

Abstract
Immune cells in the kidney are implicated in the development of hypertension and renal damage in the Dahl salt-sensitive (SS) rat. Interestingly, interleukin 6 (IL-6) mRNA is 54-fold higher in T-lymphocytes isolated from the kidney compared with circulating T-lymphocytes. The present experiments assessed the role of IL-6 in the development of SS hypertension by treating rats (n = 13–14/group) with an IL-6 neutralizing antibody or normal IgG during an 11-day period of high-salt (4.0% NaCl chow) intake. The mean arterial pressure (MAP) and urine albumin excretion rates (Ualb) were not different between the groups fed low salt (0.4% NaCl). Following 11 days of drug treatment and high salt, however, the rats receiving anti-IL-6 demonstrated a 47% reduction of IL-6 in the renal medulla compared with control SS. Moreover, the increase in MAP following 11 days of high-NaCl intake was significantly attenuated in SS administered anti-IL-6 compared with the control group (138 ± 3 vs. 149 ± 3 mmHg) as was the salt-induced increase in Ualb and glomerular and tubular damage. To investigate potential mechanisms of action, a flow cytometric analysis of immune cells in the kidney (n = 8–9/group) demonstrated that the total number of monocytes and macrophages was significantly lower in the treatment vs. the control group. The total number of T- and B-lymphocytes in the kidneys was not different between groups. These studies indicate that IL-6 production may participate in the development of SS hypertension and end-organ damage by mediating increased infiltration or proliferation of macrophages into the kidney.
Keywords: kidney, hypertension, cytokines, immune system, rats
hypertension is a leading cause of morbidity and mortality worldwide (24). Approximately 80 million people, age >20 years, in the United States have high blood pressure (>140/90 mmHg) (33). Hypertension is a major risk factor of cardiovascular disease (43), stroke (21), and chronic kidney disease (CKD) (14, 18). Chronic elevation of arterial pressure leads to reduced glomerular filtration rate, albuminuria, and eventually renal failure (40).
Several studies have implicated immune cells in clinical and experimental hypertension (15, 19, 31, 32). Multiple groups have demonstrated the presence of lymphocytes (8, 9) and macrophages (35) in the kidneys of hypertensive animal models. Other studies have shown that manipulation of the immune system via immunosuppressive therapy (3, 27), thymectomy (20), or genetic alteration of immune cells (29, 37) attenuated the development of hypertension and kidney disease. Our laboratory recently focused on the role of immune cells in the amplification of salt-sensitive hypertension in Dahl salt-sensitive (SS) rats (9, 10). Experiments have demonstrated that an elevated sodium chloride intake in SS rats is associated with increased T-lymphocytes and macrophages in the renal interstitium, a progressive elevation of arterial blood pressure, albuminuria, and renal histological damage (9, 10). These phenotypes are remarkably similar to those observed in patients (1, 13). Subsequent experiments in the Dahl SS demonstrated that pharmacological treatments or genetic manipulations that inhibit the immune system result in an attenuation of salt-sensitive hypertension. These data demonstrate that the immune system amplifies salt-sensitive hypertension and kidney damage in SS rats.
Numerous experiments have assessed the mechanisms by which immune cells may influence kidney disease and hypertension. It is known that immune cells in the kidney release a number of different cytokines, and the accumulation and action of these inflammatory molecules in target organs have been implicated in the exacerbation of hypertensive pathology (11, 32). Several inflammatory cytokines, including TNF-α (12), IL-17 (26), IFN-γ (39), and IL-6 (7, 22), have been shown to play a role in the development of hypertension. Previous data from our laboratory showed that T-lymphocytes isolated from the damaged kidneys of Dahl SS rats fed high salt expressed significantly greater mRNA of cytokines and other inflammatory molecules compared with circulating T-lymphocytes (37). Notably, interleukin-6 mRNA expression was 54-fold higher in T-cells in the kidney compared with circulating T-cells. These studies indicate a possible role of IL-6 in the development of salt-sensitive hypertension.
IL-6 is a proinflammatory cytokine produced by various cells including macrophages, T-lymphocytes, endothelial cells, and vascular smooth muscle cells (16). It plays a critical role in driving the inflammatory process in various autoimmune diseases such as rheumatoid arthritis and multiple sclerosis (34, 36). IL-6 has been implicated as causative in models of experimental hypertension including angiotensin II (ANG II) (22) and cold-induced (7) hypertension. Human data showed that IL-6 is an independent risk factor for hypertension in otherwise healthy human subjects (4) and that serum IL-6 concentration is significantly higher in hypertensive patients compared with normotensive patients (6). Experimental studies have demonstrated that IL-6−/− mice have attenuated hypertension and albuminuria when exposed to ANG II infusion and high salt, further supporting a possible role for IL-6 in the development of hypertension (5, 22). Additionally, IL-6 mRNA expression is increased in kidney biopsies of CKD patients compared with a normal control group, and is even further elevated in biopsies of CKD patients with hypertension (46). IL-6 mRNA expression was also found to be significantly elevated in the kidneys of mice after 2 wk of ANG II infusion compared with saline-infused mice (46). These findings, along with our previous observation that infiltrating renal T-cells in the Dahl SS kidney significantly upregulate IL-6 (37), suggest that IL-6 may exacerbate Dahl SS pathology. The current studies were therefore performed to test the hypothesis that IL-6 mediates salt-sensitive hypertension and renal damage in Dahl SS rats.
METHODS
Study animals.
Studies were performed on male SS/JrHsdMcwi (Dahl SS) rats obtained from a colony at the Medical College of Wisconsin. Study animals were fed purified AIN-76A rodent diet (Dyets, Bethlehem, PA) containing 0.4% NaCl from weaning. Starting at 9 wk of age, the salt content of the diet was increased to 4.0% NaCl. The Institutional Animal Care and Use Committee of the Medical College of Wisconsin approved all studies.
Study protocol.
Experiments were performed on 57 rats; for technical and logistical reasons, all measurements were not made on every animal. At 9 wk of age, the dietary salt content was increased from a low (0.4% NaCl)- to a high-salt diet (4.0% NaCl). Animals were randomized into two groups to receive a daily intraperitoneal injection of either vehicle (normal goat IgG; 4 μg/day in PBS) or goat anti-rat IL-6 neutralizing antibody (anti-rat IL-6; 4 μg/day, R&D Systems, Minneapolis, MN) during the 11-day period of high-salt intake (n = 13–16/group). In the interventional experiments, lyophilized anti-rat IL-6 antibody and goat IgG were diluted in sterile PBS, according to the manufacturer's instructions. The anti-IL-6 group received daily intraperitoneal injections of 4 μg (200 μl) of anti-rat IL-6 antibody, whereas the IgG group received injections of 4 μg (200 μl) of goat IgG (2, 41, 44).
Surgical preparation.
Surgical procedures were performed on rats deeply anesthetized with an intraperitoneal injection of ketamine (49 mg/kg), xylazine (7 mg/kg), and acepromazine (1.4 mg/kg) with supplemental inhalational anesthesia (isoflurane) administered when needed. With the use of aseptic technique, rats were instrumented with telemetry transmitters (Data Sciences International, St. Paul, MN) with the catheter implanted in the carotid artery. Both antibiotic (25 mg/kg sc Cefazolin) and analgesia (5 mg/kg sc Carpofen) were administered postsurgically. Following a week of recovery, blood pressure was continuously monitored for 5 days while the rats were maintained on the low-salt (0.4% NaCl) diet and for 11 days following the transition to the high-salt (4.0% NaCl) diet. Overnight urine collections were obtained in metabolic cages while rats were on low-salt diet and on days 7 and 11 of the high-salt diet. Urine studies were performed to assess urinary creatinine, albumin, and protein excretion rates as well as urinary sodium and potassium excretion rates. After 11 days of high-salt intake, the rats were deeply anesthetized and the kidneys were flushed with heparinized saline and removed for histology, ELISA, and immune cell isolation. A final arterial blood sample was also obtained for serum creatinine and electrolyte analysis.
Histological analysis of kidney tissues.
Kidneys (n = 6 per group) were obtained for histological analysis from rats maintained on the high-salt diet and treated with vehicle or anti-rat IL-6 antibody as described above. The rats were deeply anesthetized with pentobarbital sodium (83 mg/kg ip); the kidneys were flushed with heparinized saline and then removed, decapsulated, weighed, and placed in 10% formaldehyde. The tissue was paraffin-embedded and stained with Masson's trichrome. Slides were photographed using a Nikon E-400 fitted with a Spot Insight camera; digital micrographs were taken at different magnifications. Individual glomeruli (40 per rat) were evaluated using the semiquantitative index method and scored from 0 (best) to 4 (worst) on the basis of glomerulosclerosis and mesangial expansion as we described previously (27, 28, 30). The percentage of the outer medullary tissue containing blocked tubules filled with protein casts was quantified by determining the proportion of red-stained structures in this region using Metamorph Image Analysis software (version 4.6, Universal Imaging Systems) as we described previously (27, 28, 30). The grading of glomerular and medullary damage was performed in a blinded manner.
Immunohistochemistry was performed to localize ED1 staining in the kidneys of Dahl SS/Mcw rats fed the 4.0% NaCl diet as we previously described (27). Briefly, excised kidneys were fixed in 10% formaldehyde, paraffin-embedded, cut in 3-μm sections, and mounted as described above. Sections were then deparaffinized with xylene and ethanol and incubated with Proteinase K for antigen retrieval. Endogenous biotin and peroxidase activity were blocked by incubation with avidin and biotin, and hydrogen peroxide, respectively. The primary ED1 monoclonal antibody (MCA341R; Serotec) was used at a dilution of 1:400 and incubated at room temperature for 60 min. A biotinylated horse anti-mouse secondary antibody was used for development with avidin-biotinylated horseradish peroxidase complex (Vectastain ABC kits; Vector Laboratory, Burlingame, CA) and 0.02% H2O2 + 0.1% diaminobenzidine tetrahydrochloride (DAB). The slides were lightly counterstained with aniline blue dye to assist in tissue visualization and determination if tissue damage and macrophage infiltration were colocalized.
Quantification of IL-6.
An ELISA (Thermo Scientific, Pierce Biotechnology, Rockford, IL) was performed to assess IL-6 levels in the kidney tissue of Dahl SS rats maintained on the high-salt diet and treated with vehicle or anti-IL-6 as described above. The rats were euthanized with an overdose of pentobarbital sodium (50 mg/kg ip), and the kidneys were flushed with heparinized saline and removed, decapsulated, and separated into cortical and medullary sections, and frozen on dry ice. Isolated tissue was stored at −80°C until protein extraction. Total protein from kidneys was extracted using RIPA buffer (Thermo Scientific). Briefly, 20 mg of flash-frozen tissue were added to 1 ml of RIPA buffer; tissue was homogenized using a mortar and pestle; the homogenized solution was then incubated at 4°C for 2 h on a rocker platform. The sample was then centrifuged at 12,000 rpm for 20 min to pellet debris. The supernatant containing total protein was transferred to a new tube. The protein concentration was determined using DC Protein Assay (Bio-Rad) with albumin as a standard.
Immune cell isolation and flow cytometry.
A separate group of male rats was treated with anti-IL-6 antibody or control IgG for 11 days, starting at 9 wk of age, while maintained on 4% NaCl diet. After 11 days of high-salt intake, animals were anesthetized with pentobarbital sodium (50 mg/kg ip) and immune cells were isolated from the kidney as previously described (37, 38). Briefly, the kidneys were flushed with heparinized PBS, and the tissue was then minced and incubated in RPMI-1640 (Gibco) containing FBS (5%) and collagenase type IV (0.1%). The cell suspension was filtered through 70- and 40-μm cell strainers and mononuclear cells were separated by density gradient centrifugation over Histopaque-1083. The mononuclear cells were then counted on a hemocytometer and incubated with PE-Cy7-anti-CD45 (Biolegend) for leukocytes, PerCP-eFluor 710-anti-CD3 (eBioscience) for T-lymphocytes, APC-anti-CD45R (Becton Dickinson) for B-lymphocytes, and FITC-anti-CD11b/c (Becton Dickinson) for macrophages and monocytes. Cell viability was assessed using DAPI. The cells were analyzed by flow cytometry (FACS Diva or LSRII, Becton Dickinson) with FlowJo software (Tree Star).
Statistical analysis.
Data are expressed as means ± 1 SE. Data were assessed for significance using a t-test, a one-way repeated-measures ANOVA, or a two-way repeated-measures ANOVA as applicable. A probability value of <0.05 was considered significant.
RESULTS
Initial studies assessed the effectiveness of the anti-IL-6 treatment compared with the IgG control treatment. Tissue IL-6 concentration in the homogenized renal medulla, measured by ELISA, was significantly lower in the group treated with anti-IL-6 compared with the control group (954 ± 133 pg/100 μg of homogenized tissue protein vs. 1,802 ± 336 pg/100 μg of protein; n = 4–5/group; P = 0.04), demonstrating the effectiveness of this treatment regimen. The mean arterial pressure (MAP) values, expressed as the daily mean of continuous measurements, in conscious Dahl SS rats treated with anti-IL-6 antibody or vehicle are illustrated in Fig. 1 (n = 13–14/group). The average MAP while maintained on 0.4% NaCl low-salt diet (before anti-IL-6 or vehicle treatment) was not different between the groups (121 ± 2 vs. 121 ± 1 mmHg; P = 0.8). However, when the groups were switched to high salt (4.0% NaCl) and received vehicle or anti-IL-6 treatment, the MAP diverged. During the final 3 days (days 9–11) of high-salt diet, the average MAP was significantly lower by 6, 7, and 9 mmHg, respectively, in the anti-IL-6-treated group compared with the vehicle-treated rats (P = 0.03, 0.19, and <0.001). Furthermore, the onset of salt-sensitive hypertension was delayed in the anti-IL-6-treated group. After being switched to high-salt chow, the average MAP became significantly higher than low-salt values following 24 h of high salt in the vehicle-treated group vs. 48 h in the IL-6 inhibitor-treated group (P < 0.05). The changes in systolic and diastolic arterial pressure in the rats paralleled the increase in MAP, increasing from 136 ± 2 to 163 ± 2 and 110 ± 2 to 133 ± 5 mmHg, respectively, in the control rats from the final day of low salt to the end of the experiment. Differences in diastolic pressure between the groups were observed on the final day of the experiment, and systolic blood pressure was elevated in the vehicle-treated group compared with the anti-IL-6-treated group on the final 3 days of high-salt feeding. Heart rate was not significantly different between the groups at any time point, although heart rate significantly decreased during the high-salt period in each group; heart rate significantly decreased from 430 ± 5 to 378 ± 7 beats/min from the final day of low salt through the end of the experiment in vehicle-treated rats.
Fig. 1.
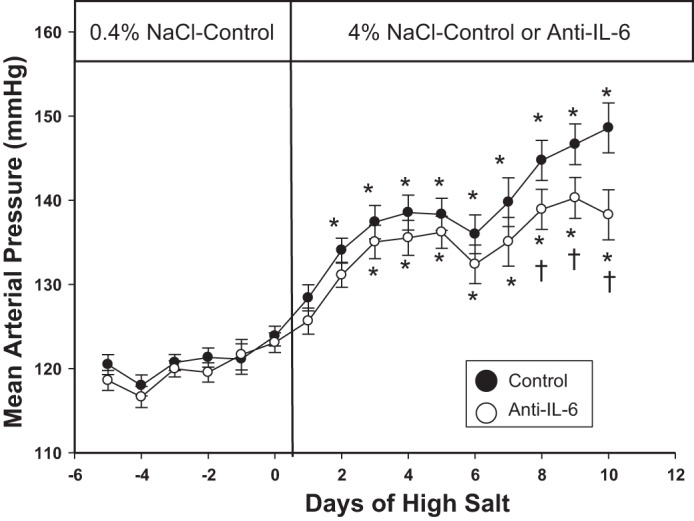
Average 24-h mean arterial pressure in Dahl salt-sensitive (SS) rats fed 0.4% NaCl chow and following transition to 4.0% NaCl chow while administered IgG control or anti-IL-6 (4 μg/day ip; n = 13–14/group). *P < 0.05 vs. values obtained on the last day of 0.4% NaCl chow. †P < 0.05 vs. control on the same diet.
Urinary albumin and protein excretion rates, as markers of renal damage, are illustrated in Fig. 2 (n = 15–16/group; these are the same rats assessed for blood pressure with the addition of four rats with transmitters that failed). The average urine albumin and protein excretion rates while the rats were maintained on the low-salt diet (before anti-IL-6 or vehicle treatment) were similar between the groups. After 6 days of high-salt intake, both urinary albumin and protein excretion rates were significantly increased compared with low-salt values, but not different between the vehicle-treated (81 ± 7 and 159 ± 13 mg/day) and anti-IL-6-treated group (61 ± 6 and 134 ± 11 mg/day; P = 0.06 and 0.13, respectively). Associated with the reduced MAP in the anti-IL-6 group, the urine albumin excretion rate was significantly lower in the anti-IL-6 group compared with the vehicle-treated group after 11 days of treatment (110 ± 10 vs. 151 ± 16 mg/day; P = 0.03). Similarly, the protein excretion rate was also significantly lower in the anti-IL-6-treated vs. the vehicle-treated group after 11 days of treatment (193 ± 17 vs. 252 ± 20 mg/day; P = 0.03).
Fig. 2.
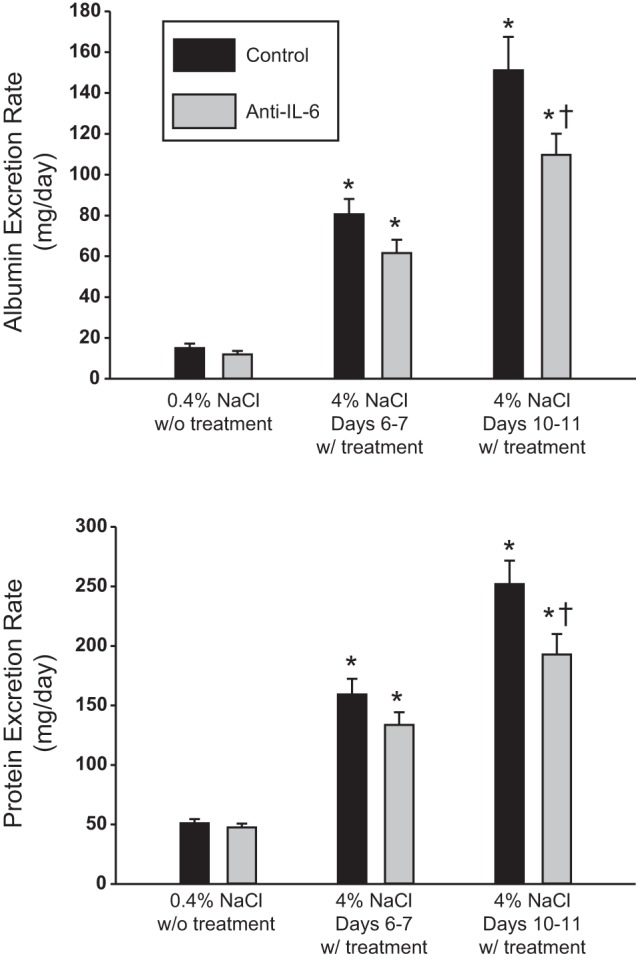
Urinary albumin (top) and protein excretion rate (bottom) in Dahl SS rats maintained on 0.4 and 4.0% NaCl while administered IgG control or anti-IL-6 (4 μg/day ip; n = 15–16/group). *P < 0.05 vs. values obtained on the last day of 0.4% NaCl chow. †P < 0.05 vs. control on the same diet.
Body weight and other parameters are summarized in Table 1. Body weight was not different between the vehicle- and anti-IL-6-treated groups, nor was total kidney weight. Urinary sodium and potassium excretion rates on the low-salt diet and after 11 days of high-salt intake were also not different between the groups. The similar body weight and steady-state sodium and potassium excretion rates indicated equivalent food consumption in the groups. Conscious creatinine clearance, measured at the conclusion of experiment, was also comparable between the vehicle- and anti-IL-6-treated groups.
Table 1.
Mean body and kidney weights
| Treatment | IgG Control | Anti-IL-6 |
|---|---|---|
| Body weight, g | 330 ± 8 | 341 ± 7 |
| Kidney weight, g | 3.2 ± 0.1 | 3.2 ± 0.1 |
| Low-salt sodium excretion rate, meq/day | 1.2 ± 0.7 | 1.1 ± 0.1 |
| Low-salt potassium excretion rate, meq/day | 1.3 ± 0.05 | 1.3 ± 0.03 |
| High-salt sodium excretion rate, meq/day | 16.2 ± 0.5 | 16.9 ± 1.4 |
| High-salt potassium excretion rate, meq/day | 1.6 ± 0.05 | 1.7 ± 0.1 |
| High-salt creatinine clearance rate, ml·min−1·g kidney wt−1 | 0.47 ± 0.02 | 0.50 ± 0.02 |
Data are means ± SE. Mean body and kidney weights are shown at the end of the experiment, sodium and potassium excretion rates when the rats were fed the low-salt (0.4% NaCl) or high-salt (4.0% NaCl) diet, and creatinine clearance rates, normalized to kidney weight (kidney wt), in salt-sensitive rats administered IgG control or anti-IL-6 (4 μg/day ip; n = 15–16/group).
Representative histological images of kidneys obtained from the vehicle- and anti-IL-6-treated rats after 11 days on the 4% NaCl diet are illustrated in Fig. 3. Glomerular and mesangial hypercellularity and tubular casts are present in both groups. The kidneys of vehicle-treated rats had glomerular sclerosis (seen as blue fibrotic tissue and collapsed capillary tufts) and a greater degree of protein casts (tubules filled with pink protein deposits), indicating tubular damage. The glomerular injury index was significantly lower in the treated vs. control group, averaging 1.9 ± 0.1 vs. 2.3 ± 0.1 (P = 0.0008). The percentage of outer medulla composed of protein casts, indicative of tubular damage, was 11.1 ± 1.2% in the treated group vs. 16.2 ± 2.5% in the control group (P = 0.09).
Fig. 3.

Light microscopy of trichrome-stained sections of renal cortex and outer medulla (×20 original magnification) of Dahl SS rats fed 4.0% NaCl while administered IgG vehicle or anti-IL-6 (4 μg/day ip) for 11 days. Right panels illustrate the calculated glomerular injury score and percentage of renal outer medulla consisting of protein casts in the rats (n = 6/group; *P < 0.05 vs. control).
Figure 4 illustrates the quantification of flow cytometric analysis of isolated immune cells from the kidneys of separate groups of vehicle and anti-IL-6-treated rats (n = 8–9/group). A reduction in the total number of infiltrating leukocytes (CD45+) in the kidney was observed in the treated compared with the control group (5.15 ± 0.28 × 106 vs. 6.24 ± 0.59 × 106 cells/kidney; P = 0.054), including a significant decrease in the total number of macrophages and monocytes (CD11b/c+) infiltrating the kidney (4.1 ± 0.26 × 106 vs. 4.97 ± 0.45 × 106 cells/kidney; P = 0.049). The total number of T-lymphocytes (CD3+) in the kidney was not significantly different between the treated and control groups (0.57 ± 0.04 × 106 vs. 0.63 ± 0.07 × 106 cells/kidney; P = 0.20). The total number of B cells (CD45R+) was also not different between treated vs. control group (0.16 ± 0.02 × 106 vs. 0.19 ± 0.03 × 106 cells/kidney; P = 0.19). To localize the potential site(s) of macrophages/monocytes in the kidneys, an immunohistochemical localization was performed in kidneys of Dahl SS rats fed 4.0% NaCl chow. Positive staining for ED1 was observed throughout the kidneys, but was localized in regions surrounding damaged glomeruli and tubules in the renal cortex and medulla (Fig. 4).
Fig. 4.

Mean number of leukocytes (CD45+), T-lymphocytes (CD3+), B-lymphocytes (CD45R+), and macrophages and monocytes (CD11b/c+) infiltrating the kidney of Dahl SS rats maintained on 4% diet for 11 days while administered IgG control or anti-IL-6 (4 μg/day ip; n = 4–5/group). *P < 0.05 vs. control (top). Immunohistochemical images of ED-1 antibody staining for macrophages/monocytes in the renal cortex (bottom left) and outer medulla (bottom right) in Dahl SS fed the 4.0% NaCl diet.
DISCUSSION
The present data demonstrate a role for IL-6 in the development of salt-sensitive hypertension and associated kidney damage in Dahl SS rats. The development of salt-sensitive hypertension and associated kidney damage was attenuated in Dahl SS rats administered anti-IL-6 antibody, which reduced renal tissue IL-6 as well as blunted the infiltration or proliferation of macrophages and monocytes into the kidneys. The mechanisms of Dahl SS hypertension are not completely understood, although immune cells have been demonstrated to amplify the extent of hypertension and renal damage. The present data, which demonstrate that IL-6 inhibition reduces immune cells in the kidney as well as hypertension and the renal damage phenotype, indicate that IL-6 may function as a chemoattractant or proliferating agent in the kidney. This concept is consistent with previous studies demonstrating that IL-6 is essential for macrophage migration in a mouse model of muscle injury (45). Although T-lymphocytes have been implicated (1, 9) in salt-sensitive hypertension, the mechanisms whereby T-cells and other immune cells exert their detrimental effects are still under investigation. A previous study from our laboratory showed that the T-lymphocytes in the kidney are activated and express several cytokines, especially IL-6, in much greater abundance compared with peripheral T-lymphocytes (37). This current study provides insight into the potential role of IL-6 in the development of salt-sensitive hypertension.
Since IL-6 is known to be a proinflammatory cytokine that is released from various cell types, including endothelial cells, vascular smooth muscle cells, lymphocytes, and macrophages (16), it is possible that inhibition of IL-6 attenuated salt-sensitive hypertension and renal damage through a variety of different mechanisms. IL-6 has been shown to be an independent risk factor for hypertension in otherwise heathy human subjects (4). Serum IL-6 level is significantly higher in hypertensive patients compared with normotensive patients (6). Additionally, IL-6 expression levels are increased in kidney biopsies of CKD patients compared with controls and further elevated in CKD patients with hypertension (46).
ANG II and salt-mediated hypertension and kidney damage are attenuated in IL-6 knock-out mice, but the deleterious mechanisms of action are not clear (22). Interestingly, the same group of investigators demonstrated that IL-6 is not required for mineralocorticoid-dependent hypertension in mice (44), which is contrary to human data that showed that ANG II induces serum IL-6 in humans through a mineralocorticoid receptor-dependent mechanism (25). Previous attempts to explore the mechanisms of IL-6 in hypertension have indicated that IL-6 does not affect ANG II-induced renal vasoconstriction and there are likely other mechanisms causing attenuation of hypertension in IL-6 knockout mice (5). Others have postulated that IL-6 impairs pressure natriuresis (23), and it was shown that genetic or pharmacological inhibition of IL-6 reduces hypertension and renal impairment and fibrosis in ANG II-infused mice. In those studies, it was postulated that IL-6 mediates these effects by inducing profibrotic genes and increasing production of endothein-1 (ET-1) mRNA (46). The effect of IL-6 inhibition on immune cell infiltration or proliferation has not been studied in hypertension models but our data are in agreement with the effects for IL-6 on tissue macrophages in different disease models (45). Although the present studies indicate a role of IL-6 to promote tissue macrophages, further studies will be necessary to determine the major site of production of IL-6 within the body and determine the mechanisms of action in hypertension and renal disease. Moreover, the potential interplay of IL-6 with other cytokines to mediate hypertension and renal damage requires further examination.
In summary, the present data demonstrate that administration of an anti-IL-6 antibody reduced renal tissue IL-6, blunted macrophage and monocyte accumulation in the kidneys, and attenuated the development of salt-sensitive hypertension and associated kidney damage in Dahl SS rats. These findings validate a role for the immune system in hypertension and renal disease. The present data, along with additional experiments, may help facilitate the future development of therapeutic targets for the treatment of hypertension.
GRANTS
This work was partially supported by National Institutes of Health Grants DK96859 and HL116264 and American Heart Association Grant 15SFRN2391002.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
S.H., S.K.V.W., and D.L.M. conception and design of research; S.H., N.P.R., H.L., and J.M.A.-B. performed experiments; S.H., N.P.R., H.L., and D.L.M. analyzed data; S.H., N.P.R., J.M.A.-B., S.K.V.W., and D.L.M. interpreted results of experiments; S.H. and D.L.M. prepared figures; S.H. and D.L.M. drafted manuscript; S.H., N.P.R., H.L., J.M.A.-B., S.K.V.W., and D.L.M. edited and revised manuscript; S.H., N.P.R., J.M.A.-B., S.K.V.W., and D.L.M. approved final version of manuscript.
REFERENCES
- 1.Alvarez V, Quiroz Y, Nava M, Pons H, Rodriguez-Iturbe B. Overload proteinuria is followed by salt-sensitive hypertension caused by renal infiltration of immune cells. Am J Physiol Renal Physiol 283: F1132–F1141, 2002. [DOI] [PubMed] [Google Scholar]
- 2.Arruda JL, Sweitzer S, Rutkowski MD, DeLeo JA. Intrathecal anti-IL-6 antibody and IgG attenuates peripheral nerve injury-induced mechanical allodynia in the rat: Possible immune modulation in neuropathic pain. Brain Res 879: 216–225, 2000. [DOI] [PubMed] [Google Scholar]
- 3.Bataillard A, Vincent M, Sassard J, Touraine JL. Antihypertensive effect of an immunosuppressive agent, cyclophosphamide, in genetically hypertensive rats of the Lyon strain. Int J Immunopharmacol 11: 377–384, 1989. [DOI] [PubMed] [Google Scholar]
- 4.Bautista LE, Vera LM, Arenas IA, Gamarra G. Independent association between inflammatory markers (c-reactive protein, interleukin-6, and TNF-alpha) and essential hypertension. J Hum Hypertens 19: 149–154, 2005. [DOI] [PubMed] [Google Scholar]
- 5.Brands MW, Banes-Berceli AK, Inscho EW, Al-Azawi H, Allen AJ, Labazi H. Interleukin 6 knockout prevents angiotensin II hypertension: Role of renal vasoconstriction and janus kinase 2/signal transducer and activator of transcription 3 activation. Hypertension 56: 879–884, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chamarthi B, Williams GH, Ricchiuti V, Srikumar N, Hopkins PN, Luther JM, Jeunemaitre X, Thomas A. Inflammation and hypertension: the interplay of interleukin-6, dietary sodium, and the renin-angiotensin system in humans. Am J Hypertens 24: 1143–1148, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Crosswhite P, Sun Z. Ribonucleic acid interference knockdown of interleukin 6 attenuates cold-induced hypertension. Hypertension 55: 1484–1491, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Crowley SD, Frey CW, Gould SK, Griffiths R, Ruiz P, Burchette JL, Howell DN, Makhanova N, Yan M, Kim HS, Tharaux PL, Coffman TM. Stimulation of lymphocyte responses by angiotensin ii promotes kidney injury in hypertension. Am J Physiol Renal Physiol 295: F515–F524, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.De Miguel C, Das S, Lund H, Mattson DL. T lymphocytes mediate hypertension and kidney damage in Dahl salt-sensitive rats. Am J Physiol Regul Integr Comp Physiol 298: R1136–R1142, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.De Miguel C, Guo C, Lund H, Feng D, Mattson DL. Infiltrating T lymphocytes in the kidney increase oxidative stress and participate in the development of hypertension and renal disease. Am J Physiol Renal Physiol 300: F734–F742, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.De Miguel C, Rudemiller NP, Abais JM, Mattson DL. Inflammation and hypertension: new understandings and potential therapeutic targets. Curr Hypertens Rep 17: 507, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elmarakby AA, Quigley JE, Imig JD, Pollock JS, Pollock DM. TNF-α inhibition reduces renal injury in Doca-salt hypertensive rats. Am J Physiol Regul Integr Comp Physiol 294: R76–R83, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grim CE, Wilson TW, Nicholson GD, Hassell TA, Fraser HS, Grim CM, Wilson DM. Blood pressure in blacks. Twin studies in Barbados. Hypertension 15: 803–809, 1990. [DOI] [PubMed] [Google Scholar]
- 14.Haroun MK, Jaar BG, Hoffman SC, Comstock GW, Klag MJ, Coresh J. Risk factors for chronic kidney disease: a prospective study of 23,534 men and women in Washington County, Maryland. J Am Soc Nephrol 14: 2934–2941, 2003. [DOI] [PubMed] [Google Scholar]
- 15.Harrison DG. The immune system in hypertension. Trans Am Clin Climatol Assoc 125: 130–138, 2014. [PMC free article] [PubMed] [Google Scholar]
- 16.Hirano T. Interleukin 6 and its receptor: ten years later. Int Rev Immunol 16: 249–284, 1998. [DOI] [PubMed] [Google Scholar]
- 17.Horowitz B, Miskulin D, Zager P. Epidemiology of hypertension in CKD. Adv Chronic Kidney Dis 22: 88–95, 2015. [DOI] [PubMed] [Google Scholar]
- 18.Hsu CY, McCulloch CE, Darbinian J, Go AS, Iribarren C. Elevated blood pressure and risk of end-stage renal disease in subjects without baseline kidney disease. Arch Intern Med 165: 923–928, 2005. [DOI] [PubMed] [Google Scholar]
- 19.Hughson MD, Gobe GC, Hoy WE, Manning RD Jr, Douglas-Denton R, Bertram JF. Associations of glomerular number and birth weight with clinicopathological features of African Americans and Whites. Am J Kidney Dis 52: 18–28, 2008. [DOI] [PubMed] [Google Scholar]
- 20.Khraibi AA, Smith TL, Hutchins PM, Lynch CD, Dusseau JW. Thymectomy delays the development of hypertension in okamoto spontaneously hypertensive rats. J Hypertens 5: 537–541, 1987. [DOI] [PubMed] [Google Scholar]
- 21.Lawes CM, Bennett DA, Feigin VL, Rodgers A. Blood pressure and stroke: an overview of published reviews. Stroke 35: 776–785, 2004. [DOI] [PubMed] [Google Scholar]
- 22.Lee DL, Sturgis LC, Labazi H, Osborne JB Jr, Fleming C, Pollock JS, Manhiani M, Imig JD, Brands MW. Angiotensin II hypertension is attenuated in interleukin-6 knockout mice. Am J Physiol Heart Circ Physiol 290: H935–H940, 2006. [DOI] [PubMed] [Google Scholar]
- 23.Li K, Guo D, Zhu H, Hering-Smith KS, Hamm LL, Ouyang J, Dong Y. Interleukin-6 stimulates epithelial sodium channels in mouse cortical collecting duct cells. Am J Physiol Regul Integr Comp Physiol 299: R590–R595, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lopez AD, Mathers CD, Ezzati M, Jamison DT, Murray CJ. Global and regional burden of disease and risk factors, 2001: systematic analysis of population health data. Lancet 367: 1747–1757, 2006. [DOI] [PubMed] [Google Scholar]
- 25.Luther JM, Gainer JV, Murphey LJ, Yu C, Vaughan DE, Morrow JD, Brown NJ. Angiotensin II induces interleukin-6 in humans through a mineralocorticoid receptor-dependent mechanism. Hypertension 48: 1050–1057, 2006. [DOI] [PubMed] [Google Scholar]
- 26.Madhur MS, Lob HE, McCann LA, Iwakura Y, Blinder Y, Guzik TJ, Harrison DG. Interleukin 17 promotes angiotensin II-induced hypertension and vascular dysfunction. Hypertension 55: 500–507, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mattson DL, James L, Berdan EA, Meister CJ. Immune suppression attenuates hypertension and renal disease in the Dahl salt-sensitive rat. Hypertension 48: 149–156, 2006. [DOI] [PubMed] [Google Scholar]
- 28.Mattson DL, Kunert MP, Kaldunski ML, Greene AS, Roman RJ, Jacob HJ, Cowley AW Jr. Influence of diet and genetics on hypertension and renal disease in Dahl salt-sensitive rats. Physiol Genomics 16: 194–203, 2004. [DOI] [PubMed] [Google Scholar]
- 29.Mattson DL, Lund H, Guo C, Rudemiller N, Geurts AM, Jacob H. Genetic mutation of recombination activating gene 1 in Dahl salt-sensitive rats attenuates hypertension and renal damage. Am J Physiol Regul Integr Comp Physiol 304: R407–R414, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mattson DL, Meister CJ, Marcelle ML. Dietary protein source determines the degree of hypertension and renal disease in the Dahl salt-sensitive rat. Hypertension 45: 736–741, 2005. [DOI] [PubMed] [Google Scholar]
- 31.Mattson DL. Infiltrating immune cells in the kidney in salt-sensitive hypertension and renal injury. Am J Physiol Renal Physiol 307: F499–F508, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McMaster WG, Kirabo A, Madhur MS, Harrison DG. Inflammation, immunity, and hypertensive end-organ damage. Circ Res 116: 1022–1033, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, de Ferranti S, Despres JP, Fullerton HJ, Howard VJ, Huffman MD, Judd SE, Kissela BM, Lackland DT, Lichtman JH, Lisabeth LD, Liu S, Mackey RH, Matchar DB, McGuire DK, Mohler ER 3rd Moy CS, Muntner P, Mussolino ME, Nasir K, Neumar RW, Nichol G, Palaniappan L, Pandey DK, Reeves MJ, Rodriguez CJ, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Willey JZ, Woo D, Yeh RW, Turner MB, American Heart Association Statistics Committee, Stroke Statistics Subcommittee. Heart disease and stroke statistics–2015 update: a report from the American Heart Association. Circulation 131: e29–322, 2015. [DOI] [PubMed] [Google Scholar]
- 34.Neurath MF, Finotto S. IL-6 signaling in autoimmunity, chronic inflammation and inflammation-associated cancer. Cytokine Growth Factor Rev 22: 83–89, 2011. [DOI] [PubMed] [Google Scholar]
- 35.Ozawa Y, Kobori H, Suzaki Y, Navar LG. Sustained renal interstitial macrophage infiltration following chronic angiotensin II infusions. Am J Physiol Renal Physiol 292: F330–F339, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Papanicolaou DA, Wilder RL, Manolagas SC, Chrousos GP. The pathophysiologic roles of interleukin-6 in human disease. Ann Intern Med 128: 127–137, 1998. [DOI] [PubMed] [Google Scholar]
- 37.Rudemiller N, Lund H, Jacob HJ, Geurts AM, Mattson DL. CD247 modulates blood pressure by altering T-lymphocyte infiltration in the kidney. Hypertension 63: 559–564, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rudemiller NP, Lund H, Priestley JRC, Endres BT, Prokop JW, Jacob HJ, Geurts AM, Cohen EP, Mattson DL. Mutation of SH2B3 (LNK), a GWAS candidate for hypertension, attenuates Dahl SS hypertension via inflammatory modulation. Hypertension 65: 1111–1117, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Saleh MA, McMaster WG, Wu J, Norlander AE, Funt SA, Thabet SR, Kirabo A, Xiao L, Chen W, Itani HA, Michell D, Huan T, Zhang Y, Takaki S, Titze J, Levy D, Harrison DG, Madhur MS. Lymphocyte adaptor protein lnk deficiency exacerbates hypertension and end-organ inflammation. J Clin Invest 125: 1189–1202, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schmieder RE. End organ damage in hypertension. Dtsch Arztebl Int 107: 866–873, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Smith C, Wilson NW, Louw A, Myburgh KH. Illuminating the interrelated immune and endocrine adaptations after multiple exposures to short immobilization stress by in vivo blocking of IL-6. Am J Physiol Regul Integr Comp Physiol 292: R1439–R1447, 2007. [DOI] [PubMed] [Google Scholar]
- 42.Sturgis LC, Cannon JG, Schreihofer DA, Brands MW. The role of aldosterone in mediating the dependence of angiotensin hypertension on IL-6. Am J Physiol Regul Integr Comp Physiol 297: R1742–R1748, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wong ND. Epidemiological studies of CHD and the evolution of preventive cardiology. Nat Rev Cardiol 11: 276–289, 2014. [DOI] [PubMed] [Google Scholar]
- 44.Yamashita S, Kato R, Kobayashi K, Hisasue S, Arai Y, Tsukamoto T. Inhibition of interleukin-6 attenuates erectile dysfunction in a rat model of nerve-sparing radical prostatectomy. J Sex Med 8: 1957–1964, 2011. [DOI] [PubMed] [Google Scholar]
- 45.Zhang C, Li Y, Wu Y, Wang L, Wang X, Du J. Interleukin-6/signal transducer and activator of transcription 3 (stat3) pathway is essential for macrophage infiltration and myoblast proliferation during muscle regeneration. J Biol Chem 288: 1489–1499, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang W, Wang W, Yu H, Zhang Y, Dai Y, Ning C, Tao L, Sun H, Kellems RE, Blackburn MR, Xia Y. Interleukin 6 underlies angiotensin II-induced hypertension and chronic renal damage. Hypertension 59: 136–144, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]