

Abstract
The relationship between soluble epoxide hydrolase (sEH) and coronary reactive hyperemia (CRH) response to a brief ischemic insult is not known. Epoxyeicosatrienoic acids (EETs) exert cardioprotective effects in ischemia/reperfusion injury. sEH converts EETs into dihydroxyeicosatrienoic-acids (DHETs). Therefore, we hypothesized that knocking out sEH enhances CRH through modulation of oxylipin profiles, including an increase in EET/DHET ratio. Compared with sEH+/+, sEH−/− mice showed enhanced CRH, including greater repayment volume (RV; 28% higher, P < 0.001) and repayment/debt ratio (32% higher, P < 0.001). Oxylipins from the heart perfusates were analyzed by LC-MS/MS. The 14,15-EET/14,15-DHET ratio was 3.7-fold higher at baseline (P < 0.001) and 5.6-fold higher post-ischemia (P < 0.001) in sEH−/− compared with sEH+/+ mice. Likewise, the baseline 9,10- and 12,13-EpOME/DiHOME ratios were 3.2-fold (P < 0.01) and 3.7-fold (P < 0.001) higher, respectively in sEH−/− compared with sEH+/+ mice. 13-HODE was also significantly increased at baseline by 71% (P < 0.01) in sEH−/− vs. sEH+/+ mice. Levels of 5-, 11-, 12-, and 15-hydroxyeicosatetraenoic acids were not significantly different between the two strains (P > 0.05), but were decreased postischemia in both groups (P = 0.02, P = 0.04, P = 0.05, P = 0.03, respectively). Modulation of CRH by peroxisome proliferator-activated receptor gamma (PPARγ) was demonstrated using a PPARγ-antagonist (T0070907), which reduced repayment volume by 25% in sEH+/+ (P < 0.001) and 33% in sEH−/− mice (P < 0.01), and a PPARγ-agonist (rosiglitazone), which increased repayment volume by 37% in both sEH+/+ (P = 0.04) and sEH−/− mice (P = 0.04). l-NAME attenuated CRH in both sEH−/− and sEH+/+. These data demonstrate that genetic deletion of sEH resulted in an altered oxylipin profile, which may have led to an enhanced CRH response.
Keywords: coronary reactive hyperemia, epoxyeicosatrienoic acids, dihydroxyeicosatrienoic acids, soluble epoxide hydrolase, oxylipins, isolated perfused heart
during reactive hyperemia (RH), blood flow increases transiently in response to ischemia or interruption of blood supply (58). In the heart, RH is referred to as coronary RH (CRH), and is associated with coronary vasodilation in response to cessation in coronary perfusion. This occlusion or ischemic insult is inherently associated with potentially detrimental effects on the heart and cardiac function. The increase in blood flow associated with CRH increases repayment volume (RV) to prevent or decrease potential injury due to ischemia. CRH speeds up functional recovery by supplying more than the usual amount of blood to nourish the deprived heart muscle, and wash away accumulated metabolic byproducts (58); therefore, CRH may be viewed as a protective mechanism. Research has shown that RH is blunted in pathologic conditions affecting blood flow, including the coronary circulation; CRH is decreased in cardiac hypertrophy (20) and metabolic syndrome (5). Moreover, higher risk for cardiovascular diseases, such as unstable angina, myocardial infarction, congestive heart failure, and cardiac death are linked to decreased CRH (18). Corrective measures, such as coronary angioplasty, have been shown to reverse or mitigate decreased CRH in patients with acute myocardial infarction and significantly improved CRH after successful coronary angioplasty (53). Although coronary vascular reserve is traditionally measured through pharmacological agents, CRH is also an accepted alternative to measure it if patients cannot tolerate the pharmacological agents (21).
The mechanisms and mediators involved in RH have been studied extensively. These mechanisms may be broadly divided into two categories: mechanosensitive and metabolic (58). The mechanosensitive mechanisms include shear stress and myogenic response. Metabolic mediators involved in RH include adenosine (4, 20), nitric oxide (NO) (20), KATP channels (20), and hydrogen peroxide (H2O2) (20, 46).
Epoxyeicosatrienoic acids (EETs) are oxidized metabolites of arachidonic acid (AA) that have numerous physiological actions. EETs are produced in endothelial cells and induce hyperpolarization and relaxation in vascular smooth muscle cells by activating large-conductance Ca2+-activated K+ channels (BKCa) (6, 12). EETs are rapidly metabolized by soluble epoxide hydrolase (sEH) to less active dihydroxyeicosatrienoic acids (DHETs) through hydration, the main catabolic pathway responsible for EETs breakdown (47); therefore, targeting sEH is associated with beneficial effects on the cardiovascular system (11, 35). Also, others (9, 22, 27) have reported that sEH−/− mice were associated with elevated EET levels compared with sEH+/+ mice, and EETs exerted cardioprotective effects against ischemia/reperfusion injury (45). Additionally, targeting sEH impacted the level of oxylipins, such as 9,–10-epoxyoctadecaenoic acids (EpOMEs), dihydroxyoctadecaenoic acids (DiHOMEs), hydroxyoctadecadienoic acid (HODEs), and hydroxyeicosatetraenoic acids (HETEs) (27, 29). Lee et al. (26) reported that EpOME/DiHOME ratio was increased in response to the sEH inhibitor 12-(3-adamantan-1-yl-ureido)-dodecanoic acid (AUDA) in vivo; this change was associated with improved renal recovery against ischemia reperfusion injury in C57BL/6 mice. De Meyer et al. (8) reported that 13-HODE was involved in canine splenic and coronary artery relaxation through PGI2 (prostacyclin) biosynthesis in both endothelial and smooth muscle cells. Mid-chain HETEs had direct chemotaxis effects on vascular tone and production of endothelial vascular factors, such as vascular endothelial growth factor, and pigment epithelium-derived factor (1, 17, 32, 50, 54). Further, EET-induced aortic relaxation in mice was mediated by peroxisome proliferator-activated receptor gamma (PPARγ) (35, 42). Also, in mouse mesenteric arteries, EET-induced vasodilatory effects were inhibited by l-NAME (a nonselective NO synthase, NOS, inhibitor) (16). Seubert et al. (45) reported that CYP2J2 overexpression in the mouse heart involved increased generation of EETs compared with their respective control and exerted cardioprotective effects against ischemia/reperfusion injury. However, the potential role of sEH deletion in coronary flow (CF) during CRH in response to short ischemia has not been investigated. Therefore, we hypothesized that knocking out sEH enhances CRH in isolated mouse heart through modulation of oxylipin profiles, including an increase in the EET/DHET ratio.
MATERIALS AND METHODS
Animals
The generation of sEH−/− mice was described by Sinal et al. (47). sEH−/− and sEH+/+ mice were provided by Dr. Darryl Zeldin, National Institute of Environmental Health Sciences/National Institutes of Health (NIH). All animal care and experimentation protocols were approved and carried out in accordance with the West Virginia University Institutional Animal Care and Use Committee and were in accordance with the principles and guidelines of the NIH's “Guide for the Care and Use of Laboratory Animals”. Both male and female mice (14–16 wk old) in equal ratio were used in our study. Mice were maintained in cages with a 12:12-h light-dark cycle and free access to standard chow and water.
Langendorff-Perfused Heart Preparation
The Langendorff perfused heart preparation is a well-established technique for studying the cardiac functions and pathologies in vitro. There are two available Langendorff technique modes to perfuse the heart: 1) constant pressure mode, and 2) constant flow mode. In our experiments, we chose the constant pressure mode. The selection between the two is especially important when trying to assess the role of coronary regulatory mechanisms in response to changing CF conditions or pathologies, such as ischemia. On the basis of that, the constant pressure mode is physiologically more relevant in experiments involving ischemia, like the model in our experiments (48).
Deletion of sEH in sEH−/− mice was confirmed by genotyping of mouse tail samples (data not shown). Also, Western blot analysis confirmed the deletion of sEH protein in sEH−/− mice aortic samples and was reported by our laboratory (35). Soluble epoxide hydrolase null (sEH−/−) and wild-type mice (sEH+/+) mice (14–16 wk) of both sexes (equal ratios) were euthanized with pentobarbital sodium (100 mg/kg body wt intraperitoneally). Hearts were excised and immediately placed into heparinized (5 U/ml) ice-cold Krebs-Henseleit buffer containing (in mM) 119.0 NaCl, 11.0 glucose, 22.0 NaHCO3, 4.7 KCl, 1.2 KH2PO4, 1.2 MgSO4, 2.5 CaCl2, 2.0 pyruvate, and 0.5 EDTA. After removal of the lung and surrounding tissue around the heart, the aorta was rapidly cannulated with a 20-gauge, blunt-ended needle and continuously perfused with 37°C buffer continuously bubbled with ([95% O2] − [5% CO2]) at a constant perfusion pressure of 80 mmHg. The left atrium was excised, and a water-filled balloon made of plastic wrap was inserted into the left ventricle through the mitral valve. The balloon was connected to a pressure transducer for continuous measurement of left ventricular developed pressure (LVDP) and heart rate (HR). The heart was then immersed in a water-jacketed perfusate bath (37°C) and left to beat spontaneously. Left ventricular diastolic pressure was adjusted to 2–5 mmHg. A flow transducer was installed above the cannulated aorta for continuous measurement of CF with an ultrasonic flow probe (Transonic Systems, Ithaca, NY). A Power-Lab Chart data acquisition system (AD Instruments, Colorado Springs, CO) was used for data acquisition. Heart was allowed to stabilize for 30–40 min before initiation of CRH. Only hearts whose peak CF increased by more than twofold after a 15-s total occlusion were included in the analysis. This demonstrated that the isolated heart preparation was intact to be included in the experiment. Hearts with persistent arrhythmias or LVDP of less than 80 mmHg were excluded. Nine mice (4 males and 5 females), out of 58 mice, were excluded from our study for the mentioned reasons.
Coronary Reactive Hyperemic Response
After stabilization for 30–40 min, baseline CF, HR, and LVDP were recorded. Hearts were subjected to 15 s of total occlusion by closing the valve directly above the cannulated heart to bring forth CRH. After CF returned to pre-CRH baseline levels, post-CRH baseline CF, CF tracing, peak hyperemic flow (PHF), HR, LVDP, RV, and repayment duration (RD) recordings were analyzed for each isolated heart. Investigation drugs were infused into the aortic perfusion line using a microinjection pump (Harvard Apparatus, Holliston, MA) for 15 min, after which another CRH was induced and the same parameters analyzed again. Drugs were infused at a rate equivalent to 1% of CF. The final concentrations, after standardization of dose (0.01, 0.1, 1, and 10 μM) response for the various drugs used in this study were 10 μM for T0070907 (PPARγ-antagonist), rosiglitazone (PPARγ− agonist), and 100 μM for l-NAME (Nω-nitro-l-arginine methyl ester hydrochloride, a nonselective nitric oxide synthase inhibitor). These concentrations were selected on the basis of our dose-response studies (for T0070907 and rosiglitazone) and on the basis of the concentrations used in previous studies [rosiglitazone, 10 μM; (40)], and [l-NAME, 100 μM; (58)]. Time-matched control experiments with WT (sEH+/+) mouse hearts, employing three consecutive inductions of CRH, showed no change in CRH response and in baseline heart functions, including CF, LVDP, and HR (data not shown).
LC-MS/MS Oxylipin Analysis
Oxylipin levels (5,6-, 8,9-, 11,12-, and 14,15-EETs; 5,6-, 8,9-, 11,12-, and 14,15-DHETs; 5-, 8-, 9-, 11-, 12-, and 15-HETEs; 9,10- and 12,13-EpOMEs; 9,10- and 12,13-DiHOME; 9- and 13-HODEs) were analyzed in pre- and post-CRH heart perfusates of sEH−/− and sEH+/+ mice through liquid chromatography, tandem mass spectroscopy (LC-MS/MS), as described previously (23). Briefly, heart perfusates were collected after the first 30 min of stabilization, and right after reperfusion for 2.5 min. Hearts were immersed in 5 ml of warm (37°C) Krebs-Henseleit buffer and 5 μl of 10 μM t-AUCB {trans-4-[4-(3-adamantan-1-yl-ureido)-cyclohexyloxy]-benzoic acid, selective sEH-inhibitor} to block further breakdown of EETs and EpOMEs by sEH. Heart perfusates were collected two times before (at baseline) and pooled together as one sample, and two times after ischemia and pooled together as another sample for LC-MS/MS analysis. Samples were stored at −80°C until processing. Samples were spiked with 30 ng PGE2-d4, 10,11-DiHN, and 10,11-EpHep (Cayman) as internal standards, mixed with 0.1 vol of 1% acetic acid in 50% methanol, and extracted by serial passage through Oasis HLB C18 3-ml columns (Waters, Milford, MA). Columns were washed twice with 0.1% acetic acid in 5% methanol, and eluted with methanol into glass tubes containing 6 μl of 30% glycerol in methanol. The methanol was then evaporated under a stream of nitrogen gas, and the dried tubes were frozen and stored at −80°C until analysis. Online LC of extracted samples was performed with an Agilent 1200 series capillary HPLC (Agilent Technologies, Santa Clara, CA). Separations were achieved using a Halo C18 column (2.7 mm, 10,062.1 mm; MAC-MOD Analytical, Chadds Ford, PA), which was held at 50°C. Mobile phase A was 85:15:0.1 water: acetonitrile: acetic acid. Mobile phase B was 70:30:0.1 acetonitrile: methanol: acetic acid. Flow rate was 400 μl/min. Gradient elution was used. Mobile phase percentage B and flow rates were varied as follows: 20% B at 0 min, ramp from 0 to 5 min to 40% B, ramp from 5 to 7 min to 55% B, ramp from 7 to 13 min to 64% B. From 13 to 19 min, the column was flushed with 100% B at a flow rate of 550 μl/min. Samples were solvated in 50 μl of 30% ethanol. The injection volume was 10 μl. Samples were analyzed in triplicate. Analyses were performed on an MDS Sciex API 3,000 equipped with a TurboIonSpray source (Applied Biosystems). Turbo desolvation gas was heated to 425°C at a flow rate of 6 l/min. Negative ion electrospray ionization tandem mass spectrometry with multiple reaction monitoring was used for detection.
Effect of PPARγ-Antagonist (T0070907) on CRH Response in sEH−/− and sEH+/+ Mice
Isolated hearts from sEH−/− and sEH+/+ mice were stabilized for 30–40 min followed by 15 s of total occlusion. Recordings of the first CRH (baseline CF, CF tracing, LVDP, HR, RV, PHF, and RD) were analyzed for each heart and averaged for each group, as mentioned previously. The PPARγ-antagonist T0070907 was infused at a final concentration of 10 μM and 1% of CF rate for 15 min, after which, another CRH was induced, and the same parameters were recorded and analyzed.
Effect of PPARγ-Agonist (Rosiglitazone) on CRH in sEH−/− and sEH+/+ Mice
After stabilization, hearts from sEH−/− and sEH+/+ mice were subjected to 15 s of total occlusion. As described above, baseline CRH was induced in each mouse heart. Then, the PPARγ-agonist rosiglitazone was infused at a final concentration of 10 μM for 15 min, followed by another CRH. CRHs before and after rosiglitazone infusion were analyzed.
Effect of l-NAME on CRH Response in sEH−/− and sEH+/+ Mice
Baseline CRH was induced in each mouse strain. The nonselective nitric oxide synthase inhibitor l-NAME was infused at a final concentration of 100 μM for 15 min, after which, another CRH was induced. CRH responses before and after l-NAME infusion were analyzed and compared.
Statistical and Data Analyses
Flow debt (baseline flow rate multiplied by occlusion duration) and repayment volume (the integral of hyperemic area above the baseline flow) were calculated using “the integral relative to baseline” function in the data pad of LabChart 7.0 software. Since absolute coronary flow rates change proportionally with heart mass, the RV and flow debt are presented as milliliters per gram wet heart weight, and baseline and peak flow rate data are presented as milliliters per minute per gram wet heart rate. Values are expressed as means ± SE; n represents the number of animals. For data analysis, two-tailed unpaired t-tests were used for unpaired data analysis, repeated-measures ANOVAs were used for populations measured 3 times, and two-way ANOVAs were used to compare data between groups. Differences were considered statistically significant when P ≤ 0.05. Post hoc power analyses for the main results in each experiment were calculated using G*Power 3.1.9.2. software (Heinrich Heine, Universität Düsseldorf, Düsseldorf, Germany).
RESULTS
CRH Response
Baseline function of sEH−/− and sEH+/+ mouse hearts: Baseline functional data of isolated mouse hearts were recorded after stabilization (between 30–40 min) before starting the experimental protocol. There was no statistically significant difference in body weight, heart weight, heart-to-body weight ratio, baseline CF, LVDP, and HR between sEH+/+ and sEH−/− mice (Table 1).
Table 1.
Baseline functional data for wild type (sEH+/+) and sEH-null (sEH−/−) isolated mouse heart
| sEH+/+ | sEH−/− | |
|---|---|---|
| Age, wk | 14.6 ± 0.3 | 14.7 ± 0.4 |
| Body weight, g | 22.7 ± 1.1 | 24.3 ± 1.5 |
| Heart weight, mg | 109 ± 7.0 | 121 ± 10.0 |
| Heart-to-body weight ratio, % | 0.48 ± 0.01 | 0.49 ± 0.01 |
| Coronary flow, ml.min−1.g−1 | 15.7 ± 0.6 | 16.5 ± 0.8 |
| LVDP, mmHg | 109.6 ± 10.0 | 115.0 ± 12.3 |
| Heart rate, beat.min−1 | 414 ± 11 | 396 ± 6 |
Values are expressed as means ± SE; n = 12 per group.
Role of sEH in CRH
Comparison of repayment volume between female and male sEH+/+ and sEH−/− mice showed no statistically significant differences between female (F) and male (M) sEH+/+mice (P > 0.05) or between female (F) and male (M) sEH−/− mice (P > 0.05, Fig. 1). However, M-sEH−/− had more enhanced (P = 0.04, Fig. 1) RV compared with M-sEH+/+. Also, F-sEH−/− had more enhanced (P = 0.01, Fig. 1) RV compared with F-sEH+/+. The post hoc power analysis for repayment volume was 77.7%. Because of the lack of sex difference in CRH response, we used equal ratios of male and female mice from each strain for the remainder of the study.
Fig. 1.
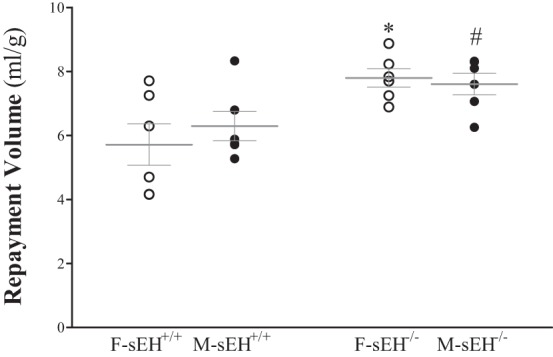
Comparison of repayment volume (RV) between female and male wild-type soluble epoxide hydrolase (sEH+/+) and sEH−/− (sEH-null) mice. Repayment volume was not significantly different between female (F-sEH+/+) and male (M-sEH+/+) WT mice (P > 0.05) and between female (F-sEH−/−) and male (M-sEH−/−) sEH-null mice (P > 0.05). However, M-sEH−/− had more enhanced (P = 0.04) repayment volume compared with M-sEH+/+. Also, F-sEH−/− had more enhanced (P = 0.01) repayment volume compared with F-sEH+/+. *P ≤ 0.05 vs. baseline F-sEH+/+. #P ≤ 0.05 vs. M-sEH+/+. n = 6 per group.
Figure 2 presents CRH comparison between sEH+/+ and sEH−/− mice. RV in sEH−/− was 28% higher compared with sEH+/+ mice (7.7 ± 0.4 and 6.0 ± 0.2 ml/g, respectively, P < 0.001, Fig. 2B), and the repayment/debt ratio was also 32% higher in sEH−/− compared with sEH+/+ mice (2.0 ± 0.1 and 1.5 ± 0.1, respectively, P < 0.001, Fig. 2C). The post hoc power analysis for the RV was 96.8%. RD (Fig. 2D), baseline CF, LVDP, and HR were not significantly different between the two groups.
Fig. 2.

Comparison of coronary reactive hyperemia (CRH) between wild type (sEH+/+) and sEH−/− (sEH-null) mice. A: tracing depicting coronary flow changes at baseline (before CRH) and after CRH was induced by 15-s no-flow ischemia in both sEH+/+ (dash line) and sEH−/− (continuous line) mice. Repayment volume (B; P < 0.001) and repayment/debt ratio (C; P = 0.0004) were enhanced in sEH−/− vs. sEH+/+ mice. Repayment duration (D) was increased in sEH−/− vs. sEH+/+, but did not reach statistical significance (P = 0.31). Peak hyperemic flow, baseline coronary flow, left-ventricular developed pressure (LVDP) and heart rate (HR) were not significantly different between the two groups (data not shown). *P ≤ 0.05 sEH+/+. n = 12 per group.
Oxylipin Analysis in Heart Perfusate Samples
Heart perfusate oxylipin levels were determined by LC-MS/MS. Perfusate samples were collected at baseline after stabilization and after the 15 s of ischemia in sEH+/+ and sEH−/− mice. Out of the four regioisomers of EETs, only 14,15-EET and its corresponding metabolite 14,15-DHET were detected. As expected, 14,15-EET was increased by 2.5 fold (P < 0.001), whereas 14,15-DHET was decreased (P < 0.001) in sEH−/− vs. sEH+/+ mice at baseline and post-ischemia (Fig. 3A). As a result, the 14,15-EET/DHET ratio was increased in sEH−/− vs. sEH+/+ mice (P < 0.001, Fig. 3B). The post hoc power analysis for the 14,15-EET/DHET ratio was 94.5%. There was no difference in either metabolite pre-ischemia or post-ischemia within the same strain (Fig. 3).
Fig. 3.

LC-MS/MS analysis for 14, 15-EETs and 14,15-DHETs in sEH+/+ and sEH−/− mice heart perfusate at baseline (preischemia) and directly after 15-s ischemia (post-ischemia). A: at baseline and post-ischemia, 14,15-EET was increased 2.5 fold in sEH−/− vs. sEH+/+ mice (P < 0.001), whereas 14,15-DHET was decreased (P < 0.001). There was no difference in either metabolite preischemia and postischemia within the same strain. B: ratio between 14,15-EET and 14,15-DHET was increased at baseline and postischemia in sEH−/− vs. sEH+/+ mice (P < 0.001). *P ≤ 0.05 vs. baseline sEH+/+. #P ≤ 0.05 vs. postischemia sEH+/+. n = 7 sEH+/+, n = 10 sEH−/−.
Our LC-MS/MS also detected 4 isomers of midchain HETEs: 5-, 11-, 12-, and 15-HETEs. All detected HETEs (5-, 11-, 12-, and 15-HETE) were significantly decreased postischemia compared with baseline in both sEH−/− and sEH+/+ mice (P = 0.02, P = 0.04, P = 0.05, and P = 0.03, respectively, Fig. 4, A–D), except for 12-HETE in sEH+/+ mice, which did not reach significance. There was no difference in any of the measured HETE metabolites between sEH−/− and sEH+/+ mice (Fig. 4, A–D). The post hoc power analysis for 5-, 11-, 12-, and 15-HETEs was 56%, 48%, 48%, and 73%, respectively.
Fig. 4.

LC-MS/MS analysis of HETEs in sEH+/+ and sEH−/− heart perfusate at baseline (preischemia) and directly after 15-s ischemia (post-ischemia). All detected HETEs (5-, 11-, 12-, and 15-HETE), were decreased postischemia (after perfusion was reinstated) compared with baseline in both sEH+/+ and sEH−/− mice (P = 0.02, P = 0.04, P = 0.05, and P = 0.03 respectively; A–D). Only 12-HETE in sEH+/+ mice did not reach statistically significant level (P = 0.09). There was no difference in any of the measured HETEs between sEH+/+ and sEH−/− mice (P > 0.05). *P ≤ 0.05 vs. baseline sEH+/+. #P ≤ 0.05 vs. baseline sEH−/−. n = 7 sEH+/+, n = 10 sEH−/−.
The 9,–10-epoxyoctadecaenoic acid (9,10-EpOME) was increased by 118% at baseline in sEH−/− vs. sEH+/+ mice (Fig. 5A, P = 0.03), but not postischemia (Fig. 5A, P > 0.05), whereas 12,13-EpOME was increased by 116% at baseline and 46% postischemia in sEH−/− vs. sEH+/+ mice (Fig. 5A, P = 0.03). However, 9,10- and 12,13-DiHOMEs, dihydroxyoctadecaenoic acids, were decreased by 59% and 77%, respectively, at baseline (P < 0.01), and by 48% and 76%, respectively, post-ischemia (Fig. 5B, P < 0.001) in sEH−/− vs. sEH+/+ mice. Interestingly, a decreasing trend in EpOMEs post-ischemia compared with baseline was observed in sEH−/− mice, but was not statistically significant (Fig. 5A, P = 0.21). As a result, 9,10-EpOME/DiHOME and 12,13-EpOME/DiHOME ratios were increased at baseline (Fig. 5C, P < 0.01), and postischemia (Fig. 5C, P < 0.01). The post hoc power analysis for 9,10-EpOME/DiHOME and 12,13-EpOME/DiHOME ratios were 88%, and 89%, respectively. Additionally, analysis of HODEs showed an increase in 13-HODE, but not 9-HODE, in sEH−/− vs. sEH+/+ mice at baseline and postischemia (Fig. 6, P < 0.01). The post hoc power analysis for 13-HODE analysis was 79%. There was no statistically significant change in EpOMEs, DiHOMEs, or HODEs preischemia and postischemia within the same group (Figs. 5 and 6, P > 0.05).
Fig. 5.

LC-MS/MS analysis of 9,–10-epoxyoctadecaenoic acids (EpOMEs) and dihydroxyoctadecaenoic acids (DiHOMEs) in sEH+/+ and sEH−/− mouse heart perfusate samples at baseline (preischemia) and directly after 15-s ischemia (postischemia). A: 9,10-EpOME was increased at baseline (P = 0.03) and postischemia, but was not statistically significant (P = 0.21), whereas 12,13-EpOME was increased both at baseline and postischemia (P = 0.03) in sEH−/− vs. sEH+/+ mice. B: 9,10- and 12,13-DiHOMEs were decreased at baseline (P = 0.001), and post-ischemia (P < 0.001) in sEH−/− vs. sEH+/+ mice. A decreasing trend in EpOMEs postischemia compared with baseline was observed in sEH−/− mice, but was not statistically significant. C: 9,10- and 12,13-EpOME/DiHOME ratios were increased at baseline (P = 0.002) and postischemia (P = 0.0008). *P ≤ 0.05 vs. baseline sEH+/+. #P ≤ 0.05 vs. postischemia sEH+/+. n = 7 sEH+/+, n = 10 sEH−/−.
Fig. 6.
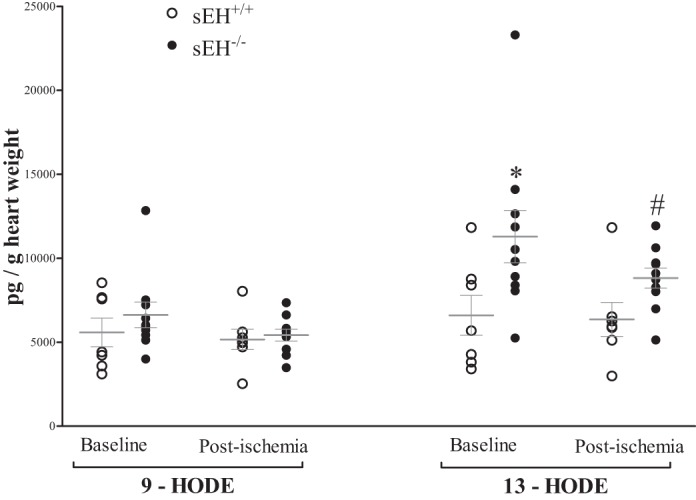
LC-MS/MS analysis of hydroxyoctadecadienoic acid (HODEs) in sEH+/+ and sEH−/− mouse heart perfusate samples at baseline (preischemia) and directly after 15-s ischemia (postischemia). Baseline and postischemia analysis 9- and 13-HODEs in sEH−/− vs. sEH+/+. 13-HODE, but not 9-HODE, was increased in sEH−/− vs. sEH+/+ mice at baseline and postischemia (P = 0.006). There was no change in either HODEs before and after ischemia in both groups (P > 0.05). *P ≤ 0.05 vs. baseline sEH+/+. #P ≤ 0.05 vs. postischemia sEH+/+. n = 7 sEH+/+, n = 10 sEH−/−.
Effect of PPARγ-Antagonist (T0070907) on CRH
Administering T0070907 decreased CRH in both sEH+/+ and sEH−/− mice (Fig. 7, A–C). In sEH+/+ mice, RV was decreased by 25% (from 5.8 ± 0.5 to 4.3 ± 0.3 ml/g, P < 0.001, Fig. 7A), repayment/debt (R/D) ratio was decreased by 14% (from 1.4 ± 0.1 to 1.2 ± 0.1, P < 0.01, Fig. 7B), repayment duration (RD) was decreased by 54% (from 2.4 ± 0.3 to 1.1 ± 0.2 min, P = 0.02 Fig. 7C). Similarly, in sEH−/− mice, repayment volume was decreased by 33% (from 7.9 ± 0.8 to 5.7 ± 0.8 ml/g, P < 0.01, Fig. 7A), repayment/debt ratio was decreased by 32% (from 2.2 ± 0.4 to 1.5 ± 0.2, P = 0.01, Fig. 7B), and repayment duration was decreased by 54% (from 2.8 ± 0.3 to 1.3 ± 0.1 min, P < 0.01, Fig. 7C). Comparing CRH responses between sEH−/− and sEH+/+ mice without T0070907: repayment volume and repayment/debt ratio were increased more in sEH−/− vs. sEH+/+ mice (P < 0.05 and P = 0.04, respectively; Fig. 7, A and B). The post hoc power analysis for the repayment volume was 76%. In both sEH+/+ and sEH−/− mice, baseline CF, peak PHF, HR, and LVDP were not changed by T0070907 (P > 0.05, Fig. 7, D–F).
Fig. 7.

Effect of PPARγ-antagonist (T0070907, 10 μM) on CRH in sEH+/+ and sEH−/− mice. Infusion of T0070907 decreased CRH in both sEH+/+and sEH−/− mice. In sEH+/+ mice, T0070907 decreased repayment volume (RV) by 25% (A; P < 0.001), repayment/debt (R/D) ratio by 14% (B; P = 0.004), and repayment duration (RD) by 54% (C; P = 0.02). In sEH−/−, T0070907 decreased RV by 33% (A; P = 0.0039), repayment/debt ratio by 32% (B; P = 0.01), and RD by 54% (C; P = 0.003). Comparing sEH−/− and sEH+/+ mice: repayment volume and repayment/debt ratio were increased more in sEH−/− vs. sEH+/+ mice (A: P = 0.048 and B: P = 0.04, respectively). In both sEH+/+ and sEH−/− mice, baseline CF (D), PHF (E), HR (F), and LVDP (data not shown) were not changed by T0070907. *P ≤ 0.05 vs. sEH+/+. #P ≤ 0.05 vs. sEH−/−. n = 6 per group.
Effect of PPARγ-Agonist (Rosiglitazone) on CRH
Infusion of rosiglitazone increased CRH in both sEH+/+ and sEH−/− mice. In sEH+/+ mice, repayment volume (RV) was increased by 37% (from 6.1 ± 0.4 to 8.4 ± 1.3 ml/g, P = 0.04, Fig. 8A), repayment duration (RD) was increased by 38% (from 2.1 ± 0.3 to 2.9 ± 0.4 min, P = 0.05, Fig. 8C), and baseline CF was increased by 13% (from 15.1 ± 0.2 to 17.1 ± 0.9 ml·min−1·g−1, P = 0.03, Fig. 8D). Although repayment/debt (R/D) ratio was increased by 25% (from 1.6 ± 0.1 to 2.0 ± 0.4), it was not statistically significant (Fig. 8B). Likewise, in sEH−/− mice, rosiglitazone increased repayment volume by 37% (from 7.6 ± 0.5 to 10.4 ± 1.1 ml/g, P = 0.04, Fig. 8A), repayment duration was increased by 50% (from 2.0 ± 0.1 to 3.0 ± 0.3 min, P = 0.01, Fig. 8C), and baseline CF was increased by 9% (from 14.0 ± 0.6 to 15.2 ± 0.7 ml·min−1·g−1, P = 0.04, Fig. 8D). Repayment/debt ratio was also increased by rosiglitazone (from 2.2 ± 0.1 to 2.7 ± 0.3), but was not statistically significant (P > 0.05, Fig. 8B). Comparison of CRH responses between sEH−/− and sEH+/+ mice without rosiglitazone: repayment volume and repayment/debt ratio were increased more in sEH−/− vs. sEH+/+ (P = 0.02 and P = 0.05, respectively, Fig. 8, A and B). The post hoc power analysis for the repayment volume was 87%. In both sEH+/+ and sEH−/− mice, PHF, HR, and LVDP were not changed by rosiglitazone (P > 0.05, Fig. 8, E and F).
Fig. 8.

Comparison CRH for wild-type (sEH+/+) mice before and after infusion of rosiglitazone (PPARγ-agonist, 10 μM); administration of rosiglitazone increased CRH in both sEH+/+and sEH−/− mice. In sEH+/+ mice, repayment volume (RV) was increased by 37% (A; P = 0.04), repayment/debt ratio (R/D) by 38% (B; P = 0.03), repayment duration (RD) by 38% (C; P = 0.05), and baseline coronary flow (CF) (D; P = 0.0280). In sEH−/− mice, rosiglitazone increased RV by 37% (A; P = 0.04), repayment/debt ratio (R/D) by 23% (B; P = 0.03), RD by 50% (C; P = 0.01), and baseline coronary flow (CF) (D; P = 0.04). Comparing sEH−/− and sEH+/+ mice: RV and R/D were increased more in sEH−/− vs. sEH+/+ mice (P = 0.02 and P = 0.05, respectively). In both sEH+/+ and sEH−/− mice, PHF (E), HR (F), and LVDP (data not shown) were not significantly changed by rosiglitazone. *P ≤ 0.05 vs. sEH+/+. #P ≤ 0.05 vs. sEH−/−. n = 6 sEH+/+, n = 7 sEH−/−.
Effect of l-NAME on CRH
Administering l-NAME decreased CRH in both sEH+/+ and sEH−/− mice. No statistically significant difference was observed between l-NAME-infused sEH+/+ and l-NAME-infused sEH−/− mice (P = 0.36, Fig. 9, A–C) In sEH+/+ mice, l-NAME decreased RV by 43% (from 6.5 ± 0.6 to 3.7 ± 0.3 ml/g, P < 0.01, Fig. 9A), decreased repayment duration (RD) by 47% (from 1.7 ± 0.2 to 0.9 ± 0.1 min, P < 0.01, Fig. 9C), decreased baseline CF by 36% (from 14.1 ± 0.7 to 9.0 ± 0.9 ml·min−1·g−1, P < 0.001, Fig. 9D), decreased PHF from 34.4 ± 0.8 to 31.2 ± 0.7 ml·min−1·g−1 (P < 0.01, Fig. 9E), and decreased HR from 366.7 ± 10.1 to 338.5 ± 18.5 beats/min (P = 0.01, Fig. 9F). R/D ratio (Fig. 9B) and LVDP (data not shown) were not different between the two groups. Similarly, in sEH−/− mice, l-NAME decreased repayment volume by 54% (from 8.9 ± 1.3 to 4.1 ± 0.3 ml/g, P < 0.01, Fig. 9A), decreased R/D ratio by 26% (from 2.7 ± 0.3 to 2.0 ± 0.1, P = 0.03, Fig. 8B), decreased repayment duration by 61% (from 2.3 ± 0.2 to 0.9 ± 0.1 min, P < 0.001, Fig. 9C), and decreased baseline CF by 36% (from 13.5 ± 0.9 to 8.6 ± 0.9 ml·min−1·g−1, P < 0.001, Fig. 9D). PHF, HR, and LVDP were not significantly changed by l-NAME (Fig. 9, E and F). Comparing CRH responses between sEH−/− and sEH+/+ mice without l-NAME treatment: repayment volume and repayment duration were increased more in sEH−/− vs. sEH+/+ mice (P = 0.01 and P = 0.01, respectively, Fig. 9, A and C). The post hoc power analysis for the repayment volume was 97%.
Fig. 9.

Comparison of the effect of Nω-nitro-l-arginine methyl ester hydrochloride (l-NAME) (nitric oxide synthase inhibitor) on CRH in sEH−/− and sEH+/+ mice before and after infusion of l-NAME (100 μM). l-NAME decreased CRH in both sEH+/+ and sEH−/− mice, and CRH responses between the two groups were not statistically significant (P = 0.36). In sEH+/+, l-NAME decreased RV by 43% (A; P = 0.002), RD by 47% (C; P = 0.002), baseline coronary flow (CF) by 36% (D; P < 0.001), peak hyperemic flow (PHF) (E; P = 0.001), and heart rate (HR) (F; P = 0.01). Repayment/debt (R/D) ratio (B) and LVDP (not shown) were not different between the two groups. In sEH−/− mice, l-NAME decreased RV by 54% (A; P = 0.005), R/D ratio by 26% (B; P = 0.03), RD by 61% (C; P < 0.001), and baseline CF by 36% (D; P < 0.001), PHF (E), HR (F) and LVPD (not shown) were not significantly changed by l-NAME. RV and RD were increased more in untreated sEH−/− vs. untreated sEH+/+ mice (A, P = 0.01 and C, P = 0.01, respectively). n = 8 per group. *P < 0.05 vs. sEH+/+. #P < 0.05 vs. sEH−/−.
We summarized our observed results into a proposed schematic diagram (Fig. 10).
Fig. 10.

A schematic diagram comparing the changes observed for selected oxylipins in response to short-lived ischemia and their possible impact on CRH between sEH−/− and sEH+/+ mice. sEH deletion enhanced CRH possibly through increased 14,15-EET/DHET ratio, increased 13-HODE, and increased EpOME/DiHOME ratio. Role of NO, PPARγ, and midchain HETEs was comparable in both sEH−/− and sEH+/+ mice.
DISCUSSION
Global deletion of soluble epoxide hydrolase (sEH−/−) in mice shows an increase in CRH in isolated heart model after a brief ischemia. The observed changes in oxylipins associated with sEH-deletion (increased EETs/DHETs, increased EpOMEs/DiHOMEs, and increased HODEs) could be involved in enhancing CRH. The relationship between sEH deletion, oxylipins, PPARγ, and CRH in isolated mouse hearts is unknown. Therefore, we designed this study to investigate the role of sEH deletion, and the associated changes in various oxylipins, in the modulation of CRH using sEH−/− and sEH+/+ mice. Our data demonstrate that, compared with sEH+/+, sEH−/− mice had 1) increased CRH, 2) increased 14,15-EET/DHET ratio, 3) increased EpOMEs/DiHOMEs ratio, and 4) increased 13-HODE. Also, both sEH+/+ and sEH−/− mice had 1) comparable role of PPARγ in mediating CRH, 2) similar response to l-NAME-mediated decrease in CRH, and 3) similar decrease in midchain HETEs postischemia.
In this study, sEH deletion in mice (sEH−/− mice) was associated with a statistically significant increase in CRH after a brief ischemia compared with sEH+/+ mice. Interestingly, there was no difference in CRH, as measured by repayment volume, between male and female mice in either strain (sEH−/− and sEH+/+). This contrasts to the sex difference in vascular response between male (M-sEH+/+) and female (F-sEH+/+) wild-type mice, where F-sEH+/+ exhibited increased flow/shear stress-induced dilator response (FID) in isolated gracilis muscle arterioles compared with M-sEH+/+ (44). In addition, Qin et al. (44) reported that sEH deletion in male (M-sEH−/−) enhanced FID to a level comparable to that observed in F-sEH−/− and female F-sEH+/+. Therefore, the authors suggested that deletion of sEH abolished the sex difference in FID response observed in wild-type mice (44). But, in our study, we did not observe any sex difference in CRH response. Although we used the same mouse strains (sEH−/− and sEH+/+) that Qin et al. used in their study, the lack of sex difference in CRH response in our study was probably due to different tissues studied [isolated mouse heart vs. leg gracilis muscle's second-order arterioles (44)] and the different triggering factor for vasodilation [CRH in response to ischemia vs. FID (44)].
CRH may be viewed as a protective mechanism and is attenuated in pathologic conditions. For example, CRH is decreased in cardiac hypertrophy (20), metabolic syndrome (5), unstable angina, myocardial infarction, congestive heart failure, and cardiac death (18). Out of the four EET isomers, only 14,15-EET was detected in the heart perfusate samples by LC-MS/MS. The other EET isomers (5,6-, 8,9-, and 11,12-EETs) were not detected, possibly because their level was lower than the level of 14,15-EET and lower than the detection power of the LC-MS/MS. In support of this speculation, Li et al. (27) reported that the level of 14,15-EET was approximately three times higher than that of 5,6-, 8,9-, and 11,12-EET isomers in mouse plasma samples. Our results demonstrate that in sEH−/− mice, the 14,15-EET/DHET ratio was increased. EETs are well established as substrates for sEH (31, 37). Also, in the presence of sEH, EETs are rapidly converted by hydration to their corresponding DHETs, resulting in loss of the beneficial cardiovascular effects of these epoxyeicosanoids (30, 35, 49, 56). Evidently, disruption of sEH causes EETs to accumulate, be retained for longer periods, and to continue to exert their beneficial effects (11, 35). Besides the main, sEH-catalyzed, breakdown pathway, EETs can be broken down through other minor pathways, such as ω-oxidation, β-oxidation, and chain elongation. The latter two pathways become more important when sEH activity is inhibited or deleted (19). None of these minor pathways has been studied or considered in our study. EETs also exert cardioprotective effects in ischemia/reperfusion injury (45). Deletion of the sEH gene increases endogenous EETs by decreasing the metabolic conversion of EETs into the less active DHETs. Our results suggest that CRH increases in mice due to sEH deletion, which is shown here to be associated with an increase in 14,15-EET/14,15-DHET ratio without changing baseline CF, HR, and LVDP.
This study also showed that CRH in sEH+/+ and sEH−/− mouse hearts was attenuated by T0070907 (PPARγ-antagonist), but enhanced by rosiglitazone (PPARγ-agonist). These results from PPARγ-antagonist and PPARγ-agonist experiments suggest that PPARγ receptors are involved in modulating CRH. Interestingly, our laboratory reported that sEH−/− mouse aorta had increased expression of PPARγ protein and enhanced vascular relaxation compared with sEH+/+ mice (35). Liu et al. (28) suggested that selective sEH inhibition potentiated the anti-inflammatory effect in endothelial cells, presumably by increasing the retention of EETs, which would activate PPARγ. These studies indicate that EETs' effects could be mediated by PPARγ receptors. However, our results did not show the same relationship; the effect of T0070907 and rosiglitazone were comparable between sEH+/+ and sEH−/− mice. Therefore, we found that CRH in sEH+/+ and sEH−/− mice may similarly be mediated by PPARγ.
Infusion of l-NAME, a nonselective NO synthase inhibitor, decreased CRH in both sEH+/+ and sEH−/− mice in the present study. These data suggest that the presence or absence of sEH does not have any specific effect on l-NAME-induced attenuation of CRH response in both sEH+/+ and sEH−/− mice. Similarly, Zhang et al. (57) described that the inhibition of the activity of sEH enzyme ameliorates endothelial dysfunction in the db/db mice, which is NO-independent, but dependent on cytochrome P-450-epoxygenase-derived metabolites (EETs). Our studies have shown that vascular relaxation through A2AAR was independent of NO and cyclooxygenase pathways (33, 34, 36). Also, others (9, 22, 27) reported that sEH−/− mice were associated with elevated EETs levels compared with sEH+/+ mice, and these EETs exerted cardioprotective effects against ischemia/reperfusion injury (45). Therefore, the enhanced CRH in sEH−/− compared with sEH+/+ mice seems to be independent of the NO pathway, but dependent on the changes associated with sEH-deletion. Although the cyclooxygenase (COX) pathway was found to be involved in myocardial reactive hyperemia in dogs, and a cross-talk between COX and nitric oxide pathways in dog coronary vessels was suggested (43), a later publication by Hellmann et al. (15) suggested that COX metabolites, prostanoids, were not involved in reactive hyperemia in human skin. Accordingly, the role of prostanoids in mouse CRH was not investigated in our study and, therefore, cannot be ruled out.
In addition to the increase in 14,15-EET/DHET ratio in sEH−/− mice, 13-hydroxylated linoleic acid (13-HODE, hydroxyoctadecadienoic acid) was also increased in sEH−/− vs. sEH+/+. 13-HODE may play an anti-inflammatory role and may act as a PPARγ-agonist in inflammatory diseases as reported by others (2, 3, 10, 13, 52). Also, De Meyer et al. (8) reported that 13-HODE was involved in canine splenic, as well as coronary artery relaxation through prostacyclin (PGI2) biosynthesis in both endothelial and smooth muscle cells. Also, in the present study, no change was observed in 9-HODE, which is considered proinflammatory, activating G protein-coupled receptor and JNK (14, 39) in both sEH−/− and sEH+/+ mice. Therefore, an increase in 13-HODE in sEH−/− mouse heart perfusate may possibly have a role in enhancing CRH.
Other linoleic acid metabolites, such as 9,10-,12,13-EpOMEs and 9,10- and 12,13-DiHOMEs were detected in sEH+/+ and sEH−/− mouse heart perfusate in the current study. The increased EpOMEs/DiHOMEs ratio was the result of increased EpOMEs and decreased DiHOMEs in sEH−/− vs. sEH+/+ mice. In support of our data, Lee et al. (26) reported that EpOMEs/DiHOMEs ratios were increased in response to sEH inhibition by AUDA in vivo and improved renal recovery against ischemia/reperfusion injury in C57BL/6 mice. Not only that, Nowak et al. (38) reported that primary cultures of rabbit renal proximal tubular cells were protected by the pretreatment of 12,13-EpOME against hypoxia/reoxygenation injury whereas, 12,13-DiHOME did not have a protective effect. On the basis of our findings, the increase in EpOME/DiHOME ratio in sEH−/− mouse heart perfusate may possibly play a role in enhancing CRH along with increased 14,15-EET/DHET ratio and 13-HODE.
Interestingly, 5-, 11-, 12-, and 15-HETEs data in the current study demonstrate a significant impact after 15 s of ischemia. Surprisingly, there was no change in the level of these mid-chain HETEs between sEH−/− and sEH+/+ mice. Midchain HETEs (5-, 11-, 12-, and 15-HETEs) are AA-derived metabolites produced by allylic oxidation of AA by the action of lipoxygenase (22). Increased formation of these midchain HETEs is involved in cardiovascular dysfunction through endothelial cells, smooth muscle cells, and monocytes (7, 41, 51, 55). These midchain HETEs had direct chemotaxis effects on vascular tone and on the production of endothelial vascular factors (17, 32, 50, 54). Our data show that 5-, 11-, 12-, and 15-HETEs were significantly decreased after a brief ischemia of 15 s in both sEH−/− and sEH+/+ mice. In contrast to our findings, Li et al. (27) reported that the plasma levels of 5-, 9-, 11-, and 15-HETEs were elevated in sEH−/− compared with sEH+/+ mice (27). This discrepancy could be explained by the different source of the samples; ours were from the isolated heart perfusate, whereas theirs were from the plasma (27). Nevertheless, CRH was enhanced more in sEH−/− compared with sEH+/+ mice. On the basis of the described effects of these midchain HETEs, CRH response to brief ischemia in both sEH−/− and sEH+/+ mice may have been enhanced by the decrease in 5-, 11-, 12-, and 15-HETEs. Also, deletion of sEH did not have an effect on the level of these HETEs in the heart perfusate samples.
In summary, the present data suggest that there is a relationship between the increase in CRH and deletion of sEH in both male and female mice. CRH is mediated, in part, by PPARγ activation in mice, as demonstrated by inhibition of CRH with a PPARγ antagonist and enhanced CRH with a PPARγ-agonist. Likewise, CRH is also dependent on nitric oxide (NO), but the enhanced CRH presented by sEH−/− mice cannot be solely explained through changes in PPARγ or NOS. However, deletion of sEH resulted in significant changes in several oxylipins, such as increased EET/DHET ratios, increased EpOME/DiHOME ratios, and increased 13-HODE. These oxylipins are reported to provide protection in other models of ischemia-reperfusion injury. Thus, our data suggest that these differences in oxylipin metabolism contribute to the enhanced CRH in sEH−/− mice. Therefore, we conclude that deletion of sEH enhances CRH through an increased 14,15-EET/DHET ratio, increased 13-HODE, and an increased EpOME/DiHOME ratio.
Perspectives and Significance
These findings demonstrate that sEH-deletion is associated with significant changes in arachidonic acid-derived metabolites (EETs), and linoleic acid-derived metabolites (EpOMEs, DiHOMEs, and HODEs). Any dysregulation in these oxylipin signaling pathways or allelic variations may possibly lead to disturbances in regular coronary circulation and may have clinical implications (24, 25), such as coronary artery disease in humans with blunted or compromised hyperemic response. Current findings suggest that deletion of sEH enhances CRH through increased 14,15-EET/DHET ratio, increased 13-HODE, and increased EpOME/DiHOME ratio. Therefore, targeting sEH through pharmacology could be a feasible option for clinical implementation in cardiovascular diseases, in which the hyperemic response is blunted or compromised.
GRANTS
This work was supported by National Institutes of Health (NIH; Grant HL-114559) to M. A. Nayeem and the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences (Z01 ES025034 to D. C. Zeldin). The West Virginia Clinical and Translational Science Institute is supported by the National Institute of General Medical Sciences of the NIH under award number U54GM104942.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
A.H. and M.A.N. conception and design of research; A.H. and M.L.E. performed experiments; A.H. and M.L.E. analyzed data; A.H. and M.A.N. interpreted results of experiments; A.H. prepared figures; A.H. drafted manuscript; A.H. and M.A.N. edited and revised manuscript; A.H., M.L.E., D.C.Z., C.M., and M.A.N. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Jianjun Zhang and the Department of Biostatistics at the School of Public Health, West Virginia University for their support with biostatistical analyses. We also thank Dr. Bunyen Teng and Dr. Xueping Zhou for their technical support and help with trouble shooting. Also, we thank Dr. Brandi Talkington and the West Virginia Clinical and Translational Science Institute for editorial support.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
REFERENCES
- 1.Al-Shabrawey M, Mussell R, Kahook K, Tawfik A, Eladl M, Sarthy V, Nussbaum J, El-Marakby A, Park SY, Gurel Z, Sheibani N, Maddipati KR. Increased expression and activity of 12-lipoxygenase in oxygen-induced ischemic retinopathy and proliferative diabetic retinopathy: implications in retinal neovascularization. Diabetes 60: 614–624, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Altmann R, Hausmann M, Spottl T, Gruber M, Bull AW, Menzel K, Vogl D, Herfarth H, Scholmerich J, Falk W, Rogler G. 13-Oxo-ODE is an endogenous ligand for PPARγ in human colonic epithelial cells. Biochem Pharmacol 74: 612–622, 2007. [DOI] [PubMed] [Google Scholar]
- 3.Belvisi MG, Mitchell JA. Targeting PPAR receptors in the airway for the treatment of inflammatory lung disease. Br J Pharmacol 158: 994–1003, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berwick ZC, Payne GA, Lynch B, Dick GM, Sturek M, Tune JD. Contribution of adenosine A2A and A2B receptors to ischemic coronary dilation: role of KV and KATP channels. Microcirculation 17: 600–607, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Borbouse L, Dick GM, Payne GA, Berwick ZC, Neeb ZP, Alloosh M, Bratz IN, Sturek M, Tune JD. Metabolic syndrome reduces the contribution of K+ channels to ischemic coronary vasodilation. Am J Physiol Heart Circ Physiol 298: H1182–H1189, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Campbell WB, Gebremedhin D, Pratt PF, Harder DR. Identification of epoxyeicosatrienoic acids as endothelium-derived hyperpolarizing factors. Circ Res 78: 415–423, 1996. [DOI] [PubMed] [Google Scholar]
- 7.Conrad DJ, Kuhn H, Mulkins M, Highland E, Sigal E. Specific inflammatory cytokines regulate the expression of human monocyte 15-lipoxygenase. Proc Natl Acad Sci USA 89: 217–221, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.De Meyer GR, Bult H, Verbeuren TJ, Herman AG. The role of endothelial cells in the relaxations induced by 13-hydroxy- and 13-hydroperoxylinoleic acid in canine arteries. Br J Pharmacol 107: 597–603, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Edin ML, Wang Z, Bradbury JA, Graves JP, Lih FB, DeGraff LM, Foley JF, Torphy R, Ronnekleiv OK, Tomer KB, Lee CR, Zeldin DC. Endothelial expression of human cytochrome P450 epoxygenase CYP2C8 increases susceptibility to ischemia-reperfusion injury in isolated mouse heart. FASEB J 25: 3436–3447, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Emerson MR, LeVine SM. Experimental allergic encephalomyelitis is exacerbated in mice deficient for 12/15-lipoxygenase or 5-lipoxygenase. Brain Res 17: 140–145, 2004. [DOI] [PubMed] [Google Scholar]
- 11.Fang X, Kaduce TL, Weintraub NL, Harmon S, Teesch LM, Morisseau C, Thompson DA, Hammock BD, Spector AA. Pathways of epoxyeicosatrienoic acid metabolism in endothelial cells. Implications for the vascular effects of soluble epoxide hydrolase inhibition. J Biol Chem 276: 14,867–14,874, 2001. [DOI] [PubMed] [Google Scholar]
- 12.Fleming I. The pharmacology of the cytochrome P450 epoxygenase/soluble epoxide hydrolase axis in the vasculature and cardiovascular disease. Pharmacol Rev 66: 1106–1140, 2014. [DOI] [PubMed] [Google Scholar]
- 13.Fritsche KL. Too much linoleic acid promotes inflammation-doesn't it? Prostaglandins Leukot Essent Fatty Acids 79: 173–175, 2008. [DOI] [PubMed] [Google Scholar]
- 14.Hattori T, Obinata H, Ogawa A, Kishi M, Tatei K, Ishikawa O, Izumi T. G2A plays proinflammatory roles in human keratinocytes under oxidative stress as a receptor for 9-hydroxyoctadecadienoic acid. J Invest Dermatol 128: 1123–1133, 2008. [DOI] [PubMed] [Google Scholar]
- 15.Hellmann M, Gaillard-Bigot F, Roustit M, Cracowski JL. Prostanoids are not involved in postocclusive reactive hyperaemia in human skin. Fundam Clin Pharmacol 29: 510–516, 2015. [DOI] [PubMed] [Google Scholar]
- 16.Hercule HC, Schunck WH, Gross V, Seringer J, Leung FP, Weldon SM, da Costa Goncalves A, Huang Y, Luft FC, Gollasch M. Interaction between P450 eicosanoids and nitric oxide in the control of arterial tone in mice. Arterioscler Thromb Vasc Biol 29: 54–60, 2009. [DOI] [PubMed] [Google Scholar]
- 17.Honda HM, Leitinger N, Frankel M, Goldhaber JI, Natarajan R, Nadler JL, Weiss JN, Berliner JA. Induction of monocyte binding to endothelial cells by MM-LDL: role of lipoxygenase metabolites. Arterioscler Thromb Vasc Biol 19: 680–686, 1999. [DOI] [PubMed] [Google Scholar]
- 18.Huang AL, Silver AE, Shvenke E, Schopfer DW, Jahangir E, Titas MA, Shpilman A, Menzoian JO, Watkins MT, Raffetto JD, Gibbons G, Woodson J, Shaw PM, Dhadly M, Eberhardt RT, Keaney JF Jr, Gokce N, Vita JA. Predictive value of reactive hyperemia for cardiovascular events in patients with peripheral arterial disease undergoing vascular surgery. Arterioscler Thromb Vasc Biol 27: 2113–2119, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Imig JD. Epoxides and soluble epoxide hydrolase in cardiovascular physiology. Physiol Rev 92: 101–130, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kingsbury MP, Turner MA, Flores NA, Bovill E, Sheridan DJ. Endogenous and exogenous coronary vasodilatation are attenuated in cardiac hypertrophy: a morphological defect? J Mol Cell Cardiol 32: 527–538, 2000. [DOI] [PubMed] [Google Scholar]
- 21.Klein LW, Agarwal JB, Schneider RM, Hermann G, Weintraub WS, Helfant RH. Effects of previous myocardial infarction on measurements of reactive hyperemia and the coronary vascular reserve. J Am Coll Cardiol 8: 357–363, 1986. [DOI] [PubMed] [Google Scholar]
- 22.Konkel A, Schunck WH. Role of cytochrome P450 enzymes in the bioactivation of polyunsaturated fatty acids. Biochim Biophys Acta 1: 210–222, 2011. [DOI] [PubMed] [Google Scholar]
- 23.Lee CR, Imig JD, Edin ML, Foley J, DeGraff LM, Bradbury JA, Graves JP, Lih FB, Clark J, Myers P, Perrow AL, Lepp AN, Kannon MA, Ronnekleiv OK, Alkayed NJ, Falck JR, Tomer KB, Zeldin DC. Endothelial expression of human cytochrome P450 epoxygenases lowers blood pressure and attenuates hypertension-induced renal injury in mice. FASEB J 24: 3770–3781, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee CR, North KE, Bray MS, Couper DJ, Heiss G, Zeldin DC. CYP2J2 and CYP2C8 polymorphisms and coronary heart disease risk: the Atherosclerosis Risk in Communities (ARIC) study. Pharmacogenet Genomics 17: 349–358, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee CR, North KE, Bray MS, Fornage M, Seubert JM, Newman JW, Hammock BD, Couper DJ, Heiss G, Zeldin DC. Genetic variation in soluble epoxide hydrolase (EPHX2) and risk of coronary heart disease: The Atherosclerosis Risk in Communities (ARIC) study. Hum Mol Genet 15: 1640–1649, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee JP, Yang SH, Lee HY, Kim B, Cho JY, Paik JH, Oh YJ, Kim DK, Lim CS, Kim YS. Soluble epoxide hydrolase activity determines the severity of ischemia-reperfusion injury in kidney. PLoS One 7: 10, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li L, Li N, Pang W, Zhang X, Hammock BD, Ai D, Zhu Y. Opposite effects of gene deficiency and pharmacological inhibition of soluble epoxide hydrolase on cardiac fibrosis. PLoS One 9, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu Y, Zhang Y, Schmelzer K, Lee TS, Fang X, Zhu Y, Spector AA, Gill S, Morisseau C, Hammock BD, Shyy JY. The antiinflammatory effect of laminar flow: the role of PPARγ, epoxyeicosatrienoic acids, and soluble epoxide hydrolase. Proc Natl Acad Sci USA 102: 16,747–16,752, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Luria A, Weldon SM, Kabcenell AK, Ingraham RH, Matera D, Jiang H, Gill R, Morisseau C, Newman JW, Hammock BD. Compensatory mechanism for homeostatic blood pressure regulation in Ephx2 gene-disrupted mice. J Biol Chem 282: 2891–2898, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moghaddam MF, Grant DF, Cheek JM, Greene JF, Williamson KC, Hammock BD. Bioactivation of leukotoxins to their toxic diols by epoxide hydrolase. Nat Med 3: 562–566, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morisseau C, Hammock BD. Epoxide hydrolases: mechanisms, inhibitor designs, and biological roles. Annu Rev Pharmacol Toxicol 45: 311–333, 2005. [DOI] [PubMed] [Google Scholar]
- 32.Nakao J, Ooyama T, Ito H, Chang WC, Murota S. Comparative effect of lipoxygenase products of arachidonic acid on rat aortic smooth muscle cell migration. Atherosclerosis 44: 339–342, 1982. [DOI] [PubMed] [Google Scholar]
- 33.Nayeem MA, Poloyac SM, Falck JR, Zeldin DC, Ledent C, Ponnoth DS, Ansari HR, Mustafa SJ. Role of CYP epoxygenases in A2A AR-mediated relaxation using A2A AR-null and wild-type mice. Am J Physiol Heart Circ Physiol 295: H2068–H2078, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nayeem MA, Ponnoth DS, Boegehold MA, Zeldin DC, Falck JR, Mustafa SJ. High-salt diet enhances mouse aortic relaxation through adenosine A2A receptor via CYP epoxygenases. Am J Physiol Regul Integr Comp Physiol 296: R567–R574, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nayeem MA, Pradhan I, Mustafa SJ, Morisseau C, Falck JR, Zeldin DC. Adenosine A2A receptor modulates vascular response in soluble epoxide hydrolase-null mice through CYP-epoxygenases and PPARγ. Am J Physiol Regul Integr Comp Physiol 304: R23–R32, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nayeem MA, Zeldin DC, Boegehold MA, Falck JR. Salt modulates vascular response through adenosine A2A receptor in eNOS-null mice: role of CYP450 epoxygenase and soluble epoxide hydrolase. Mol Cell Biochem 350: 101–111, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Newman JW, Morisseau C, Hammock BD. Epoxide hydrolases: their roles and interactions with lipid metabolism. Prog Lipid Res 44: 1–51, 2005. [DOI] [PubMed] [Google Scholar]
- 38.Nowak G, Grant DF, Moran JH. Linoleic acid epoxide promotes the maintenance of mitochondrial function and active Na+ transport following hypoxia. Toxicol Lett 147: 161–175, 2004. [DOI] [PubMed] [Google Scholar]
- 39.Obinata H, Izumi T. G2A as a receptor for oxidized free fatty acids. Prostagl Other Lipid Mediat 89: 66–72, 2009. [DOI] [PubMed] [Google Scholar]
- 40.Park S, Lim S, Chang W, Song H, Lee S, Song BW, Kim HJ, Cha MJ, Choi E, Jang Y, Chung N, Cho SY, Hwang KC. The inhibition of insulin-stimulated proliferation of vascular smooth muscle cells by rosiglitazone is mediated by the Akt-mTOR-P70S6K pathway. Yonsei Med J 49: 592–600, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Patricia MK, Kim JA, Harper CM, Shih PT, Berliner JA, Natarajan R, Nadler JL, Hedrick CC. Lipoxygenase products increase monocyte adhesion to human aortic endothelial cells. Arterioscler Thromb Vasc Biol 19: 2615–2622, 1999. [DOI] [PubMed] [Google Scholar]
- 42.Pradhan I, Ledent C, Mustafa SJ, Morisseau C, Nayeem MA. High salt diet modulates vascular response in AAR and A AR mice: role of sEH, PPARγ, and K channels. Mol Cell Biochem 404: 87–96, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Puybasset L, Bea ML, Ghaleh B, Giudicelli JF, Berdeaux A. Coronary and systemic hemodynamic effects of sustained inhibition of nitric oxide synthesis in conscious dogs. Evidence for cross talk between nitric oxide and cyclooxygenase in coronary vessels. Circ Res 79: 343–357, 1996. [DOI] [PubMed] [Google Scholar]
- 44.Qin J, Kandhi S, Froogh G, Jiang H, Luo M, Sun D, Huang A. Sexually dimorphic phenotype of arteriolar responsiveness to shear stress in soluble epoxide hydrolase-knockout mice. Am J Physiol Heart Circ Physiol 309: H1860–H1866, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Seubert J, Yang B, Bradbury JA, Graves J, Degraff LM, Gabel S, Gooch R, Foley J, Newman J, Mao L, Rockman HA, Hammock BD, Murphy E, Zeldin DC. Enhanced postischemic functional recovery in CYP2J2 transgenic hearts involves mitochondrial ATP-sensitive K+ channels and p42/p44 MAPK pathway. Circ Res 95: 506–514, 2004. [DOI] [PubMed] [Google Scholar]
- 46.Sharifi-Sanjani M, Zhou X, Asano S, Tilley S, Ledent C, Teng B, Dick GM, Mustafa SJ. Interactions between A2A adenosine receptors, hydrogen peroxide, and KATP channels in coronary reactive hyperemia. Am J Physiol Heart Circ Physiol 304: H1294–H1301, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sinal CJ, Miyata M, Tohkin M, Nagata K, Bend JR, Gonzalez FJ. Targeted disruption of soluble epoxide hydrolase reveals a role in blood pressure regulation. J Biol Chem 275: 40,504–40,510, 2000. [DOI] [PubMed] [Google Scholar]
- 48.Skrzypiec-Spring M, Grotthus B, Szelag A, Schulz R. Isolated heart perfusion according to Langendorff—still viable in the new millennium. J Pharmacol Toxicol Methods 55: 113–126, 2007. [DOI] [PubMed] [Google Scholar]
- 49.Spector AA, Fang X, Snyder GD, Weintraub NL. Epoxyeicosatrienoic acids (EETs): metabolism and biochemical function. Prog Lipid Res 43: 55–90, 2004. [DOI] [PubMed] [Google Scholar]
- 50.Stern N, Natarajan R, Tuck ML, Laird E, Nadler JL. Selective inhibition of angiotensin-II-mediated aldosterone secretion by 5-hydroxyeicosatetraenoic acid. Endocrinology 125: 3090–3095, 1989. [DOI] [PubMed] [Google Scholar]
- 51.Stern N, Yanagawa N, Saito F, Hori M, Natarajan R, Nadler J, Tuck M. Potential role of 12 hydroxyeicosatetraenoic acid in angiotensin II-induced calcium signal in rat glomerulosa cells. Endocrinology 133: 843–847, 1993. [DOI] [PubMed] [Google Scholar]
- 52.Stoll LL, Morland MR, Spector AA. 13-HODE increases intracellular calcium in vascular smooth muscle cells. Am J Physiol Cell Physiol 266: C990–C996, 1994. [DOI] [PubMed] [Google Scholar]
- 53.Suryapranata H, Zijlstra F, MacLeod DC, van den Brand M, de Feyter PJ, Serruys PW. Predictive value of reactive hyperemic response on reperfusion on recovery of regional myocardial function after coronary angioplasty in acute myocardial infarction. Circulation 89: 1109–1117, 1994. [DOI] [PubMed] [Google Scholar]
- 54.Tang DG, Renaud C, Stojakovic S, Diglio CA, Porter A, Honn KV. 12(S)-HETE is a mitogenic factor for microvascular endothelial cells: its potential role in angiogenesis. Biochem Biophys Res Commun 211: 462–468, 1995. [DOI] [PubMed] [Google Scholar]
- 55.Wen Y, Nadler JL, Gonzales N, Scott S, Clauser E, Natarajan R. Mechanisms of ANG II-induced mitogenic responses: role of 12-lipoxygenase and biphasic MAP kinase. Am J Physiol Cell Physiol 271: C1212–C1220, 1996. [DOI] [PubMed] [Google Scholar]
- 56.Zeldin DC. Epoxygenase pathways of arachidonic acid metabolism. J Biol Chem 276: 36,059–36,062, 2001. [DOI] [PubMed] [Google Scholar]
- 57.Zhang LN, Vincelette J, Chen D, Gless RD, Anandan SK, Rubanyi GM, Webb HK, MacIntyre DE, Wang YX. Inhibition of soluble epoxide hydrolase attenuates endothelial dysfunction in animal models of diabetes, obesity and hypertension. Eur J Pharmacol 654: 68–74, 2011. [DOI] [PubMed] [Google Scholar]
- 58.Zhou X, Teng B, Tilley S, Mustafa SJ. A1 adenosine receptor negatively modulates coronary reactive hyperemia via counteracting A2A-mediated H2O2 production and KATP opening in isolated mouse hearts. Am J Physiol Heart Circ Physiol 305: H1668–H1679, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]