal gland duct openings to the airway lumen.
Meyerholz et al. (17) previously reported that the tracheas of pigs with CF were significantly more narrow than those of non-CF pigs. We also observed this in the pigs with gut-corrected CF in the present study. Adams and coworkers (1) showed that the airways distal to the trachea, including the mainstem bronchi and proximal airways, were also narrow relative to non-CF airways. This airway narrowing is the most likely cause of the hyperpnea during and after exercise we observed in the two surviving pits with gut-corrected CF. We also noted that the ends of the cartilage rings in the tracheas curled inward into the tracheal lumen, which also would obstruct airflow and create turbulence in the airstream.
In conclusion, we found in this study that the CFTR gut-corrected pig model of CF significantly mollified the lethal meconium ileus complication that plagues the porcine CFTR gene-knockout model of CF. We also found that the CFTR gut-corrected CF pig model exhibits several important clinical signs of human CF disease including reduced mucociliary clearance from the airways; reduced capacity for secretion of Cl−, HCO3−, and airway liquid from tracheobronchial airways; and secretion by the tracheobronchial airways of an abnormally thick mucus containing a high percentage of nonvolatile solids. Unfortunately, this model was plagued by ∼90% neonatal mortality, which appears to go beyond what would be expected with only loss of CFTR activity. Unexpected complications included extremely low level of plasma proteins, extremely high plasma level of hepatic enzymes, and severe acidosis. We speculate that these complications inherent to this porcine CF model are in some way related to the gut-correction maneuver to express normal CFTR in the gastrointestinal organs and were not related simply to loss of CFTR expression in other organs.
GRANTS
Support for this study was provided by Cystic Fibrosis Foundation Therapeutics Grant BALLAR07XX0 and by an appropriation from the Alabama Department of Public Health to S. T. Ballard and J. W. Evans. H. S. Drag was supported by National Institutes of HealthTraining Grant T32 NIHHL-076125.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
S.T.B. and M.S. conception and design of research; S.T.B., J.W.E., H.S.D., and M.S. performed experiments; S.T.B., J.W.E., H.S.D., and M.S. analyzed data; S.T.B., J.W.E., and M.S. interpreted results of experiments; S.T.B. prepared figures; S.T.B. drafted manuscript; S.T.B., J.W.E., and M.S. edited and revised manuscript; S.T.B., J.W.E., H.S.D., and M.S. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Lawrence Sindel, who graciously helped us understand the clinical aspects of human CF disease and its treatment. We thank Stephanie and Scott Cleveland and Tonya Gollotte of Making Miracles for Cystic Fibrosis, and the Honorable Jim Barton, for their tireless support of our research efforts. We also thank Dr. Steven Rowe, University of Alabama-Birmingham, Dr. Lane Clarke, University of Missouri at Columbia, and Dr. Jonathan Scammell, Chair of Comparative Medicine at the University of South Alabama, for their helpful comments and discussions. This project would not have been possible without the dedicated 24-h care provided by the vivarial staff at the University of South Alabama; in particular, Leigh Ann Wiggins, Craig Youngman, Francelina Nores, and Benjamin Gumbs.
REFERENCES
- 1.Adams RJ, Michalski AS, Bauer C, Abou Alaiwa MH, Gross TJ, Awadalla MS, Bouzek DC, Gansemer ND, Taft PJ, Hoegger MJ, Diwakar A, Ochs M, Reinhardt JM, Hoffman EA, Beichel RR, Meyerholz DK, Stoltz DA. Air trapping and airflow obstruction in newborn cystic fibrosis piglets. Am J Respir Crit Care Med 188: 1434–1441, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ballard ST, Parker JC, Hamm CR. Restoration of mucociliary transport in the fluid-depleted trachea by surface-active instillates. Am J Respir Cell Mol Biol 34: 500–504, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ballard ST, Trout L, Bebök Z, Sorscher EJ, Crews A. CFTR involvement in chloride, bicarbonate, and liquid secretion by airway submucosal glands. Am J Physiol Lung Cell Mol Physiol 277: L694–L699, 1999. [DOI] [PubMed] [Google Scholar]
- 4.Ballard ST, Trout L, Mehta A, Inglis SK. Liquid secretion inhibitors reduce mucociliary transport in glandular airways. Am J Physiol Lung Cell Mol Physiol 283: L329–L335, 2002. [DOI] [PubMed] [Google Scholar]
- 5.Borowitz D, Gelfond D. Intestinal complications of cystic fibrosis. Curr Opin Pulm Med 19: 676–680, 2013. [DOI] [PubMed] [Google Scholar]
- 6.Boucher RC. Cystic Fibrosis. In: Harrison's Principles of Internal Medicine (17th ed), edited by Fauci AS, Braunwald E, Kasper DL, Hauser SL, Longo DL, Jameson JL, and Loscalzo J. New York: McGraw-Hill, 2008, pp. 1632–1635. [Google Scholar]
- 7.Brooks CC, Davis JW. Changes in hematology of the perinatal pig. J Anim Sci 28: 517–522, 1969. [DOI] [PubMed] [Google Scholar]
- 8.Clarke LL, Gawenis LR, Franklin CL, Harline MC. Increased survival of CFTR knockout mice with an oral osmotic laxative. Lab Anim Sci 46: 612–618, 1996. [PubMed] [Google Scholar]
- 9.Hoegger MJ, Fischer AJ, McMenimen JD, Ostedgaard LS, Tucker AJ, Awadalla MA, Moninger TO, Michalski AS, Hoffman EA, Joseph Zabner J, Stoltz DA, Welsh MJ. Impaired mucus detachment disrupts mucociliary transport in a piglet model of cystic fibrosis. Science 345: 818–822, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Inglis SK, Corboz MR, Ballard ST. Effect of anion secretion inhibitors on mucin content of airway submucosal gland ducts. Am J Physiol Lung Cell Mol Physiol 274: L762–L766, 1998. [DOI] [PubMed] [Google Scholar]
- 11.Inglis SK, Corboz MR, Taylor AE, Ballard ST. In situ visualization of bronchial submucosal glands and their secretory response to acetylcholine. Am J Physiol Lung Cell Mol Physiol 272: L203–L210, 1997. [DOI] [PubMed] [Google Scholar]
- 12.Jackson PG, Cockcroft PD. Handbook of Pig Medicine (1st ed), Philadelphia: Saunders Elsevier, 2007. [Google Scholar]
- 13.Kerem B, Rommens JM, Buchanan JA, Markiewicz D, Cox TK, Chakravarti A, Buchwald M, Tsui LC. Identification of the cystic fibrosis gene: genetic analysis. Science 45: 1073–1080, 1989. [DOI] [PubMed] [Google Scholar]
- 14.Latimer KS, Mahaffey EA, Prasse KW. Duncan & Prasse's Veterinary Laboratory Medicine: Clinical Pathology (4th ed), Ames: Iowa State Press, 2003. [Google Scholar]
- 15.Martens CJ, Inglis SK, Valentine VG, Garrison J, Conner GE, Ballard ST. Mucous solids and liquid secretion by airways: studies with normal pig, cystic fibrosis human, and non-cystic fibrosis human bronchi. Am J Physiol Lung Cell Mol Physiol 301: L236–L246, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Massie RJ, Curnow L, Glazner J, Armstrong DS, Francis I. Lessons learned from 20 years of newborn screening for cystic fibrosis. Med J Aust 196: 67–70, 2012. [DOI] [PubMed] [Google Scholar]
- 17.Meyerholz DK, Stoltz DA, Namati E, Ramachandran S, Pezzulo AA, Smith AR, Rector MV, Suter MJ, Kao S, McLennan G, Tearney GJ, Zabner J, McCray PB, Welsh MJ. Loss of cystic fibrosis transmembrane conductance regulator function produces abnormalities in tracheal development in neonatal pigs and young children. Am J Respir Crit Care Med 182: 1251–1261, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meyerholz DK, Stoltz DA, Pezzulo AA, Welsh MJ. Pathology of gastrointestinal organs in a porcine model of cystic fibrosis. Am J Pathol 176: 1377–1389, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Poulsen JH, Fischer H, Illek B, Machen TE. Bicarbonate conductance and pH regulatory capability of cystic fibrosis transmembrane conductance regulator. Proc Natl Acad Sci USA 91: 5340–5344, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Quinton PM. Chloride impermeability in cystic fibrosis. Nature 301: 421–422, 1983. [DOI] [PubMed] [Google Scholar]
- 21.Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou JL, Drumm ML, Iannuzzi MC, Collins FS, Tsui LC. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 245: 1066–1073, 1989. [DOI] [PubMed] [Google Scholar]
- 22.Rogers CR, Stoltz DA, Meyerholz DK, Ostedgaard LS, Rokhlina T, Taft PJ, Rogan MP, Pezzulo AA, Karp PH, Itani OA, Kabel AC, Wohlford-Lenane CL, Davis GJ, Hanfland RA, Smith TL, Samuel M, Wax D, Murphy CN, Rieke A, Whitworth K, Uc A, Starner TD, Brogden KA, Shilyansky J, McCray PB, Zabner J, Prather RS, Welsh MJ. Disruption of the CFTR gene produces a model of cystic fibrosis in newborn pigs. Science 321: 1837–1841, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rommens JM, Iannuzzi MC, Kerem B, Drumm ML, Melmer G, Dean M, Rozmahel R, Cole JL, Kennedy D, Hidaka N, Zsiga M, Buchwald M, Riordan JR, Tsui L-C, Collins FS. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science 245: 1059–1065, 1989. [DOI] [PubMed] [Google Scholar]
- 24.Smith JJ, Welsh MJ. cAMP stimulates bicarbonate secretion across normal, but not cystic fibrosis airway epithelia. J Clin Invest 89: 1148–1153, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Snouwaert JN, Brigman KK, Latour AM, Malouf NN, Boucher RC, Smithies O, Koller BH. An animal model for cystic fibrosis made by gene targeting. Science 257: 1083–1088, 1992. [DOI] [PubMed] [Google Scholar]
- 26.Snouwaert JN, Brigman KK, Latour AM, Iraj E, Schwab U, Gilmour MI, Koller BH. A murine model of cystic fibrosis. Am J Respir Crit Care Med 151: S59–S64, 1995. [DOI] [PubMed] [Google Scholar]
- 27.Stoltz DA, Rokhlina T, Ernst SE, Pezzulo AA, Ostedgaard LS, Karp PH, Samuel MS, Reznikov LR, Rector MV, Gansemer ND, Bouzek DC, Abou Alaiwa MH, Hoegger MJ, Ludwig PS, Taft PJ, Wallen TJ, Wohlford-Lenane C, McMenimen JD, Chen JH, Bogan KL, Adam RJ, Hornick EE, Nelson GA 4th, Hoffman EA, Chang EH, Zabner J, McCray PB Jr, Prather RS, Meyerholz DK, Welsh MJ. Intestinal CFTR expression alleviates meconium ileus in cystic fibrosis pigs. J Clin Invest 123: 2685–2693, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sun X, Olivier AK, Yi Y, Pope CE, Hayden HS, Liang B, Sui H, Zhou W, Hager KR, Zhang Y, Liu X, Yan Z, Fisher JT, Keiser NW, Song Y, Tyler SR, Goeken JA, Kinyon JM, Radey MC, Fligg D, Wang X, Xie W, Lynch TJ, Kaminsky PM, Brittnacher MJ, Miller SI, Parekh K, Meyerholz DK, Hoffman LR, Frana T, Stewart ZA, Engelhardt JF. Gastrointestinal pathology in juvenile and adult CFTR-knockout ferrets. Am J Pathol 184: 1309–1322, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sun X, Sui H, Fisher JT, Yan Z, Liu X, Cho HJ, Joo NS, Zhang Y, Zhou W, Yi Y, Kinyon JM, Lei-Butters DC, Griffin MA, Naumann P, Luo M, Ascher J, Wang K, Frana T, Wine JJ, Meyerholz DK, Engelhardt JF. Disease phenotype of a ferret CFTR-knockout model of cystic fibrosis. J Clin Invest 120: 3149–3160, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Trout L, Gatzy JT, Ballard ST. Acetylcholine-induced liquid secretion by bronchial epithelium: role of Cl− and HCO3− transport. Am J Physiol Lung Cell Mol Physiol 275: L1095–L1099, 1998. [DOI] [PubMed] [Google Scholar]
- 31.Tuggle KL, Birket SE, Cui X, Hong J, Warren J, Reid L, Chambers A, Ji D, Gamber K, Chu KK, Tearney G, Tang LP, Fortenberry JA, Du M, Cadillac JM, Bedwell DM, Rowe SM, Sorscher EJ, Fanucchi MV. Characterization of defects in ion transport and tissue development in cystic fibrosis transmembrane conductance regulator (CFTR)-knockout rats. PLoS One 9: e91253, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhou L, Dey CR, Wert SE, DuVall MD, Frizzell RA, Whitsett JA. Correction of lethal intestinal defect in a mouse model of cystic fibrosis by human CFTR. Science 266: 1705–1708, 1994. [DOI] [PubMed] [Google Scholar]