

Abstract
Mechanical ventilation (MV) and oxygen therapy (hyperoxia; HO) comprise the cornerstones of life-saving interventions for patients with acute respiratory distress syndrome (ARDS). Unfortunately, the side effects of MV and HO include exacerbation of lung injury by barotrauma, volutrauma, and propagation of lung inflammation. Despite significant improvements in ventilator technologies and a heightened awareness of oxygen toxicity, besides low tidal volume ventilation few if any medical interventions have improved ARDS outcomes over the past two decades. We are lacking a comprehensive understanding of mechanotransduction processes in the healthy lung and know little about the interactions between simultaneously activated stretch-, HO-, and cytokine-induced signaling cascades in ARDS. Nevertheless, as we are unraveling these mechanisms we are gathering increasing evidence for the importance of stretch-activated ion channels (SACs) in the activation of lung-resident and inflammatory cells. In addition to the discovery of new SAC families in the lung, e.g., two-pore domain potassium channels, we are increasingly assigning mechanosensing properties to already known Na+, Ca2+, K+, and Cl− channels. Better insights into the mechanotransduction mechanisms of SACs will improve our understanding of the pathways leading to ventilator-induced lung injury and lead to much needed novel therapeutic approaches against ARDS by specifically targeting SACs. This review 1) summarizes the reasons why the time has come to seriously consider SACs as new therapeutic targets against ARDS, 2) critically analyzes the physiological and experimental factors that currently limit our knowledge about SACs, and 3) outlines the most important questions future research studies need to address.
Keywords: ARDS, hyperoxia, ion channels, lung injury, stretch
Current Knowledge Gaps and Obstacles in Studying Stretch-Activated Ion Channels in ARDS
The biophysical characterization of ion channel properties in living biological systems represents one of the most difficult challenges in modern medical research. The multitude of different ion channels expressed on a given cell makes it notoriously difficult to isolate the functional and biophysical characteristics of a single channel. To circumvent this problem, electrophysiologists routinely overexpress channels of interest in heterologous expression systems such as the Xenopus laevis oocyte. This approach allows for a detailed characterization of biophysical channel properties in a controlled, well-defined environment but the native intra- and extracellular milieus of the channel are lost. Conversely, the study of a given channel in a native cell is often limited by low expression levels and interference from the variety of other channels present on any cell. Although many of these limitations apply to ion channel research in general, they are particularly relevant to stretch-activated ion channels (SACs), since not only the intra- and extracellular environments but also the amount and direction of mechanical forces that SACs are exposed to in vivo are difficult to standardize.
Nevertheless, the central role of SACs in mechanotransduction processes in the lung is nowadays undisputed. Understanding the biophysical properties and contribution of SACs to both the development and resolution of ARDS is particularly important, since by definition the lungs of all ARDS patients are exposed to mechanical stretch induced by positive pressure ventilation. Decades of intense research have substantially expanded our knowledge to the point that we can now confidently assign a role to SACs in the development of the two main clinical features of ARDS: 1) alveolar inflammation and 2) loss of alveolar-capillary barrier function. To advance this field, it is vitally important to combine our knowledge from three different areas of research: 1) the detailed understanding of biophysical SAC properties, unfortunately often obtained in rather artificial expression systems; 2) our knowledge of ARDS pathophysiology derived from in vitro and in vivo lung injury models; and 3) changes in ARDS biomarker profiles and physiological variables obtained from clinical studies, e.g., oxygenation and ventilation indexes, plasma and bronchoalveolar lavage (BAL) pH, BAL cytokines and cell counts, lung compliance and resistance measurements, and morbidity/mortality data. The creation of unifying ARDS models that integrate all of these research areas represents an extremely challenging task given the complexity of each individual field. This review highlights the most relevant topics in each field and unravels common areas of interest to facilitate collaborations between investigators from these different, often even opposing fields. Combining our knowledge will undoubtedly unify these fields, and together we can target SACs as a very promising new therapeutic approach against ARDS.
Background
Acute respiratory distress syndrome (ARDS) was first described in 1967 and to this day remains a devastating illness characterized by diffuse alveolar injury and inflammation caused by neutrophil recruitment to the lungs, macrophage activation, inflammatory cytokine release, and loss of alveolar-capillary barrier function (177). Despite our best efforts, mortality rates for ARDS patients remain high, and more patients die from ARDS than any other illness in our intensive care units (ICUs), accounting for 196,600 cases, 74,500 deaths, and 2.2 million ICU days per year for adults (163), and 7,700 cases, 1,400 deaths, and 62,000 ICU days per year for children in the US (225). Current treatment regimens for ARDS primarily consist of oxygen supplementation therapy (hyperoxia; HO) and mechanical ventilation (MV) (65). Unfortunately, the combined effects of HO and MV cause even more lung damage than each stimulus alone (119, 176). For example, HO exposure of alveolar epithelial cells (AECs) alters their mechanical properties, resulting in increased cell detachment during mechanical stretch (159). Although the signaling cascades employed by HO and inflammatory cytokines are relatively well established, less is known about “mechanosensing” and “mechanotransduction” processes in the lung, including the mechanisms responsible for the simultaneous integration of HO-, MV- and cytokine-mediated signaling during epithelial, endothelial, inflammatory, and immune cell activation. Nevertheless, our knowledge in this field is rapidly expanding. The combined effects of HO and MV activate c-Jun NH2-terminal kinase (JNK) and apoptosis signal-regulating kinase (ASK)-1 (117, 118), but other mitogen-activated protein kinases, tyrosine kinases, transcription factors (including NF-κB), and protein kinase C also mediate mechanical coupling processes in ARDS lungs (7, 61, 126). Therefore, the identification of specific targets within these complex and interrelated signaling pathways constitutes a major challenge in the development of new therapeutic strategies against ARDS. This is further evidenced by the fact that, besides the introduction of “lung-protective,” low tidal volume ventilation strategies and possibly prone positioning, few if any interventions have improved the outcomes or survival rates of ARDS patients over the past two decades (16, 51). Importantly, in tissues other than the lung, SACs are well known to function as so-called “signal integrators” and it is becoming increasingly evident that in the lung these channels could constitute the link between HO, MV, and cytokine effects (76, 166, 174).
SACs Are Mechanosensors and Mechanotransducers in the Lung
The effects of mechanical stretch on many biological lung functions, including fetal lung growth, surfactant metabolism, extracellular matrix and cytoskeleton turnover, cell proliferation, apoptosis, mediator release, and alveolar-capillary permeability, have been recognized for over two decades (3, 38, 43, 47, 48). Lansman et al. (104) described the first stretch-activated cation channel in 1987 in the vascular endothelium. Advances in nanotechnology techniques now enable us to image and measure changes in physical forces at subcellular level, improving our understanding of membrane deformation processes in health and disease. These advances led to a renewed interest in the molecular mechanisms underlying mechanotransduction processes in living organisms (91). SACs are among the best-studied examples of mechanosensors and mechanotransducers in nature and can mediate both mechanoelectrical and mechanochemical signals (139). Although mechanosensitive ion channels were originally identified and cloned in bacteria (25), in humans various types of SACs can be found in almost any body tissue and dysregulation of mechanical responses often results in major pathology (137, 191). SACs are usually involved in the earliest stage of mechanosensing and can alter the cell membrane potential within micro- to milliseconds. Such a rapid perturbation can lead to either membrane depolarization via Na+, Ca2+, or nonselective cation channels (such as Piezo 1 and 2), or to membrane hyperpolarization via K+ channels. To add further complexity to this system, in mammalian cells not only stretch-activated but also some stretch-inactivated ion channels have been characterized (166). While certain SACs can be directly activated by membrane tension, others require force transduction via anchors in the extracellular matrix, the cytoskeleton, or both (139) (Fig. 1), and any damage to the cortical cytoskeleton, as induced by MV, can severely affect SAC gating and function (199).
Fig. 1.
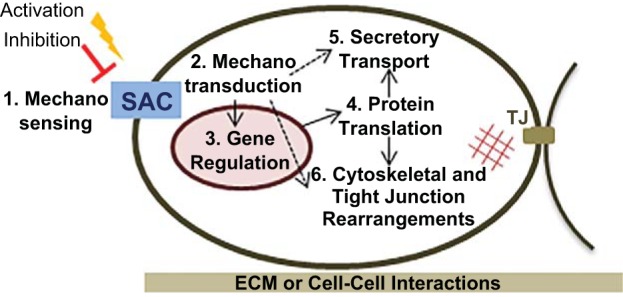
SACs are stretch sensors and stretch integrators. Mechanical stretch can either activate or inhibit SAC function (1) and mobilize intracellular signaling cascades (2) affecting gene transcription (3) and protein translation (4), which ultimately regulate secretory transport pathways (5), cytoskeletal rearrangements, and capillary-alveolar barrier function (6). In addition, SACs may also directly modulate nongenomic downstream effector functions (dotted arrows). The amount and vector of stretch forces applied to a plasma membrane depend on the integrity of tight junctions (TJ), cell adhesion to the extracellular matrix (ECM), and cell-cell interactions (epithelial/endothelial-to-inflammatory cell interactions). Adapted from Ref. 115.
To date, no unique protein structure has been found to be inherent to SACs. The term SAC does not apply to a specific ionic conductance but includes Na+ (221), K+ (158), Ca2+ (112), Cl− (75), and nonselective cation channels (35) (Fig. 2). The main unifying feature of SACs is their ability to respond to membrane stretch with a configuration change between open and closed states (122) and, with few exceptions, their inhibition by gadolinium (Gd3+) (215). However, just because mechanical stretch changes the open probability of a given channel, this does not imply any physiological relevance. Therefore, to understand the pathophysiological implications of changes in SAC expression and function in ARDS lungs, each channel of interest has to be characterized in the appropriate microenvironment consisting of HO, mechanical stretch, and inflammatory cytokines.
Fig. 2.
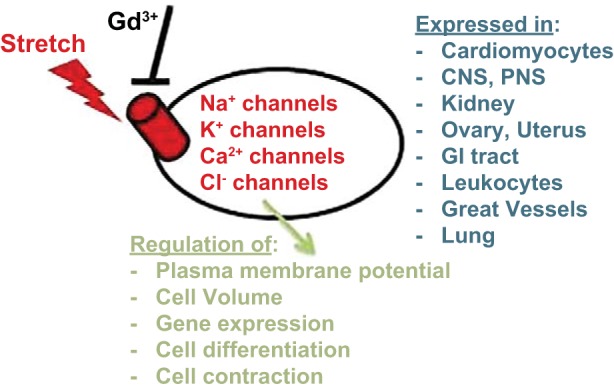
Expression and role of SACs. A major unifying feature of SACs is a configuration change upon mechanical stretch exposure and inhibition by gadolinium (Gd3+). All major classes of ion channels contain members that are stretch-sensitive. SACs are almost ubiquitously expressed in body tissues and regulate a variety of cellular functions. CNS, central nervous system; PNS, peripheral nervous system.
The ability of SACs to sense changes in membrane shear stress, tension, and curvature, which are all features associated with MV, makes SACs the ideal candidates for mechanotransducers and signal integrators in ARDS, even though the specific mechanisms of channel activation (or inhibition) by stretch may vary substantially between channels. Stretch can activate a given SAC via 1) direct tension on the lipid bilayer, 2) transduction of stretch via a tethering mechanism from cytoskeletal or extracellular matrix structures, or 3) release or binding of another molecule that in turn activates a channel (e.g., a phosphokinase), as has been proposed for the epithelial Na+ channel (ENaC) (20). However, to assign a role to SACs in the pathogenesis of ARDS, it is important to recognize that SACs are not exclusively activated by mechanical stretch but also by a variety of other stimuli that are altered in ARDS lungs, including changes in pH or temperature, drugs (e.g., volatile anesthetics), biological ligands (e.g., ATP, lipids), and changes in the membrane potential (voltage dependency) (40, 103). Over the past decade, we accumulated evidence that pharmacological and molecular modulation of SACs alters inflammatory processes in the lung. Nevertheless, some of the specific mechanisms that link SAC activation (or inhibition) to inflammatory mediator release and alveolar-capillary barrier function, the two hallmarks of ARDS, are still under investigation. Due to our limited knowledge about SAC expression and function in the human lung, many of our mechanistic hypotheses rely on SAC characteristics described in other organs and on data extrapolated from animal models. In rat lungs, so-called rapidly adapting receptors (RARs) transduce mechanical stimuli to nerve fibers via activation of voltage-dependent Na+ and K+ channels, N-type Ca2+ channels, and Cl− channels (GABA A- and glycine-activated) (36). The complex interplay between these channels excites the afferent nerve terminal and regulates airway tone and fluid homeostasis (36). This is only one example of a well-defined pathway in rodents that, if confirmed in the human lung, could become tremendously useful in designing new therapies against ARDS. Other exciting candidates are so-called 2-pore domain K+ channels (K2P), which are not only stretch sensitive (73, 223) but are also regulated by HO (172) and affect cytokine secretion from mouse ARDS lungs (174). We are now finally at a stage where many of the characteristics assigned to individual SACs can be exploited in the context of ventilator-induced lung injury (VILI), which allows us to specifically target these channels as new therapeutic options against ARDS.
The Role of SACs in Inflammatory Mediator Release and Loss of Capillary-Alveolar Barrier Function
Two hallmarks of ARDS are 1) increased levels of inflammatory mediators in the BAL fluid and plasma (19) and 2) the loss of alveolar-capillary barrier function (33), both of which correlate with patient mortality rates (56, 125). Historically, ion channel activity has been closely linked to mediator release, particularly in neuronal cells where cell depolarization leads to Ca2+ influx followed by neurotransmitter secretion (134), but it is still controversial whether this model also applies to nonneuronal cells. Today we know that ion channels regulate mediator secretion in many other cell types, including lung-resident, inflammatory, and immune cells (94, 152), although the secretory mechanisms employed by nonexcitatory cells are not as clearly defined. In general, an increase in intracellular Ca2+ as the final common pathway precedes mediator release (160). K+ and Cl− currents affect Ca2+ entry via alteration of the membrane potential, thereby increasing or decreasing the electrical driving force for Ca2+ to enter a cell (53, 121). In many tissues K2P channels, including the TREK subfamily, are important stabilizers of the resting membrane potential and prevent cell depolarization (59). In contrast, inhibition of K2P channels results in decreased “leak currents” and accumulation of positive K+ charges inside the cell resulting in cell depolarization (18). Importantly, in lung epithelial, inflammatory, and immune cells not all secretory pathways are Ca2+ dependent (173, 190). Furthermore, certain meditators such as macrophage-inflammatory protein-2 (MIP-2) are secreted upon stretch exposure without activation of any particular SACs, as suggested by a lack of effect of the SAC blocker gadolinium on stretch-induced MIP-2 secretion (130).
From both in vitro and in vivo ARDS studies we learned that mechanical forces, HO exposure, and proinflammatory cytokines such as TNF-α alter transepithelial ion transport. The net result is a decrease in alveolar fluid clearance (68, 154, 206), which is directly associated with an increased length of MV and patient mortality (83, 177). Undoubtedly, SACs are important in both alveolar edema formation and resolution, but their regulation in the complex inflammatory environment of ARDS lungs is somewhat difficult to predict (Fig. 3). Nevertheless, in rat lungs, Gd3+ (an inhibitor of stretch-activated ion channels) prevents stretch-induced loss of capillary-alveolar barrier function (142), and HO, stretch, and TNF-α all regulate barrier function by altering the phosphorylation status of tight junction (TJ) proteins (58). In general, epithelial TJ phosphorylation is a Ca2+-dependent process and several Ca2+ channels, including transient receptor potential (TRP) channels, contribute to alveolar-capillary barrier dysfunction in ARDS (78, 129) but other conductances such as the CFTR Cl− channel may be involved as well (108). Besides ventilator-induced stretch, mechanosensitive ion channels in the lung are also exposed to stretch forces evoked by cell swelling, a process underlying cell apoptosis, detachment, proliferation, and repair processes in ARDS lungs. Na+, K+, Ca2+, and Cl− channel families all include members that are activated by cell swelling-induced membrane stretch (180).
Fig. 3.
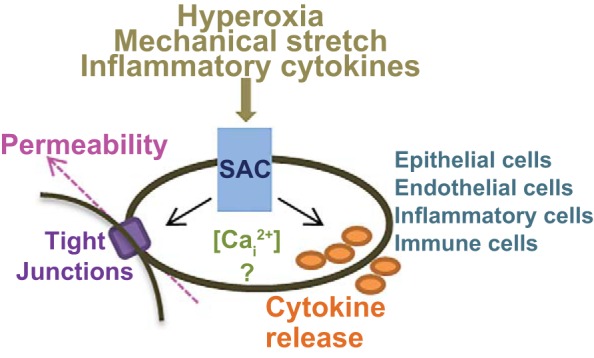
SACs as orchestrators of alveolar inflammation in ARDS. Hyperoxia, mechanical stretch, and inflammatory cytokines alter SAC expression and function, which impacts the pathogenesis of ARDS through downstream effects on cytokine secretion from epithelial, endothelial, inflammatory, and immune cells and alveolar-capillary barrier function (epithelial and endothelial cells). While some of these processes are mediated via changes in intracellular Ca2+ concentrations ([Cai2+]), others appear Ca2+ independent.
The enthusiasm about targeting SACs as mechanosensors and mechanotransducers in ARDS stems from the possibility of specifically altering stretch-induced lung injury processes without interfering with innate immune mechanisms responsible for host defenses and lung repair.
Stretch-Activated Na+ Channels
Stretch-activated Na+ channels are important regulators of alveolar fluid clearance and inflammatory mediator secretion by regulating the plasma membrane potential of lung-resident and inflammatory cells. One of the earliest models of a mechanotransducing complex was described in the nematode Caenorhabditis elegans, where the key molecules (so-called MEC and DEG proteins) forming a mechano-gated ion channel are related to the α, β, and γ subunits of the mammalian epithelial Na+ channel ENaC (54, 114, 185). Despite some controversy in the 1990s over the mechanosensitivity of ENaC, which derived from differences in biophysical channel properties between lipid bilayer and oocyte experiments (161), nowadays the stretch-activated nature of ENaC is widely accepted (4, 20, 66, 67, 175). In fact, the channel appears intrinsically mechanosensitive since it can be directly activated by mechanical stretch without cytoskeletal interactions (100). Besides direct ENaC activation by membrane tension, other models propose stretch-mediated ENaC activation via a tethering mechanism to the cytoskeleton or extracellular matrix, or via stretch-mediated release of another molecule from the cell that in turn activates ENaC. Evidence actually exists to support all three mechanisms (44, 77, 209). Importantly, stretch activation of ENaC plays a role not only for lung epithelial but also for inflammatory cells, likely via channel interactions with cytoskeletal structures (1). Interestingly, in lymphocytes (116) and osteoblasts (101) only the α subunit is required to form a functional mechanosensitive channel.
ENaC plays a central role in ARDS pathogenesis as a key regulator of alveolar fluid clearance, as required during the exudative phase of ARDS (5, 6, 69, 110). Furthermore, in the developing lung stretch increases epithelial ENaC expression via p38, MAPK, and JNK pathways and improves alveolar fluid clearance (14, 102, 133), which could accelerate recovery from the exudative phase of ARDS. In contrast, elevated serotonin levels, as commonly observed in ARDS lungs, inhibit ENaC activity and correlate with worsening pulmonary edema and ARDS outcomes (72, 128). In an inhalation (hydrogen sulfide) lung injury model, downregulation of α-ENaC also correlated with worsening pulmonary edema, which was prevented by steroid therapy (97). In airway neurons, activation of an ENaC-like, mechanosensitive Na+ channel contributes to the propagation of lung inflammation (36). ENaC is also expressed in B lymphocytes and can be activated by membrane stretch, but, in contrast to epithelial ENaC, in lymphocytes an association of ENaC with the cytoskeleton is mandatory for current activation (1, 116). ENaC activity is also sensitive to stretch induced by cell swelling and shrinkage (96), as observed during cell death or due to osmotic changes caused by protein-rich alveolar exudate in ARDS lungs, and could thus affect both the early exudative phase and late fibrotic phase of ARDS. In addition to stretch, ENaC can also be activated by reactive oxygen species (ROS) (10, 188), which are routinely elevated in ARDS lungs due to HO and MV exposure. In a murine LPS-induced ARDS model, ROS increased ENaC activity and alveolar fluid clearance (71). Therefore, ROS-induced upregulation of ENaC activity may constitute a protective mechanism to counteract increased pulmonary edema formation during the exudative phase of ARDS.
Besides ENaC, stretch can also activate voltage-sensitive Na+ channels (Nav1.5–1.9) (23, 179, 201). In the lung, Nav channels are expressed in bronchial smooth muscle (29) and afferent nerve endings in the airways (132). Interestingly, calpain, which is elevated in lungs exposed to MV and promotes lung injury via neutrophil recruitment (113) and loss of epithelial barrier function (203), can cleave Nav channels (197). Nav channels should be considered as potential targets during the exudative and proliferative phases of ARDS, since stretch activation of Nav channels in colonic epithelial cells promotes cell migration and steroid metabolism (85), while in neurons it leads to Ca2+ influx-mediated cell death (201).
Furthermore, the recent characterization of slowly and rapidly adapting stretch receptors (SARs and RARs, respectively) in the lung has greatly improved our understanding of mechanosensing (30). SAR and RAR activation is regulated by Na+ channels and is responsible for adaptation of hemodynamic responses to breathing patterns. Importantly, SAR and RAR activity is also regulated by hypoxia, a key feature of ARDS pathology. Harvesting the potential of SARs and RARs could be particularly important in light of the increasing utilization rates of airway pressure release ventilation (APRV) for ARDS. This ventilation mode exposes lungs to prolonged inflation and deflation pressures, which is the main stimulus for SAR and RAR activation. In the future, modulation of these receptors could not only provide us with novel ways to target neuronal regulation of airway inflammation but also lead to new lung recruitment strategies to improve ventilation-perfusion mismatching in ARDS.
Besides ion channels, several Na+ transporters and exchangers (Na+/Ca2+, Na+/H+, Na+/K+ ATPase) are mechanosensitive and thus attractive targets for new ARDS therapies (123, 196, 206). In fact, the Na+/K+ ATPase promotes alveolar edema clearance in ARDS and may be stimulated by Na+ influx via stretch-activated Na+ channels, but the results of clinical trials remain disappointing (32).
Stretch-Activated K+ Channels
K+ channels are typically classified according to their number of transmembrane domains (TMD): voltage-dependent (Kv) and Ca2+-activated (KCa) channels have six TMDs; K2P have four TMDs; inwardly rectifying K+ channels (Kir; including KATP) have two TMDs. Although each class includes channels with stretch-sensitive characteristics, K2P channels are particularly well-known for their mechanosensing and mechanotransducing capabilities (107). The K2P channel family consists of 15 members and is divided into six subfamilies (TWIK, THIK, TREK, TASK, TALK, TRESK) (144). What distinguishes K2P channels from almost any other SAC is that their intrinsic mechanosensitivity, independent of an intact cytoskeleton. Evidence from our group suggests that K2P channels, especially TREK-1, are important regulators of the inflammatory processes observed in ARDS since they are expressed in lung epithelial cells and macrophages and are regulated by both stretch and HO (172). In fact, stimulation of TREK-1-deficient (TREK-1 ko) AECs with TNF-α, the main cytokine in the BAL fluid of ARDS patients, decreases IL-6 and RANTES secretion but increases MCP-1 secretion, while KC/IL-8 release is not affected. Although several TNF-α-mediated signaling pathways including p38 kinase, PKC, and c-Jun NH2-terminal kinases (JNK1/2/3) are altered in TREK-1 ko AECs, decreased IL-6 secretion is associated with impaired PKCθ phosphorylation, whereas increased MCP-1 secretion occurs unrelated to the increased JNK1/2/3 phosphorylation found in TREK-1 ko AECs (171, 173).
Based on these in vitro findings we expected that TREK-1 ko mice would be protected from HO- and MV-induced lung injury. To our surprise, however, exposure of TREK-1 ko mice to HO increases lung injury as evidenced by gross lung anatomy, hematoxylin and eosin staining of lung sections (increased inflammatory cell infiltration and hemorrhage), increased lung injury scores, decreased lung compliance, and increased macrophage and neutrophil accumulation in the BAL fluid of TREK-1 ko mice (174). Interestingly, exposure of these mice to HO+MV results in no further injury when compared with HO exposure alone. In accordance with our in vitro findings (173), IL-6 levels decrease in HO+MV-treated TREK-1 ko mice, but, in contrast to our in vitro findings, MCP-1 levels also decrease in HO+MV treated TREK-1 ko mice, while TNF-α levels are unchanged between control and TREK-1 ko mice (174). Therefore, in our mouse ARDS model alterations in BAL cytokine concentrations are unlikely the cause for the increased lung injury observed in TREK-1 ko mice. We cannot, however, exclude that intraparenchymal or plasma cytokine levels are altered in TREK-1 ko mice contributing to the observed compliance and lung injury changes. Interestingly, alveolar barrier function appears uncompromised by TREK-1 deficiency since BAL protein levels are similar between control and TREK-1 ko mice (174). One explanation for the increased lung damage observed in HO-exposed TREK-1 ko mice could be an increase in alveolar type II (AT I) and AT II cell apoptosis as evidenced by TUNEL staining and corroborated by increased PARP-1 cleavage (174).
Importantly, TREK channel gating is strongly affected by pH changes with intracellular acidification leading to TREK activation, whereas extracellular acidification results in TREK inhibition (49). Patients with severe ARDS suffer from a significant metabolic acidosis caused by multiorgan failure, which is also the main cause of mortality in these patients. In addition, although never documented in ARDS, the pH of exhaled water vapor of asthmatic subjects is 5.2 compared with a pH of 7.6 in healthy controls (89). Therefore, the acidotic extracellular environment in ARDS could easily inhibit TREK-1 channels and potentially worsen ARDS consistent with the increased lung injury seen in TREK-1 ko mice (174).
Besides, K2P channels, other stretch-sensitive K+ channels cannot be overlooked. For instance, KATP channels can directly be activated by local bilayer tension and cortical F-actin (87). It is also known that cyclic stretch depletes intracellular ATP stores in AECs, which in turn could inhibit KATP activation (37). In addition to the epithelium, KATP channels are also mechanosensors in lung endothelial cells where they regulate ROS formation, Ca2+ influx, and inflammatory cytokine secretion (39). Although KATP channels decrease lymphocyte TNF-α production (208), they promote neutrophil influx and TNF-α and IL-6 release in the lung (149). A variety of Kv channels promote lymphocyte activation and proliferation, and Kv1.3 currents promote LPS- and TNF-α-induced macrophage activation (192). Large conductance KCa channels (BKCa) are sensitive to both oxygen and stretch and regulate pulmonary blood flow (15), potentially contributing to ventilation-perfusion mismatching in ARDS.
Therefore, KATP, Kv, and KCa channels may all play important roles in the different stages of ARDS. During the exudative phase, the Kir6.1/6.2 subunits of KATP channels can upregulate transepithelial Na+ transport via ENaC and Cl− transport via CFTR in AECs (106). In primary lung epithelial cells, KATP openers (pinacidil, YM934) and blockers (glibenclamide) can regulate fluid clearance by activation or inhibition of ENaC and cAMP-activated Cl− channels, respectively (106, 168), and inhibition of stretch-activated KATP channels impairs fluid clearance in frog lungs (26). Besides KATP channels, activation of KvLQT1 and KCa3.1 channels also has a positive effect on alveolar fluid clearance (12). Similar to stretch-activated Na+ channels, several of the mechanosensitive K+ channels linked to alveolar fluid clearance are also activated by cell swelling, including the K2P channel TASK-2 (136), KCa/IK (200), and KATP (39) channels. During the proliferative and fibrotic phases of ARDS, activation of KATP currents in lung epithelial cells by both physiological (lipoxin A4) and pharmacological (pinacidil) stimuli can promote cell migration, proliferation, and epithelial repair via ERK-MAP kinase phosphorylation (31). Furthermore, deficiency of Kir7.1 in the lung epithelium delays lung development (194) and KCa3.1-anti-β1 integrin interactions regulate alveolar epithelial wound healing (70).
Stretch-Activated Ca2+ Channels
Stretch-activated Ca2+ conductances gained increasing attention in the 1980s as endothelial regulators of vascular tone (104), but their contribution to inflammatory processes was not much explored until the 1990s. Initially the molecular identity of these Ca2+ conductances remained largely elusive. These channels were simply termed “stretch-activated channels” and thought to activate KCa channels, which then would hyperpolarize the cell membrane and promote further Ca2+ influx (210). In fact, this mechanism was later recognized as an important regulator of the pulmonary vascular tone (24) and of several immune cell functions, including lymphocyte adhesion (184) and endothelial MCP-1 secretion (212), which is a key neutrophil attractant in ARDS.
Unfortunately, the regulation of mediator secretion from AECs is not as well understood as in neuronal or inflammatory cells, but the activation of stretch-activated Ca2+ channels is a common trigger for cytokine release (204). In our search for new therapeutic approaches against ARDS, the TRP superfamily of cation channels has emerged as one of the most promising targets. TRP channels are key regulators and integrators of several major features of ARDS, including mechanosensing and mechanotransduction (217), O2 sensing (186), inflammatory cell activation and mediator release (111), alveolar-capillary barrier function (8), epithelial wound healing and fibrosis (214), and cell apoptosis and necrosis (79). Although TRP channels are nonselective cation channels, they perform their effector functions largely via Ca2+ inward currents (147). To date, the mammalian TRP superfamily comprises seven subfamilies (TRPC, TRPM, TRPV, TRPA, TRPP, TRPML, and TRPN) with a total of 33 members (147). Although several TRP channels are expressed in lung-resident, immune, and inflammatory cells, only four channelopathies have been associated with human disease (137). In the lung, members of the TRPA, TRPC, TRPM, and TRPV subfamilies have been linked to asthma and COPD based on studies in knockout mouse models where these channels regulate bronchoconstriction, airway hyperreactivity, fibrosis, airway remodeling, chronic cough, and mucus hypersecretion (9, 50, 109). Evolutionarily, TRP channels are among the oldest mechanosensors and TRP-4 was identified in C. elegans where it acts as the pore-forming subunit of a native mechanotransduction channel (99). Of all TRPs, the mechanosensing apparatus of TRPC1 and TRPC6 is particularly well characterized (74, 143). However, most studies were performed in cardiac and smooth muscle cells (205), and the applicability of these findings to lung injury models needs to be determined. Of note, while TRP channels can be quite insensitive to stretch in heterologous expression systems, in vivo their low threshold to stretch is well documented (74).
Interestingly, the gating mechanism of several TRP channels is oxygen sensitive and TRP channels themselves can promote ROS production. In fact, TRPC3 and -C4 are endothelial redox sensors (205). In macrophages and endothelial cells, oxidative stress activates Ca2+ influx via TRPC1, -C4, and TRPM2 channels causing endothelial barrier disruption (42, 79). Similarly, oxidative stress-induced activation of TRPM2 induces zonula occludens and claudin-2 degradation, as well as neutrophil sequestration, resulting in loss of epithelial tight junctions (78, 213). In contrast, in lung macrophages activation of TRPM2 inhibits further ROS production and prevents endotoxin-induced lung injury (52). In lung nerve fibers, activation of TRPV1 and TRPA1 channels by ROS results in apnea (162). On the other hand, hypoxia, a key feature of ARDS, causes TRPC6- and TRPV4-mediated pulmonary vasoconstriction (120). TRPC1 activation contributes to alveolar-capillary barrier dysfunction and TRPC1 protein is highly upregulated by TNF-α (141), which is routinely elevated in the patients with ARDS. TRPA1 has received attention in ARDS research since its inhibition in vagal nerve afferents decreases ventilator-induced inflammation in a rat model of ARDS (202). TRPM8 activation is associated with IL-1α, IL-1β, IL-4, IL-6, IL-8, IL-13, GMCSF, and TNF-α release from human bronchial epithelial cells (164, 165), all of which contribute to the pathogenesis of ARDS. TRPC1 activation counteracts the development of sepsis-induced ARDS by promoting bacterial clearance and reducing proinflammatory cytokine secretion from macrophages and epithelial cells (224), while TRPM2 deficiency correlates with increased mortality rates in sepsis patients (151). On the other hand, TRPV1 activation promotes somatostatin release from sensory nerve endings and attenuates LPS-induced lung injury (81, 189) but also triggers IL-6 and IL-8 secretion from bronchial epithelial cells (155). TRPM7 induces mast cell degranulation (88), while TRPML2 causes chemokine release from macrophages upon TLR stimulation (182). Clearly, TRP channels affect a variety of mechanisms that regulate barrier function and cytokine secretion in ARDS, and we are in the midst of unraveling these complex networks. It appears, therefore, that while several TRP channels are important regulators of host defense mechanisms, they may also trigger downstream effects that ultimately worsen lung inflammation and barrier function.
Of all TRP channels, TRPV4 has received the most attention as a potential therapeutic target against ARDS. In mouse aspiration and inhalation models of ARDS, TRPV4 inhibition decreases neutrophil and macrophage influx and inflammatory cytokine release, reduces lung injury scores, prevents epithelial and endothelial cell detachment (193), improves lung compliance, and increases blood O2 saturations, even if administered after acid instillation (8). Recently, another group showed in a similar hydrochloric acid-induced ARDS model that genetic deficiency of TRPV4 and prophylactic administration of a TRPV4 inhibitor prevented neutrophil activation and disease development, including impairment of gas exchange and lung function, inflammation, lung edema, and histological lung damage (218). In contrast to the previous study, in this model the protective effect was no longer observed when the TRPV4 inhibitor was administered after acid instillation. The potential of preserving alveolar-capillary barrier function and reducing macrophage-derived reactive oxygen and nitrogen species by inhibiting TRPV4 was also confirmed in other inhalation- (129) and ventilator-induced (76) ARDS models. Further support for a role of TRPV4 in ARDS pathology is provided by a recent report showing TRPV4 activation by ROS and subsequent Src kinase-dependent endothelial barrier disruption (183). Interestingly, TRPV4 inhibition also prevents heart failure-induced pulmonary edema formation (187), which is important because most ARDS patients die from multiorgan failure, not respiratory failure.
In addition, TRPs may also play a role in adaptive connective tissue responses and epithelial wound healing in ARDS. Inflammatory stimuli found in ARDS, such as TNF-α, LPS, and IL-1α, induce TRPV1 expression in fibroblasts (167), whereas TRPM7 and TRPV4 inhibition decreases lung fibroblast proliferation, differentiation, and fibrogenesis (153, 219). Interestingly, blocking TRPC1 inhibits lung epithelial cell proliferation, while blocking TRPM8 and TRPA1 promotes epithelial cell proliferation (55, 220). Furthermore, several TRPs, including TRPC, TRPV, TRPM, TRPA, and TRPP, play important roles in the regulation of cell apoptosis and necrosis as they promote Ca2+ influx during cell shrinkage and cell swelling (146). Cell death via both of these mechanisms plays an important role in ARDS. Interactions of TRPM2 with its splice variant TRPM2-S induces apoptosis in endothelial cells via ROS-induced PKC activation (79), whereas degradation of TRPC1 by caspase-11 controls IL-1β release from macrophages (150).
Stretch-Activated, Nonselective Cation Channels
Piezo 1 and Piezo 2.
Mechanosensitive, stretch-activated, nonselective cation channels (NSCCs) assemble as tetramers with a Ca2+>Na+=K+ selectivity and constitute the largest ion channels complexes in nature (MmPiezo1 assembles as a 1.2 million Dalton homo-oligodimer). This fairly new group of Piezo 1 and 2 channels conducts large whole cell currents upon pressure stimulation, is inhibited by Gd3+ and ruthenium red, and shows rapid activation kinetics and conformational change-dependent gating, which are all typical features of SACs (46). Their role in mechanosensing and mechanotransduction is nowadays widely recognized and their activation leads to a 17- to 300-fold increase in mechanically activated whole cell currents (45). In lung epithelial cells, Piezo 1 deficiency decreased cell adhesion and increased cell migration, which could impact epithelial wound healing in ARDS (124, 195).
Other Ca2+-permeable NSCCs.
Mechanosensitive, Ca2+-permeable NSCCs act as volume sensors and volume regulators in many body tissues and are blocked by Gd3+, a typical feature of SACs (86). Inflammatory and epithelial cell volume changes, commonly seen in ARDS, can trigger cellular dysfunction, apoptosis, necrosis, cell detachment, and abnormal cell proliferation (86, 127, 131, 198). Inhibition of stretch-activated NSCCs could be particularly important in the exudative phase of ARDS since in rat lungs Ca2+ influx via these channels regulates microvascular permeability (142). Other features relevant to ARDS pathophysiology include their activation by oxidative stress resulting in cell necrosis (17) and their role as mechanotransducers (via stretch-induced cell adhesion kinase-β/CAK-β) in vascular smooth muscle cells (92).
Na2+- and K+-permeable-permeable NSCCs.
In AT II cells high level expression of the classical, stretch-sensitive, and Na+-selective ENaC channel depends heavily on culture conditions, including the presence of steroid hormones, permeable supports, and an air-liquid interface (93). Without these culture conditions this highly Na+-selective channel (HSC; Na+:K+ selectivity ratio >80, conductance 4–6pS) is not observed in AT II cells and instead a 21pS-conductance, nonselective cation channel (NSC; Na+:K+ selectivity ratio ∼1) can be detected. Furthermore, the ENaC α subunit alone (without β and γ) can act as a stretch-activated, nonselective cation channel and mechanosensor (101). The main importance of these channels in ARDS could relate to the exudative phase and alveolar fluid clearance and may support steroid therapy for ARDS patients. In fact, dexamethasone reduces pulmonary edema formation by upregulating α-ENaC in hydrogen sulfide-induced ARDS (97).
Stretch-Activated Cl− Channels
Mechanosensitive Cl− channels were described in the late 1990s in vascular endothelial cells and neurons where they counteract K+ efflux-induced membrane hyperpolarization and promote cell spreading (11, 57). Nowadays the cystic fibrosis transmembrane conductance regulator (CFTR) is the best characterized mechanosensing ion channel and is known to be expressed in AT I, AT II, and immune cells (63, 98, 135). Since its discovery in 1989 by Riordan et al. (156), an incredible amount of information has been published about the functional properties of CFTR and its regulation. However, the field entered a new era in 2010 with the discovery of the stretch-activated gating properties of CFTR (221). Zhang et al. (221) showed in non-CF, human airway epithelial cells (Calu-3) that the normal cellular machinery that controls CFTR can be completely bypassed simply by changes in membrane tension. This mechanosensitivity was confirmed in cell-free, excised membrane patches, demonstrating that neither an intact cell nor ATP was required for this effect. However, since membrane tension was less potent at activating CFTR in these cell-free experiments, an intracellular factor or cytoskeletal contributions to the mechanosensing properties could not be excluded (75). This discovery sparked new enthusiasm in studying CFTR as a regulator of acute lung injury and ARDS, as this channel could affect not only the exudative phase of ARDS by regulating alveolar fluid clearance but also the proliferative and fibrotic phases via regulation of inflammatory cell activation. In fact, deficiency of CFTR in neutrophils (181) and platelets (222) accelerates LPS-induced ARDS development. In contrast, upregulation of CFTR by lipoxin A4, an endogenous “breaking signal” for inflammation, improves alveolar fluid clearance (216). Of importance, the regulation of alveolar fluid clearance by CFTR is a highly complex process (21). Although most would agree that in general CFTR activation promotes fluid clearance (62), Solymosi et al. (178) showed that under conditions of hydrostatic pressure CFTR-dependent fluid flux can reverse from an absorptive into a secretory mode, thus promoting pulmonary edema formation. These phenomena may, at least in part, be explained by the differential expression of cotransporters in AT I vs. AT II cells, which may determine the ultimate direction of fluid flux (82). AT I cells preferentially express the K+-Cl− cotransporter, resulting in apical to basolateral Cl− movement and fluid absorption, while AT II cells mostly express the Na+-K+-2Cl− cotransporter, resulting in Cl− internalization, which creates the driving force for Cl− (and subsequent fluid) secretion upon CFTR activation. In ARDS, destruction of AT I cells could thus contribute to decreased alveolar fluid clearance (105), while expansion of the AT II cell pool in an effort to replace injured epithelial areas may lead to further fluid secretion. Interestingly, in influenza models of ARDS, CFTR inhibition improves pulmonary edema formation and prevents ARDS development by upregulating macrophage and IL-6 responses (2, 211), although CFTR deficiency at the same time can also activate proinflammatory JAK3-dependent pathways (34). The astonishing complexity of CFTR regulation is further evidenced by studies reporting that CFTR inhibition promotes lung development and protects lung epithelial cells from ROS-induced damage by regulating intracellular glutathione levels, but also increases p38- and ERK-mediated IL-6 and IL-8 release and stimulate neutrophil infiltration (22, 27). Therefore, CFTR acts as an oxygen-sensitive mechanosensor and mechanotransducer in the lung with wide-ranging effects on alveolar fluid clearance and immune cell function. In addition, CFTR can directly regulate other Cl− channels (ClC3B, VRAC, CaCl), K+ channels (Kir, KvLQT1, ROMK2), Na+ channels (ENaC), water channels (aquaporins), ATP transporters, and antiporters (Na+/H+ and Cl−/HCO−) (138, 170). Volume-regulated anion channels (VRACs) are other mechanosensitive Cl− channels that partially overlap with the ClC Cl− channel family (95, 138). The importance of VRACs in ARDS is supported by their role as mechanosensors in endothelial cells, cell apoptosis, and ATP, lactate, and HCO3− transport (145).
Limitations, Challenges, and Future Research
Challenges inherent to ARDS pathophysiology.
Similar to all other treatment options for ARDS, the multifactorial etiology and pathophysiological heterogeneity of ARDS represents therapeutic challenges (157, 207). Nonuniform alveolar expansion creates multiple different biophysical microenvironments within an ARDS lung with potentially opposing stretch forces (atelectatic vs. emphysematous areas, edematous vs. nonedematous areas, fibrotic vs. nonfibrotic areas, more vs. fewer cellular infiltrates) (148). Although in ARDS alveolar cells are simultaneously exposed to a combination of stretch forces produced by MV, pulmonary blood flow, and local changes in surface tension caused by surfactant inactivation, the ramifications of this complex, heterogeneous microenvironment (mechanical stretch, hyperoxia, and inflammatory mediators) on SAC expression and function are now being addressed (13, 76, 174). In addition, clinical interventions such as neuromuscular blockade or prone positioning of patients can also affect stretch forces in a patient's lung. Future studies should also explore the possibility of inhibiting the ATP-driven inflammasome activation by modulating SAC expression and activity. We have already found TREK-1 expression on alveolar and interstitial macrophages (174) and inhibition of TRP and ENaC channels abolishes stretch-induced ATP release from epithelial cells (140). Another important area for research is the effect of metabolic acidosis on SAC function, which can be profound in ARDS patients suffering from multiorgan failure. In fact, gating of many SACs is pH dependent as seen with K2P and BKCa channels (41, 84). In addition, the role of calpain, which is increased during MV (113), in cleaving Nav should be further explored. It is conceivable that stabilization or overexpression of Nav could inhibit calpain-induced lung injury. As with all known ion channel modulators, drug specificity issues will continue to constitute challenges for future studies since many SACs are quite ubiquitously expressed. Nevertheless, specific inhibitors for some of the most promising SAC targets such as TREK-1 and TRPV4 are already commercially available and have been tested in humans, although not in the context of ARDS. The TREK-1 inhibitor Spadin is currently being used as an antidepressant (28), while TRPV4 inhibitors (GSK193874, GSK2263095) are used for prevention and resolution of cardiogenic pulmonary edema (187). Given the varying degrees of effect of TRPV4 inhibitors in different ARDS models (8, 218), future studies will need to determine which ARDS patients (possibly based on ARDS etiology) may benefit most from TRPV4 inhibition.
Difficulties in measuring stretch forces in vivo.
Despite tremendous technological advances, we are still limited in our ability to quantify alveolar wall stress in situ, especially when it comes to measuring stretch transduction between interacting epithelial/endothelial and inflammatory/immune cells (115). These are important considerations when translating the stretch forces required to activate a SAC in vitro to an in vivo system. If supraphysiological stretch forces are necessary to alter a given channel's function, then stretch activation may well be a biophysical property of this channel but potentially of no physiological relevance. Another important question for future ARDS research is how SACs react to compression forces, such as hydrostatic pressure from pulmonary edema or alveolar compression from atelectasis, since SAC activation by hydrostatic pressure has been reported in other organs (90, 169). In addition to cyclic stretch and compression forces, the effects of tonic stretch on SACs in the lung need further investigation. High positive end-expiratory pressure is an important component of ARDS treatment and exposes lungs to high tonic stretch levels. Tonic stretch forces are also caused by APRV-type MV, often employed in ARDS patients (60). Importantly, in AECs tonic stretch induces unfolding and insertion of additional phospholipids into the plasma membrane and reduces membrane tension, which could modify SAC activation during MV (64).
Patch-clamp controversies.
Whole cell and single channel recordings using multiple variations of the patch-clamping technique are the gold standard of defining the biophysical profiles of SACs. Of importance, since a patch recording is derived from a small area of a single cell under gigaohm seal conditions (necessary to minimize leak currents), it is challenging to extrapolate such a one-time measurement to the highly dynamic and heterogeneous microenvironment of ARDS lungs. Furthermore, during patch formation the outer part of the lipid bilayer is more stretched than the inner part and, depending on the location of the gating sensor within a channel, it may affect its mechanosensitivity (166). Nevertheless, despite the recent expansion of fluorescent tracer probes to detect ion currents, patch clamp will remain the gold standard for ion current measurements for the foreseeable future due to its vast superiority in isolating and delineating the biophysical properties of a channel. Further technological advances allowing to patch single cells within a native lung tissue slice have already opened exciting new opportunities to study cell currents in more physiological (and pathological) environments (80).
From heterologous expression systems to in vivo ARDS models.
Heterologous expression systems, such as the Xenopus laevis oocyte, remain an invaluable tool for electrophysiologists to study the details of an ion channel's biophysical properties in a controlled environment, especially to initially characterize newer groups of ion channels such as K2P channels or Piezos. Species differences in that regard are of limited concern since ion channel sequences are usually well preserved throughout nature. Limitations of the oocyte system include the potential lack of channel insertion into the cytoskeleton and cytoskeletal rearrangements caused by channel overexpression (26). Heterologous expression systems have been indispensable in determining the biophysical properties of ENaC but its subunit composition in lung-resident and inflammatory cells needs to be confirmed. In most cell types, the α subunit forms the core of the channel and γ-ENaC seems important for alveolar fluid clearance (14). The contribution and combination of other subunits (β, γ, δ) to create a mechanosensitive channel in the lung are important for the design of targeted therapeutic strategies against specific ENaC subunits rather than altering global fluid homeostasis by modifying all ENaC channels in ARDS lungs. The development of genetic technology such as the CRISPR systems now allows us to develop lung cell type-specific conditional knockout models in a much cheaper and less time-consuming manner. These approaches clearly aid us in clarifying some of the inconsistencies in SAC characteristics found between cultured cell models and in vivo systems, as described for example for TREK-1 (171, 174).
Conclusion
As efficient mechanosensors, mechanotransducers, and signal integrators, SACs are prime candidates for much-needed new therapeutic approaches against ARDS. A better integration of the vast amounts of basic science and clinical knowledge concerning SAC physiology and ARDS pathology will undoubtedly revitalize old and spark new collaborations between electrophysiologists and clinician-scientists. The fruits of this work will eventually allow us to counteract the life-threatening inflammatory processes in ARDS lungs at the earliest stages, rather than attempting to ameliorate already existing lung damage.
GRANTS
This research was supported by National Heart, Lung, and Blood Institute Grant 7KO8HL118118-03.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
A.S. prepared figures; A.S. drafted manuscript; A.S. edited and revised manuscript; A.S. approved final version of manuscript.
REFERENCES
- 1.Achard JM, Bubien JK, Benos DJ, Warnock DG. Stretch modulates amiloride sensitivity and cation selectivity of sodium channels in human B lymphocytes. Am J Physiol Cell Physiol 270: C224–C234, 1996. [DOI] [PubMed] [Google Scholar]
- 2.Aeffner F, Abdulrahman B, Hickman-Davis JM, Janssen PM, Amer A, Bedwell DM, Sorscher EJ, Davis IC. Heterozygosity for the F508del mutation in the cystic fibrosis transmembrane conductance regulator anion channel attenuates influenza severity. J Infect Dis 208: 780–789, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ali MH, Schumacker PT. Endothelial responses to mechanical stress: where is the mechanosensor? Crit Care Med 30: S198–S206, 2002. [DOI] [PubMed] [Google Scholar]
- 4.Althaus M, Bogdan R, Clauss WG, Fronius M. Mechanosensitivity of epithelial sodium channels (ENaCs): laminar shear stress increases ion channel open probability. FASEB J 21: 2389–2399, 2007. [DOI] [PubMed] [Google Scholar]
- 5.Althaus M, Clauss WG, Fronius M. Amiloride-sensitive sodium channels and pulmonary edema. Pulm Med 2011: 830320, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Althaus M, Fronius M, Buchackert Y, Vadasz I, Clauss WG, Seeger W, Motterlini R, Morty RE. Carbon monoxide rapidly impairs alveolar fluid clearance by inhibiting epithelial sodium channels. Am J Respir Cell Mol Biol 41: 639–650, 2009. [DOI] [PubMed] [Google Scholar]
- 7.Aschner Y, Zemans RL, Yamashita CM, Downey GP. Matrix metalloproteinases and protein tyrosine kinases: potential novel targets in acute lung injury and ARDS. Chest 146: 1081–1091, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Balakrishna S, Song W, Achanta S, Doran SF, Liu B, Kaelberer MM, Yu Z, Sui A, Cheung M, Leishman E, Eidam HS, Ye G, Willette RN, Thorneloe KS, Bradshaw HB, Matalon S, Jordt SE. TRPV4 inhibition counteracts edema and inflammation and improves pulmonary function and oxygen saturation in chemically induced acute lung injury. Am J Physiol Lung Cell Mol Physiol 307: L158–L172, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Banner KH, Igney F, Poll C. TRP channels: emerging targets for respiratory disease. Pharmacol Ther 130: 371–384, 2011. [DOI] [PubMed] [Google Scholar]
- 10.Bao HF, Song JZ, Duke BJ, Ma HP, Denson DD, Eaton DC. Ethanol stimulates epithelial sodium channels by elevating reactive oxygen species. Am J Physiol Cell Physiol 303: C1129–C1138, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barakat AI, Leaver EV, Pappone PA, Davies PF. A flow-activated chloride-selective membrane current in vascular endothelial cells. Circ Res 85: 820–828, 1999. [DOI] [PubMed] [Google Scholar]
- 12.Bardou O, Prive A, Migneault F, Roy-Camille K, Dagenais A, Berthiaume Y, Brochiero E. K+ channels regulate ENaC expression via changes in promoter activity and control fluid clearance in alveolar epithelial cells. Biochim Biophys Acta 1818: 1682–1690, 2012. [DOI] [PubMed] [Google Scholar]
- 13.Bardou O, Trinh NT, Brochiero E. Molecular diversity and function of K+ channels in airway and alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol 296: L145–L155, 2009. [DOI] [PubMed] [Google Scholar]
- 14.Barker PM, Nguyen MS, Gatzy JT, Grubb B, Norman H, Hummler E, Rossier B, Boucher RC, Koller B. Role of gammaENaC subunit in lung liquid clearance and electrolyte balance in newborn mice. Insights into perinatal adaptation and pseudohypoaldosteronism. J Clin Invest 102: 1634–1640, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barman SA, Zhu S, White RE. Hypoxia modulates cyclic AMP activation of BkCa channels in rat pulmonary arterial smooth muscle. Lung 183: 353–361, 2005. [DOI] [PubMed] [Google Scholar]
- 16.Baron RM, Levy BD. Recent advances in understanding and treating ARDS. F1000Res 5: 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barros LF, Stutzin A, Calixto A, Catalan M, Castro J, Hetz C, Hermosilla T. Nonselective cation channels as effectors of free radical-induced rat liver cell necrosis. Hepatology 33: 114–122, 2001. [DOI] [PubMed] [Google Scholar]
- 18.Bayliss DA, Barrett PQ. Emerging roles for two-pore-domain potassium channels and their potential therapeutic impact. Trends Pharmacol Sci 29: 566–575, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Belperio JA, Keane MP, Lynch JP 3rd, Strieter RM. The role of cytokines during the pathogenesis of ventilator-associated and ventilator-induced lung injury. Semin Respir Crit Care Med 27: 350–364, 2006. [DOI] [PubMed] [Google Scholar]
- 20.Benos DJ. Sensing tension: recognizing ENaC as a stretch sensor. Hypertension 44: 616–617, 2004. [DOI] [PubMed] [Google Scholar]
- 21.Berthiaume Y, Matthay MA. Alveolar edema fluid clearance and acute lung injury. Respir Physiol Neurobiol 159: 350–359, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Berube J, Roussel L, Nattagh L, Rousseau S. Loss of cystic fibrosis transmembrane conductance regulator function enhances activation of p38 and ERK MAPKs, increasing interleukin-6 synthesis in airway epithelial cells exposed to Pseudomonas aeruginosa. J Biol Chem 285: 22299–22307, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beyder A, Rae JL, Bernard C, Strege PR, Sachs F, Farrugia G. Mechanosensitivity of Nav1.5, a voltage-sensitive sodium channel. J Physiol 588: 4969–4985, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bialecki RA, Kulik TJ, Colucci WS. Stretching increases calcium influx and efflux in cultured pulmonary arterial smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 263: L602–L606, 1992. [DOI] [PubMed] [Google Scholar]
- 25.Blount P, Sukharev SI, Moe PC, Martinac B, Kung C. Mechanosensitive channels of bacteria. Methods Enzymol 294: 458–482, 1999. [DOI] [PubMed] [Google Scholar]
- 26.Bogdan R, Veith C, Clauss W, Fronius M. Impact of mechanical stress on ion transport in native lung epithelium (Xenopus laevis): short-term activation of Na+, Cl− and K+ channels. Pflügers Arch 456: 1109–1120, 2008. [DOI] [PubMed] [Google Scholar]
- 27.Boncoeur E, Criq VS, Bonvin E, Roque T, Henrion-Caude A, Gruenert DC, Clement A, Jacquot J, Tabary O. Oxidative stress induces extracellular signal-regulated kinase 1/2 mitogen-activated protein kinase in cystic fibrosis lung epithelial cells: potential mechanism for excessive IL-8 expression. Int J Biochem Cell Biol 40: 432–446, 2008. [DOI] [PubMed] [Google Scholar]
- 28.Borsotto M, Veyssiere J, Moha Ou Maati H, Devader C, Mazella J, Heurteaux C. Targeting two-pore domain K+ channels TREK-1 and TASK-3 for the treatment of depression: a new therapeutic concept. Br J Pharmacol 172: 771–784, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bradley E, Webb TI, Hollywood MA, Sergeant GP, McHale NG, Thornbury KD. The cardiac sodium current Nav1.5 is functionally expressed in rabbit bronchial smooth muscle cells. Am J Physiol Cell Physiol 305: C427–C435, 2013. [DOI] [PubMed] [Google Scholar]
- 30.Brouns I, Pintelon I, Timmermans JP, Adriaensen D. Novel insights in the neurochemistry and function of pulmonary sensory receptors. Adv Anat Embryol Cell Biol 211: 1–115, vii, 2012. [PubMed] [Google Scholar]
- 31.Buchanan PJ, McNally P, Harvey BJ, Urbach V. Lipoxin A4-mediated KATP potassium channel activation results in cystic fibrosis airway epithelial repair. Am J Physiol Lung Cell Mol Physiol 305: L193–L201, 2013. [DOI] [PubMed] [Google Scholar]
- 32.Budinger GR, Mutlu GM. beta2-agonists and acute respiratory distress syndrome. Am J Respir Crit Care Med 189: 624–625, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Budinger GR, Sznajder JI. The alveolar-epithelial barrier: a target for potential therapy. Clin Chest Med 27: 655–669; abstract ix, 2006. [DOI] [PubMed] [Google Scholar]
- 34.Cao K, Chen M, Jie X, Wang Y, Li Q, Xu J. H5N1 virus hemagglutinin inhibition of cAMP-dependent CFTR via TLR4-mediated Janus tyrosine kinase 3 activation exacerbates lung inflammation. Mol Med 21: 134–142, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carlson BE, Beard DA. Mechanical control of cation channels in the myogenic response. Am J Physiol Heart Circ Physiol 301: H331–H343, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Carr MJ, Undem BJ. Ion channels in airway afferent neurons. Respir Physiol 125: 83–97, 2001. [DOI] [PubMed] [Google Scholar]
- 37.Cavanaugh KJ Jr, Oswari J, Margulies SS. Role of stretch on tight junction structure in alveolar epithelial cells. Am J Respir Cell Mol Biol 25: 584–591, 2001. [DOI] [PubMed] [Google Scholar]
- 38.Chapman KE, Sinclair SE, Zhuang D, Hassid A, Desai LP, Waters CM. Cyclic mechanical strain increases reactive oxygen species production in pulmonary epithelial cells. Am J Physiol Lung Cell Mol Physiol 289: L834–L841, 2005. [DOI] [PubMed] [Google Scholar]
- 39.Chatterjee S, Levitan I, Wei Z, Fisher AB. KATP channels are an important component of the shear-sensing mechanism in the pulmonary microvasculature. Microcirculation 13: 633–644, 2006. [DOI] [PubMed] [Google Scholar]
- 40.Chemin J, Patel AJ, Delmas P, Sachs F, Lazdunski M, Honore E. Regulation of the mechano-gated K2P channel TREK-1 by membrane phospholipids. Curr Top Membr 59: 155–170, 2007. [DOI] [PubMed] [Google Scholar]
- 41.Cho YE, Ahn DS, Kim YH, Taggart MJ, Lee YH. Changes in stretch-induced tone induced by intracellular acidosis in rabbit basilar artery: effects on BKCa channel activity. Vascul Pharmacol 47: 74–82, 2007. [DOI] [PubMed] [Google Scholar]
- 42.Cioffi DL. Redox regulation of endothelial canonical transient receptor potential channels. Antioxid Redox Signal 15: 1567–1582, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Copland IB, Post M. Stretch-activated signaling pathways responsible for early response gene expression in fetal lung epithelial cells. J Cell Physiol 210: 133–143, 2007. [DOI] [PubMed] [Google Scholar]
- 44.Corey DP, Garcia-Anoveros J. Mechanosensation and the DEG/ENaC ion channels. Science 273: 323–324, 1996. [DOI] [PubMed] [Google Scholar]
- 45.Coste B, Mathur J, Schmidt M, Earley TJ, Ranade S, Petrus MJ, Dubin AE, Patapoutian A. Piezo1 and Piezo2 are essential components of distinct mechanically activated cation channels. Science 330: 55–60, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Coste B, Xiao B, Santos JS, Syeda R, Grandl J, Spencer KS, Kim SE, Schmidt M, Mathur J, Dubin AE, Montal M, Patapoutian A. Piezo proteins are pore-forming subunits of mechanically activated channels. Nature 483: 176–181, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Crosby LM, Luellen C, Zhang Z, Tague LL, Sinclair SE, Waters CM. Balance of life and death in alveolar epithelial type II cells: proliferation, apoptosis, and the effects of cyclic stretch on wound healing. Am J Physiol Lung Cell Mol Physiol 301: L536–L546, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.D'Angelo E, Koutsoukou A, Della Valle P, Gentile G, Pecchiari M. Cytokine release, small airway injury, and parenchymal damage during mechanical ventilation in normal open-chest rats. J Appl Physiol 104: 41–49, 2008. [DOI] [PubMed] [Google Scholar]
- 49.Decher N, Kiper AK, Rolfes C, Schulze-Bahr E, Rinne S. The role of acid-sensitive two-pore domain potassium channels in cardiac electrophysiology: focus on arrhythmias. Pflügers Arch 467: 1055–1067, 2015. [DOI] [PubMed] [Google Scholar]
- 50.Deering-Rice CE, Shapiro D, Romero EG, Stockmann C, Bevans TS, Phan QM, Stone BL, Fassl B, Nkoy F, Uchida DA, Ward RM, Veranth JM, Reilly CA. Activation of transient receptor potential ankyrin-1 by insoluble particulate material and association with asthma. Am J Respir Cell Mol Biol 53: 893–901, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Delong P, Murray JA, Cook CK. Mechanical ventilation in the management of acute respiratory distress syndrome. Semin Dial 19: 517–524, 2006. [DOI] [PubMed] [Google Scholar]
- 52.Di A, Gao XP, Qian F, Kawamura T, Han J, Hecquet C, Ye RD, Vogel SM, Malik AB. The redox-sensitive cation channel TRPM2 modulates phagocyte ROS production and inflammation. Nat Immunol 13: 29–34, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dietrich J, Lindau M. Chloride channels in mast cells: block by DIDS and role in exocytosis. J Gen Physiol 104: 1099–1111, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Drummond HA, Grifoni SC, Jernigan NL. A new trick for an old dogma: ENaC proteins as mechanotransducers in vascular smooth muscle. Physiology (Bethesda) 23: 23–31, 2008. [DOI] [PubMed] [Google Scholar]
- 55.Du GJ, Li JH, Liu WJ, Liu YH, Zhao B, Li HR, Hou XD, Li H, Qi XX, Duan YJ. The combination of TRPM8 and TRPA1 expression causes an invasive phenotype in lung cancer. Tumour Biol 35: 1251–1261, 2014. [DOI] [PubMed] [Google Scholar]
- 56.Dushianthan A, Grocott MP, Postle AD, Cusack R. Acute respiratory distress syndrome and acute lung injury. Postgrad Med J 87: 612–622, 2011. [DOI] [PubMed] [Google Scholar]
- 57.Eder C, Klee R, Heinemann U. Involvement of stretch-activated Cl− channels in ramification of murine microglia. J Neurosci 18: 7127–7137, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.El-Hashash AH, Turcatel G, Varma S, Berika M, Al Alam D, Warburton D. Eya1 protein phosphatase regulates tight junction formation in lung distal epithelium. J Cell Sci 125: 4036–4048, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 59.Es-Salah-Lamoureux Z, Steele DF, Fedida D. Research into the therapeutic roles of two-pore-domain potassium channels. Trends Pharmacol Sci 31: 587–595, 2010. [DOI] [PubMed] [Google Scholar]
- 60.Facchin F, Fan E. Airway pressure release ventilation and high-frequency oscillatory ventilation: potential strategies to treat severe hypoxemia and prevent ventilator-induced lung injury. Respir Care 60: 1509–1521, 2015. [DOI] [PubMed] [Google Scholar]
- 61.Fanelli V, Ranieri VM. Mechanisms and clinical consequences of acute lung injury. Ann Am Thorac Soc 12, Suppl 1: S3–S8, 2015. [DOI] [PubMed] [Google Scholar]
- 62.Fang X, Fukuda N, Barbry P, Sartori C, Verkman AS, Matthay MA. Novel role for CFTR in fluid absorption from the distal airspaces of the lung. J Gen Physiol 119: 199–207, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fang X, Song Y, Hirsch J, Galietta LJ, Pedemonte N, Zemans RL, Dolganov G, Verkman AS, Matthay MA. Contribution of CFTR to apical-basolateral fluid transport in cultured human alveolar epithelial type II cells. Am J Physiol Lung Cell Mol Physiol 290: L242–L249, 2006. [DOI] [PubMed] [Google Scholar]
- 64.Fisher JL, Levitan I, Margulies SS. Plasma membrane surface increases with tonic stretch of alveolar epithelial cells. Am J Respir Cell Mol Biol 31: 200–208, 2004. [DOI] [PubMed] [Google Scholar]
- 65.Frank JA, Matthay MA. Science review: mechanisms of ventilator-induced injury. Crit Care 7: 233–241, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fronius M, Bogdan R, Althaus M, Morty RE, Clauss WG. Epithelial Na+ channels derived from human lung are activated by shear force. Respir Physiol Neurobiol 170: 113–119, 2010. [DOI] [PubMed] [Google Scholar]
- 67.Fronius M, Clauss WG. Mechanosensitivity of ENaC: may the (shear) force be with you. Pflügers Arch 455: 775–785, 2008. [DOI] [PubMed] [Google Scholar]
- 68.Fronius M, Clauss WG, Althaus M. Why do we have to move fluid to be able to breathe? Front Physiol 3: 146, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ghofrani HA, Kohstall MG, Weissmann N, Schmehl T, Schermuly RT, Seeger W, Grimminger F. Alveolar epithelial barrier functions in ventilated perfused rabbit lungs. Am J Physiol Lung Cell Mol Physiol 280: L896–L904, 2001. [DOI] [PubMed] [Google Scholar]
- 70.Girault A, Chebli J, Prive A, Trinh NT, Maille E, Grygorczyk R, Brochiero E. Complementary roles of KCa3.1 channels and beta1-integrin during alveolar epithelial repair. Respir Res 16: 100, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Goodson P, Kumar A, Jain L, Kundu K, Murthy N, Koval M, Helms MN. Nadph oxidase regulates alveolar epithelial sodium channel activity and lung fluid balance in vivo via O2− signaling. Am J Physiol Lung Cell Mol Physiol 302: L410–L419, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Goolaerts A, Roux J, Ganter MT, Shlyonsky V, Chraibi A, Stephane R, Mies F, Matthay MA, Naeije R, Sariban-Sohraby S, Howard M, Pittet JF. Serotonin decreases alveolar epithelial fluid transport via a direct inhibition of the epithelial sodium channel. Am J Respir Cell Mol Biol 43: 99–108, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Goonetilleke L, Quayle J. TREK-1 K+ channels in the cardiovascular system: their significance and potential as a therapeutic target. Cardiovasc Ther 30: e23–e29, 2012. [DOI] [PubMed] [Google Scholar]
- 74.Gottlieb P, Folgering J, Maroto R, Raso A, Wood TG, Kurosky A, Bowman C, Bichet D, Patel A, Sachs F, Martinac B, Hamill OP, Honore E. Revisiting TRPC1 and TRPC6 mechanosensitivity. Pflügers Arch 455: 1097–1103, 2008. [DOI] [PubMed] [Google Scholar]
- 75.Gray MA. CFTR is a mechanosensitive anion channel: a real stretch? Cellscience 7: 1–7, 2010. [PMC free article] [PubMed] [Google Scholar]
- 76.Hamanaka K, Jian MY, Townsley MI, King JA, Liedtke W, Weber DS, Eyal FG, Clapp MM, Parker JC. TRPV4 channels augment macrophage activation and ventilator-induced lung injury. Am J Physiol Lung Cell Mol Physiol 299: L353–L362, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hamill OP, Martinac B. Molecular basis of mechanotransduction in living cells. Physiol Rev 81: 685–740, 2001. [DOI] [PubMed] [Google Scholar]
- 78.Hecquet CM, Ahmmed GU, Malik AB. TRPM2 channel regulates endothelial barrier function. Adv Exp Med Biol 661: 155–167, 2010. [DOI] [PubMed] [Google Scholar]
- 79.Hecquet CM, Zhang M, Mittal M, Vogel SM, Di A, Gao X, Bonini MG, Malik AB. Cooperative interaction of trp melastatin channel transient receptor potential (TRPM2) with its splice variant TRPM2 short variant is essential for endothelial cell apoptosis. Circ Res 114: 469–479, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Helms MN, Jain L, Self JL, Eaton DC. Redox regulation of epithelial sodium channels examined in alveolar type 1 and 2 cells patch-clamped in lung slice tissue. J Biol Chem 283: 22875–22883, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Helyes Z, Elekes K, Nemeth J, Pozsgai G, Sandor K, Kereskai L, Borzsei R, Pinter E, Szabo A, Szolcsanyi J. Role of transient receptor potential vanilloid 1 receptors in endotoxin-induced airway inflammation in the mouse. Am J Physiol Lung Cell Mol Physiol 292: L1173–L1181, 2007. [DOI] [PubMed] [Google Scholar]
- 82.Hollenhorst MI, Richter K, Fronius M. Ion transport by pulmonary epithelia. J Biomed Biotechnol 2011: 174306, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Holter JF, Weiland JE, Pacht ER, Gadek JE, Davis WB. Protein permeability in the adult respiratory distress syndrome. Loss of size selectivity of the alveolar epithelium. J Clin Invest 78: 1513–1522, 1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Honore E, Maingret F, Lazdunski M, Patel AJ. An intracellular proton sensor commands lipid- and mechano-gating of the K+ channel TREK-1. EMBO J 21: 2968–2976, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.House CD, Vaske CJ, Schwartz AM, Obias V, Frank B, Luu T, Sarvazyan N, Irby R, Strausberg RL, Hales TG, Stuart JM, Lee NH. Voltage-gated Na+ channel SCN5A is a key regulator of a gene transcriptional network that controls colon cancer invasion. Cancer Res 70: 6957–6967, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hua SZ, Gottlieb PA, Heo J, Sachs F. A mechanosensitive ion channel regulating cell volume. Am J Physiol Cell Physiol 298: C1424–C1430, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Huang H, Liang L, Liu P, Wei H, Sachs F, Niu W, Wang W. Mechanical effects on KATP channel gating in rat ventricular myocytes. PLoS One 8: e63337, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Huang L, Ng NM, Chen M, Lin X, Tang T, Cheng H, Yang C, Jiang S. Inhibition of TRPM7 channels reduces degranulation and release of cytokines in rat bone marrow-derived mast cells. Int J Mol Sci 15: 11817–11831, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hunt JF, Fang K, Malik R, Snyder A, Malhotra N, Platts-Mills TA, Gaston B. Endogenous airway acidification. Implications for asthma pathophysiology. Am J Respir Crit Care Med 161: 694–699, 2000. [DOI] [PubMed] [Google Scholar]
- 90.Isenberg G, Kazanski V, Kondratev D, Gallitelli MF, Kiseleva I, Kamkin A. Differential effects of stretch and compression on membrane currents and [Na+]c in ventricular myocytes. Progr Biophys Mol Biol 82: 43–56, 2003. [DOI] [PubMed] [Google Scholar]
- 91.Iskratsch T, Wolfenson H, Sheetz MP. Appreciating force and shape—the rise of mechanotransduction in cell biology. Nat Rev Mol Cell Biol 15: 825–833, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Iwasaki H, Yoshimoto T, Sugiyama T, Hirata Y. Activation of cell adhesion kinase beta by mechanical stretch in vascular smooth muscle cells. Endocrinology 144: 2304–2310, 2003. [DOI] [PubMed] [Google Scholar]
- 93.Jain L, Chen XJ, Ramosevac S, Brown LA, Eaton DC. Expression of highly selective sodium channels in alveolar type II cells is determined by culture conditions. Am J Physiol Lung Cell Mol Physiol 280: L646–L658, 2001. [DOI] [PubMed] [Google Scholar]
- 94.Janiszewski J, Huizinga JD, Blennerhassett MG. Mast cell ionic channels: significance for stimulus-secretion coupling. Can J Physiol Pharmacol 70: 1–7, 1992. [DOI] [PubMed] [Google Scholar]
- 95.Jentsch TJ, Stein V, Weinreich F, Zdebik AA. Molecular structure and physiological function of chloride channels. Physiol Rev 82: 503–568, 2002. [DOI] [PubMed] [Google Scholar]
- 96.Ji HL, Fuller CM, Benos DJ. Osmotic pressure regulates αβγ-rENaC expressed in Xenopus oocytes. Am J Physiol Cell Physiol 275: C1182–C1190, 1998. [DOI] [PubMed] [Google Scholar]
- 97.Jiang L, Wang J, Su C, Qian W, Chen J, Zhu B, Zhang H, Xiao H, Zhang J. α-ENaC, a therapeutic target of dexamethasone on hydrogen sulfide induced acute pulmonary edema. Environ Toxicol Pharmacol 38: 616–624, 2014. [DOI] [PubMed] [Google Scholar]
- 98.Johnson M, Allen L, Dobbs L. Characteristics of Cl− uptake in rat alveolar type I cells. Am J Physiol Lung Cell Mol Physiol 297: L816–L827, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kang L, Gao J, Schafer WR, Xie Z, Xu XZ. C. elegans TRP family protein TRP-4 is a pore-forming subunit of a native mechanotransduction channel. Neuron 67: 381–391, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Karpushev AV, Ilatovskaya DV, Staruschenko A. The actin cytoskeleton and small G protein RhoA are not involved in flow-dependent activation of ENaC. BMC Res Notes 3: 210, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kizer N, Guo XL, Hruska K. Reconstitution of stretch-activated cation channels by expression of the alpha-subunit of the epithelial sodium channel cloned from osteoblasts. Proc Natl Acad Sci USA 94: 1013–1018, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Koshy S, Beard LL, Kuzenko SR, Li T, Folkesson HG. Lung fluid absorption is induced in preterm guinea pigs ventilated with low tidal volumes. Exp Lung Res 37: 44–56, 2011. [DOI] [PubMed] [Google Scholar]
- 103.Laitko U, Juranka PF, Morris CE. Membrane stretch slows the concerted step prior to opening in a Kv channel. J Gen Physiol 127: 687–701, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lansman JB, Hallam TJ, Rink TJ. Single stretch-activated ion channels in vascular endothelial cells as mechanotransducers? Nature 325: 811–813, 1987. [DOI] [PubMed] [Google Scholar]
- 105.Lee JW, Fang X, Dolganov G, Fremont RD, Bastarache JA, Ware LB, Matthay MA. Acute lung injury edema fluid decreases net fluid transport across human alveolar epithelial type II cells. J Biol Chem 282: 24109–24119, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Leroy C, Dagenais A, Berthiaume Y, Brochiero E. Molecular identity and function in transepithelial transport of KATP channels in alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol 286: L1027–L1037, 2004. [DOI] [PubMed] [Google Scholar]
- 107.Lesage F, Lazdunski M. Molecular and functional properties of two-pore-domain potassium channels. Am J Physiol Renal Physiol 279: F793–F801, 2000. [DOI] [PubMed] [Google Scholar]
- 108.LeSimple P, Liao J, Robert R, Gruenert DC, Hanrahan JW. Cystic fibrosis transmembrane conductance regulator trafficking modulates the barrier function of airway epithelial cell monolayers. J Physiol 588: 1195–1209, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Li S, Westwick J, Poll C. Transient receptor potential (TRP) channels as potential drug targets in respiratory disease. Cell Calcium 33: 551–558, 2003. [DOI] [PubMed] [Google Scholar]
- 110.Li T, Folkesson HG. RNA interference for α-ENaC inhibits rat lung fluid absorption in vivo. Am J Physiol Lung Cell Mol Physiol 290: L649–L660, 2006. [DOI] [PubMed] [Google Scholar]
- 111.Lindemann O, Strodthoff C, Horstmann M, Nielsen N, Jung F, Schimmelpfennig S, Heitzmann M, Schwab A. TRPC1 regulates fMLP-stimulated migration and chemotaxis of neutrophil granulocytes. Biochim Biophys Acta 1853: 2122–2130, 2015. [DOI] [PubMed] [Google Scholar]
- 112.Lindsey SH, Tribe RM, Songu-Mize E. Cyclic stretch decreases TRPC4 protein and capacitative calcium entry in rat vascular smooth muscle cells. Life Sci 83: 29–34, 2008. [DOI] [PubMed] [Google Scholar]
- 113.Liu D, Yan Z, Minshall RD, Schwartz DE, Chen Y, Hu G. Activation of calpains mediates early lung neutrophilic inflammation in ventilator-induced lung injury. Am J Physiol Lung Cell Mol Physiol 302: L370–L379, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Liu J, Schrank B, Waterston RH. Interaction between a putative mechanosensory membrane channel and a collagen. Science 273: 361–364, 1996. [DOI] [PubMed] [Google Scholar]
- 115.Liu M, Tanswell AK, Post M. Mechanical force-induced signal transduction in lung cells. Am J Physiol Lung Cell Mol Physiol 277: L667–L683, 1999. [DOI] [PubMed] [Google Scholar]
- 116.Ma HP, Al-Khalili O, Ramosevac S, Saxena S, Liang YY, Warnock DG, Eaton DC. Steroids and exogenous gamma-ENaC subunit modulate cation channels formed by alpha-ENaC in human B lymphocytes. J Biol Chem 279: 33206–33212, 2004. [DOI] [PubMed] [Google Scholar]
- 117.Makena PS, Gorantla VK, Ghosh MC, Bezawada L, Balazs L, Luellen CL, Parthasarathi K, Waters CM, Sinclair SE. Lung injury caused by high tidal volume mechanical ventilation and hyperoxia is dependent on oxidant-mediated c-Jun NH2-terminal kinase (JNK) activation. J Appl Physiol 111: 1467–1476, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Makena PS, Gorantla VK, Ghosh MC, Bezawada L, Kandasamy K, Balazs L, Luellen CL, Thompson KE, Parthasarathi K, Ichijo H, Waters CM, Sinclair SE. Deletion of apoptosis signal-regulating kinase-1 prevents ventilator-induced lung injury in mice. Am J Respir Cell Mol Biol 46: 461–469, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Makena PS, Luellen CL, Balazs L, Ghosh MC, Parthasarathi K, Waters CM, Sinclair SE. Preexposure to hyperoxia causes increased lung injury and epithelial apoptosis in mice ventilated with high tidal volumes. Am J Physiol Lung Cell Mol Physiol 299: L711–L719, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Malczyk M, Veith C, Schermuly RT, Gudermann T, Dietrich A, Sommer N, Weissmann N, Pak O. NADPH oxidases—do they play a role in TRPC regulation under hypoxia? Pflügers Arch 468: 23–51, 2016. [DOI] [PubMed] [Google Scholar]
- 121.Mark Duffy S, Berger P, Cruse G, Yang W, Bolton SJ, Bradding P. The K+ channel iKCA1 potentiates Ca2+ influx and degranulation in human lung mast cells. J Allergy Clin Immunol 114: 66–72, 2004. [DOI] [PubMed] [Google Scholar]
- 122.Markin VS, Martinac B. Mechanosensitive ion channels as reporters of bilayer expansion. A theoretical model. Biophys J 60: 1120–1127, 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Matsumoto S, Nishikawa T, Yoshida S, Ikeda M, Tanimoto T, Saiki C, Takeda M. Effects of potassium channel and Na+-Ca2+ exchange blockers on the responses of slowly adapting pulmonary stretch receptors to hyperinflation in flecainide-treated rats. Br J Pharmacol 134: 682–690, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.McHugh BJ, Murdoch A, Haslett C, Sethi T. Loss of the integrin-activating transmembrane protein Fam38A (Piezo1) promotes a switch to a reduced integrin-dependent mode of cell migration. PLoS One 7: e40346, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Meduri GU, Headley S, Kohler G, Stentz F, Tolley E, Umberger R, Leeper K. Persistent elevation of inflammatory cytokines predicts a poor outcome in ARDS. Plasma IL-1 beta and IL-6 levels are consistent and efficient predictors of outcome over time. Chest 107: 1062–1073, 1995. [DOI] [PubMed] [Google Scholar]
- 126.Mondrinos MJ, Kennedy PA, Lyons M, Deutschman CS, Kilpatrick LE. Protein kinase C and acute respiratory distress syndrome. Shock 39: 467–479, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Mongin AA, Orlov SN. Mechanisms of cell volume regulation and possible nature of the cell volume sensor. Pathophysiology 8: 77–88, 2001. [DOI] [PubMed] [Google Scholar]
- 128.Morel DR, Dargent F, Bachmann M, Suter PM, Junod AF. Pulmonary extraction of serotonin and propranolol in patients with adult respiratory distress syndrome. Am Rev Respir Dis 132: 479–484, 1985. [DOI] [PubMed] [Google Scholar]
- 129.Morty RE, Kuebler WM. TRPV4: an exciting new target to promote alveolocapillary barrier function. Am J Physiol Lung Cell Mol Physiol 307: L817–L821, 2014. [DOI] [PubMed] [Google Scholar]
- 130.Mourgeon E, Isowa N, Keshavjee S, Zhang X, Slutsky AS, Liu M. Mechanical stretch stimulates macrophage inflammatory protein-2 secretion from fetal rat lung cells. Am J Physiol Lung Cell Mol Physiol 279: L699–L706, 2000. [DOI] [PubMed] [Google Scholar]
- 131.Munaron L, Antoniotti S, Lovisolo D. Intracellular calcium signals and control of cell proliferation: how many mechanisms? J Cell Mol Med 8: 161–168, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Muroi Y, Undem BJ. Targeting voltage gated sodium channels NaV1.7, Na V18, and Na V19 for treatment of pathological cough. Lung 192: 15–20, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Mustafa SB, Isaac J, Seidner SR, Dixon PS, Henson BM, DiGeronimo RJ. Mechanical stretch induces lung alpha-epithelial Na+ channel expression. Exp Lung Res 40: 380–391, 2014. [DOI] [PubMed] [Google Scholar]
- 134.Myoga MH, Regehr WG. Calcium microdomains near R-type calcium channels control the induction of presynaptic long-term potentiation at parallel fiber to purkinje cell synapses. J Neurosci 31: 5235–5243, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Ng HP, Valentine VG, Wang G. CFTR targeting during activation of human neutrophils. J Leukoc Biol 2016. July 12 pii: jlb.4A0316-130RR. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 136.Niemeyer MI, Cid LP, Barros LF, Sepulveda FV. Modulation of the two-pore domain acid-sensitive K+ channel TASK-2 (KCNK5) by changes in cell volume. J Biol Chem 276: 43166–43174, 2001. [DOI] [PubMed] [Google Scholar]
- 137.Nilius B. TRP channels in disease. Biochim Biophys Acta 1772: 805–812, 2007. [DOI] [PubMed] [Google Scholar]
- 138.Nilius B, Droogmans G. Amazing chloride channels: an overview. Acta Physiol Scand 177: 119–147, 2003. [DOI] [PubMed] [Google Scholar]
- 139.Nilius B, Honore E. Sensing pressure with ion channels. Trends Neurosci 35: 477–486, 2012. [DOI] [PubMed] [Google Scholar]
- 140.Olsen SM, Stover JD, Nagatomi J. Examining the role of mechanosensitive ion channels in pressure mechanotransduction in rat bladder urothelial cells. Ann Biomed Eng 39: 688–697, 2011. [DOI] [PubMed] [Google Scholar]
- 141.Paria BC, Vogel SM, Ahmmed GU, Alamgir S, Shroff J, Malik AB, Tiruppathi C. Tumor necrosis factor-α-induced TRPC1 expression amplifies store-operated Ca2+ influx and endothelial permeability. Am J Physiol Lung Cell Mol Physiol 287: L1303–L1313, 2004. [DOI] [PubMed] [Google Scholar]
- 142.Parker JC, Ivey CL, Tucker JA. Gadolinium prevents high airway pressure-induced permeability increases in isolated rat lungs. J Appl Physiol 84: 1113–1118, 1998. [DOI] [PubMed] [Google Scholar]
- 143.Patel A, Sharif-Naeini R, Folgering JR, Bichet D, Duprat F, Honore E. Canonical TRP channels and mechanotransduction: from physiology to disease states. Pflügers Arch 460: 571–581, 2010. [DOI] [PubMed] [Google Scholar]
- 144.Patel AJ, Lazdunski M, Honore E. Lipid and mechano-gated 2P domain K+ channels. Curr Opin Cell Biol 13: 422–428, 2001. [DOI] [PubMed] [Google Scholar]
- 145.Pedersen SF, Klausen TK, Nilius B. The identification of a volume-regulated anion channel: an amazing Odyssey. Acta Physiol (Oxf) 213: 868–881, 2015. [DOI] [PubMed] [Google Scholar]
- 146.Pedersen SF, Nilius B. Transient receptor potential channels in mechanosensing and cell volume regulation. Methods Enzymol 428: 183–207, 2007. [DOI] [PubMed] [Google Scholar]
- 147.Pedersen SF, Owsianik G, Nilius B. TRP channels: an overview. Cell Calcium 38: 233–252, 2005. [DOI] [PubMed] [Google Scholar]
- 148.Perlman CE, Bhattacharya J. Alveolar expansion imaged by optical sectioning microscopy. J Appl Physiol 103: 1037–1044, 2007. [DOI] [PubMed] [Google Scholar]
- 149.Pompermayer K, Amaral FA, Fagundes CT, Vieira AT, Cunha FQ, Teixeira MM, Souza DG. Effects of the treatment with glibenclamide, an ATP-sensitive potassium channel blocker, on intestinal ischemia and reperfusion injury. Eur J Pharmacol 556: 215–222, 2007. [DOI] [PubMed] [Google Scholar]
- 150.Py BF, Jin M, Desai BN, Penumaka A, Zhu H, Kober M, Dietrich A, Lipinski MM, Henry T, Clapham DE, Yuan J. Caspase-11 controls interleukin-1beta release through degradation of TRPC1. Cell Rep 6: 1122–1128, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Qian X, Numata T, Zhang K, Li C, Hou J, Mori Y, Fang X. Transient receptor potential melastatin 2 protects mice against polymicrobial sepsis by enhancing bacterial clearance. Anesthesiology 121: 336–351, 2014. [DOI] [PubMed] [Google Scholar]
- 152.Qiu MR, Campbell TJ, Breit SN. A potassium ion channel is involved in cytokine production by activated human macrophages. Clin Exp Immunol 130: 67–74, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Rahaman SO, Grove LM, Paruchuri S, Southern BD, Abraham S, Niese KA, Scheraga RG, Ghosh S, Thodeti CK, Zhang DX, Moran MM, Schilling WP, Tschumperlin DJ, Olman MA. TRPV4 mediates myofibroblast differentiation and pulmonary fibrosis in mice. J Clin Invest 124: 5225–5238, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Ramsingh R, Grygorczyk A, Solecki A, Cherkaoui LS, Berthiaume Y, Grygorczyk R. Cell deformation at the air-liquid interface induces Ca2+-dependent ATP release from lung epithelial cells. Am J Physiol Lung Cell Mol Physiol 300: L587–L595, 2011. [DOI] [PubMed] [Google Scholar]
- 155.Reilly CA, Johansen ME, Lanza DL, Lee J, Lim JO, Yost GS. Calcium-dependent and independent mechanisms of capsaicin receptor (TRPV1)-mediated cytokine production and cell death in human bronchial epithelial cells. J Biochem Mol Toxicol 19: 266–275, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou JL, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 245: 1066–1073, 1989. [DOI] [PubMed] [Google Scholar]
- 157.Roan E, Waters CM. What do we know about mechanical strain in lung alveoli? Am J Physiol Lung Cell Mol Physiol 301: L625–L635, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Roan E, Waters CM, Teng B, Ghosh M, Schwingshackl A. The 2-pore domain potassium channel TREK-1 regulates stretch-induced detachment of alveolar epithelial cells. PLoS One 9: e89429, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Roan E, Wilhelm K, Bada A, Makena PS, Gorantla VK, Sinclair SE, Waters CM. Hyperoxia alters the mechanical properties of alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol 302: L1235–L1241, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Rose F, Dahlem G, Guthmann B, Grimminger F, Maus U, Hanze J, Duemmer N, Grandel U, Seeger W, Ghofrani HA. Mediator generation and signaling events in alveolar epithelial cells attacked by S. aureus α-toxin. Am J Physiol Lung Cell Mol Physiol 282: L207–L214, 2002. [DOI] [PubMed] [Google Scholar]
- 161.Rossier BC. Mechanosensitivity of the epithelial sodium channel (ENaC): controversy or pseudocontroversy? J Gen Physiol 112: 95–96, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Ruan T, Lin YJ, Hsu TH, Lu SH, Jow GM, Kou YR. Sensitization by pulmonary reactive oxygen species of rat vagal lung C-fibers: the roles of the TRPV1, TRPA1, and P2X receptors. PLoS One 9: e91763, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Rubenfeld GD, Caldwell E, Peabody E, Weaver J, Martin DP, Neff M, Stern EJ, Hudson LD. Incidence and outcomes of acute lung injury. N Engl J Med 353: 1685–1693, 2005. [DOI] [PubMed] [Google Scholar]
- 164.Sabnis AS, Reilly CA, Veranth JM, Yost GS. Increased transcription of cytokine genes in human lung epithelial cells through activation of a TRPM8 variant by cold temperatures. Am J Physiol Lung Cell Mol Physiol 295: L194–L200, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Sabnis AS, Shadid M, Yost GS, Reilly CA. Human lung epithelial cells express a functional cold-sensing TRPM8 variant. Am J Respir Cell Mol Biol 39: 466–474, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Sachs F. Stretch-activated ion channels: what are they? Physiology 25: 50–56, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Sadofsky LR, Ramachandran R, Crow C, Cowen M, Compton SJ, Morice AH. Inflammatory stimuli up-regulate transient receptor potential vanilloid-1 expression in human bronchial fibroblasts. Exp Lung Res 38: 75–81, 2012. [DOI] [PubMed] [Google Scholar]
- 168.Sakuma T, Takahashi K, Ohya N, Nakada T, Matthay MA. Effects of ATP-sensitive potassium channel opener on potassium transport and alveolar fluid clearance in the resected human lung. Pharmacol Toxicol 83: 16–22, 1998. [DOI] [PubMed] [Google Scholar]
- 169.Satlin LM, Sheng S, Woda CB, Kleyman TR. Epithelial Na+ channels are regulated by flow. Am J Physiol Renal Physiol 280: F1010–F1018, 2001. [DOI] [PubMed] [Google Scholar]
- 170.Schwiebert EM, Benos DJ, Egan ME, Stutts MJ, Guggino WB. CFTR is a conductance regulator as well as a chloride channel. Physiol Rev 79: S145–S166, 1999. [DOI] [PubMed] [Google Scholar]
- 171.Schwingshackl A, Teng B, Ghosh M, Waters CM. Regulation of monocyte chemotactic protein-1 secretion by the two-pore-domain potassium (K2P) channel TREK-1 in human alveolar epithelial cells. Am J Transl Res 5: 530–542, 2013. [PMC free article] [PubMed] [Google Scholar]
- 172.Schwingshackl A, Teng B, Ghosh M, West AN, Makena P, Gorantla V, Sinclair SE, Waters CM. Regulation and function of the two-pore-domain (K2P) potassium channel Trek-1 in alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol 302: L93–L102, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Schwingshackl A, Teng B, Ghosh MC, Lim KG, Tigyi GJ, Narayanan D, Jaggar JH, Waters CM. Regulation of interleukin-6 secretion by the two-pore-domain potassium (K2P) channel trek-1 in alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol 304: L276–L286, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Schwingshackl A, Teng B, Makena P, Ghosh M, Sinclair SE, Luellen C, Balasz L, Rovnaghi C, Bryan RM, Lloyd EE, Fitzpatrick E, Saravia JS, Cormier SA, Waters CM. Deficiency of the two-pore-domain potassium channel TREK-1 promotes hyperoxia-induced lung injury. Crit Care Med 42: e692–e701, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Simon A, Shenton F, Hunter I, Banks RW, Bewick GS. Amiloride-sensitive channels are a major contributor to mechanotransduction in mammalian muscle spindles. J Physiol 588: 171–185, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Sinclair SE, Altemeier WA, Matute-Bello G, Chi EY. Augmented lung injury due to interaction between hyperoxia and mechanical ventilation. Crit Care Med 32: 2496–2501, 2004. [DOI] [PubMed] [Google Scholar]
- 177.Slutsky AS, Ranieri VM. Ventilator-induced lung injury. N Engl J Med 370: 980, 2014. [DOI] [PubMed] [Google Scholar]
- 178.Solymosi EA, Kaestle-Gembardt SM, Vadasz I, Wang L, Neye N, Chupin CJ, Rozowsky S, Ruehl R, Tabuchi A, Schulz H, Kapus A, Morty RE, Kuebler WM. Chloride transport-driven alveolar fluid secretion is a major contributor to cardiogenic lung edema. Proc Natl Acad Sci USA 110: E2308–E2316, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Strege P, Beyder A, Bernard C, Crespo-Diaz R, Behfar A, Terzic A, Ackerman M, Farrugia G. Ranolazine inhibits shear sensitivity of endogenous Na+ current and spontaneous action potentials in HL-1 cells. Channels (Austin) 6: 457–462, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Stutzin A, Hoffmann EK. Swelling-activated ion channels: functional regulation in cell-swelling, proliferation and apoptosis. Acta Physiol (Oxf) 187: 27–42, 2006. [DOI] [PubMed] [Google Scholar]
- 181.Su X, Looney MR, Su HE, Lee JW, Song Y, Matthay MA. Role of CFTR expressed by neutrophils in modulating acute lung inflammation and injury in mice. Inflamm Res 60: 619–632, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Sun L, Hua Y, Vergarajauregui S, Diab HI, Puertollano R. Novel role of TRPML2 in the regulation of the innate immune response. J Immunol 195: 4922–4932, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Suresh K, Servinsky L, Reyes J, Baksh S, Undem C, Caterina M, Pearse DB, Shimoda LA. Hydrogen peroxide-induced calcium influx in lung microvascular endothelial cells involves TRPV4. Am J Physiol Lung Cell Mol Physiol 309: L1467–L1477, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Svensson L, McDowall A, Giles KM, Stanley P, Feske S, Hogg N. Calpain 2 controls turnover of LFA-1 adhesions on migrating T lymphocytes. PLoS One 5: e15090, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Tavernarakis N, Driscoll M. Molecular modeling of mechanotransduction in the nematode Caenorhabditis elegans. Annu Rev Physiol 59: 659–689, 1997. [DOI] [PubMed] [Google Scholar]
- 186.Taylor-Clark TE. Oxidative stress as activators of sensory nerves for cough. Pulm Pharmacol Ther 35: 94–99, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Thorneloe KS, Cheung M, Bao W, Alsaid H, Lenhard S, Jian MY, Costell M, Maniscalco-Hauk K, Krawiec JA, Olzinski A, Gordon E, Lozinskaya I, Elefante L, Qin P, Matasic DS, James C, Tunstead J, Donovan B, Kallal L, Waszkiewicz A, Vaidya K, Davenport EA, Larkin J, Burgert M, Casillas LN, Marquis RW, Ye G, Eidam HS, Goodman KB, Toomey JR, Roethke TJ, Jucker BM, Schnackenberg CG, Townsley MI, Lepore JJ, Willette RN. An orally active TRPV4 channel blocker prevents and resolves pulmonary edema induced by heart failure. Sci Transl Med 4: 159ra148, 2012. [DOI] [PubMed] [Google Scholar]
- 188.Trac D, Liu B, Pao AC, Thomas SV, Park M, Downs CA, Ma HP, Helms MN. Fulvene-5 inhibition of Nadph oxidases attenuates activation of epithelial sodium channels in A6 distal nephron cells. Am J Physiol Renal Physiol 305: F995–F1005, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Tsuji F, Murai M, Oki K, Inoue H, Sasano M, Tanaka H, Inagaki N, Aono H. Effects of SA13353, a transient receptor potential vanilloid 1 agonist, on leukocyte infiltration in lipopolysaccharide-induced acute lung injury and ovalbumin-induced allergic airway inflammation. J Pharm Sci 112: 487–490, 2010. [DOI] [PubMed] [Google Scholar]
- 190.Vennekens R, Olausson J, Meissner M, Bloch W, Mathar I, Philipp SE, Schmitz F, Weissgerber P, Nilius B, Flockerzi V, Freichel M. Increased IgE-dependent mast cell activation and anaphylactic responses in mice lacking the calcium-activated nonselective cation channel TRPM4. Nat Immunol 8: 312–320, 2007. [DOI] [PubMed] [Google Scholar]
- 191.Verma P, Kumar A, Goswami C. TRPV4-mediated channelopathies. Channels (Austin) 4: 319–328, 2010. [DOI] [PubMed] [Google Scholar]
- 192.Vicente R, Escalada A, Coma M, Fuster G, Sanchez-Tillo E, Lopez-Iglesias C, Soler C, Solsona C, Celada A, Felipe A. Differential voltage-dependent K+ channel responses during proliferation and activation in macrophages. J Biol Chem 278: 46307–46320, 2003. [DOI] [PubMed] [Google Scholar]
- 193.Villalta PC, Rocic P, Townsley MI. Role of MMP2 and MMP9 in TRPV4-induced lung injury. Am J Physiol Lung Cell Mol Physiol 307: L652–L659, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Villanueva S, Burgos J, Lopez-Cayuqueo KI, Lai KV, Valenzuela DM, Cid LP, Sepulveda FV. Cleft palate, moderate lung developmental retardation and early postnatal lethality in mice deficient in the Kir7.1 inwardly rectifying K+ channel. PLoS One 10: e0139284, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Volkers L, Mechioukhi Y, Coste B. Piezo channels: from structure to function. Pflügers Arch 467: 95–99, 2015. [DOI] [PubMed] [Google Scholar]
- 196.von Lewinski D, Kockskamper J, Pieske B. Stretch-induced slow force response in mammalian ventricular myocardium. In: Mechanosensitivity in Cells and Tissues, edited by Kamkin A and Kiseleva I. Moscow: Academia, 2005. [PubMed] [Google Scholar]
- 197.von Reyn CR, Mott RE, Siman R, Smith DH, Meaney DF. Mechanisms of calpain mediated proteolysis of voltage gated sodium channel alpha-subunits following in vitro dynamic stretch injury. J Neurochem 121: 793–805, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Waldegger S, Steuer S, Risler T, Heidland A, Capasso G, Massry S, Lang F. Mechanisms and clinical significance of cell volume regulation. Nephrol Dial Transplant 13: 867–874, 1998. [DOI] [PubMed] [Google Scholar]
- 199.Wan X, Juranka P, Morris CE. Activation of mechanosensitive currents in traumatized membrane. Am J Physiol Cell Physiol 276: C318–C327, 1999. [DOI] [PubMed] [Google Scholar]
- 200.Wang J, Morishima S, Okada Y. IK channels are involved in the regulatory volume decrease in human epithelial cells. Am J Physiol Cell Physiol 284: C77–C84, 2003. [DOI] [PubMed] [Google Scholar]
- 201.Wang JA, Lin W, Morris T, Banderali U, Juranka PF, Morris CE. Membrane trauma and Na+ leak from Nav1.6 channels. Am J Physiol Cell Physiol 297: C823–C834, 2009. [DOI] [PubMed] [Google Scholar]
- 202.Wang S, Shi P, Wang Y. TRPA1 ion channels in vagal afferent nerves contribute to ventilator-induced lung injury in a rat model. Gen Physiol Biophys 32: 389–394, 2013. [DOI] [PubMed] [Google Scholar]
- 203.Wang Y, Minshall RD, Schwartz DE, Hu G. Cyclic stretch induces alveolar epithelial barrier dysfunction via calpain-mediated degradation of p120-catenin. Am J Physiol Lung Cell Mol Physiol 301: L197–L206, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Wang Z, Yang Y, Yang H, Capo-Aponte JE, Tachado SD, Wolosin JM, Reinach PS. NF-kappaB feedback control of JNK1 activation modulates TRPV1-induced increases in IL-6 and IL-8 release by human corneal epithelial cells. Mol Vis 17: 3137–3146, 2011. [PMC free article] [PubMed] [Google Scholar]
- 205.Watanabe H, Murakami M, Ohba T, Takahashi Y, Ito H. TRP channel and cardiovascular disease. Pharmacol Ther 118: 337–351, 2008. [DOI] [PubMed] [Google Scholar]
- 206.Waters CM, Ridge KM, Sunio G, Venetsanou K, Sznajder JI. Mechanical stretching of alveolar epithelial cells increases Na+-K+-ATPase activity. J Appl Physiol 87: 715–721, 1999. [DOI] [PubMed] [Google Scholar]
- 207.Waters CM, Roan E, Navajas D. Mechanobiology in lung epithelial cells: measurements, perturbations, and responses. Compr Physiol 2: 1–29, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Wei XM, Heywood GJ, Di Girolamo N, Thomas PS. Nicorandil inhibits the release of TNFalpha from a lymphocyte cell line and peripheral blood lymphocytes. Int Immunopharmacol 3: 1581–1588, 2003. [DOI] [PubMed] [Google Scholar]
- 209.Welsh MJ, Price MP, Xie J. Biochemical basis of touch perception: mechanosensory function of degenerin/epithelial Na+ channels. J Biol Chem 277: 2369–2372, 2002. [DOI] [PubMed] [Google Scholar]
- 210.Wiltink A, Nijweide PJ, Scheenen WJ, Ypey DL, Van Duijn B. Cell membrane stretch in osteoclasts triggers a self-reinforcing Ca2+ entry pathway. Pflügers Arch 429: 663–671, 1995. [DOI] [PubMed] [Google Scholar]
- 211.Woods PS, Tazi MF, Chesarino NM, Amer AO, Davis IC. TGF-β-induced IL-6 prevents development of acute lung injury in influenza A virus-infected F508del CFTR-heterozygous mice. Am J Physiol Lung Cell Mol Physiol 308: L1136–L1144, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Wung BS, Cheng JJ, Chao YJ, Lin J, Shyy YJ, Wang DL. Cyclical strain increases monocyte chemotactic protein-1 secretion in human endothelial cells. Am J Physiol Heart Circ Physiol 270: H1462–H1468, 1996. [DOI] [PubMed] [Google Scholar]
- 213.Xu R, Li Q, Zhou XD, Perelman JM, Kolosov VP. Oxidative stress mediates the disruption of airway epithelial tight junctions through a TRPM2-PLCgamma1-PKCalpha signaling pathway. Int J Mol Sci 14: 9475–9486, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Yamada T, Ueda T, Ugawa S, Ishida Y, Imayasu M, Koyama S, Shimada S. Functional expression of transient receptor potential vanilloid 3 (TRPV3) in corneal epithelial cells: involvement in thermosensation and wound healing. Exp Eye Res 90: 121–129, 2010. [DOI] [PubMed] [Google Scholar]
- 215.Yang XC, Sachs F. Block of stretch-activated ion channels in Xenopus oocytes by gadolinium and calcium ions. Science 243: 1068–1071, 1989. [DOI] [PubMed] [Google Scholar]
- 216.Yang Y, Cheng Y, Lian QQ, Yang L, Qi W, Wu DR, Zheng X, Liu YJ, Li WJ, Jin SW, Smith FG. Contribution of CFTR to alveolar fluid clearance by lipoxin A4 via PI3K/Akt pathway in LPS-induced acute lung injury. Med Inf (Lond) 2013: 862628, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Yin J, Kuebler WM. Mechanotransduction by TRP channels: general concepts and specific role in the vasculature. Cell Biochem Biophys 56: 1–18, 2010. [DOI] [PubMed] [Google Scholar]
- 218.Yin J, Michalick L, Tang C, Tabuchi A, Goldenberg N, Dan Q, Awwad K, Wang L, Erfinanda L, Nouailles G, Witzenrath M, Vogelzang A, Lv L, Lee WL, Zhang H, Rotstein O, Kapus A, Szaszi K, Fleming I, Liedtke WB, Kuppe H, Kuebler WM. Role of transient receptor potential vanilloid 4 in neutrophil activation and acute lung injury. Am J Respir Cell Mol Biol 54: 370–383, 2016. [DOI] [PubMed] [Google Scholar]
- 219.Yu M, Huang C, Huang Y, Wu X, Li X, Li J. Inhibition of TRPM7 channels prevents proliferation and differentiation of human lung fibroblasts. Inflamm Res 62: 961–970, 2013. [DOI] [PubMed] [Google Scholar]
- 220.Zhang J, Zhu Z, Li XW, Zhang ZF. Knocking down TRPC1 expression by siRNA inhibits proliferation and invasiveness of human lung adenocarcinoma cell A549 in vitro. Zhonghua Yi Xue Za Zhi 93: 2241–2243, 2013. [PubMed] [Google Scholar]
- 221.Zhang WK, Wang D, Duan Y, Loy MM, Chan HC, Huang P. Mechanosensitive gating of CFTR. Nat Cell Biol 12: 507–512, 2010. [DOI] [PubMed] [Google Scholar]
- 222.Zhao C, Su EM, Yang X, Gao Z, Li L, Wu H, Jiang Y, Su X. Important role of platelets in modulating endotoxin-induced lung inflammation in CFTR-deficient mice. PLoS One 8: e82683, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Zhao F, Dong L, Cheng L, Zeng Q, Su F. Effects of acute mechanical stretch on the expression of mechanosensitive potassium channel TREK-1 in rat left ventricle. J Huazhong Univ Sci Technolog Med Sci 27: 385–387, 2007. [DOI] [PubMed] [Google Scholar]
- 224.Zhou X, Ye Y, Sun Y, Li X, Wang W, Privratsky B, Tan S, Zhou Z, Huang C, Wei YQ, Birnbaumer L, Singh BB, Wu M. Transient receptor potential channel 1 deficiency impairs host defense and proinflammatory responses to bacterial infection by regulating protein kinase cα signaling. Mol Cell Biol 35: 2729–2739, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Zimmerman JJ, Akhtar SR, Caldwell E, Rubenfeld GD. Incidence and outcomes of pediatric acute lung injury. Pediatrics 124: 87–95, 2009. [DOI] [PubMed] [Google Scholar]