

Abstract
The current study examined the effect of obesity on the development of renal injury within the genetic background of the Dahl salt-sensitive rat with a dysfunctional leptin receptor derived from zinc-finger nucleases (SSLepRmutant strain). At 6 wk of age, body weight was 35% higher in the SSLepRmutant strain compared with SSWT rats and remained elevated throughout the entire study. The SSLepRmutant strain exhibited impaired glucose tolerance and increased plasma insulin levels at 6 wk of age, suggesting insulin resistance while SSWT rats did not. However, blood glucose levels were normal throughout the course of the study. Systolic arterial pressure (SAP) was similar between the two strains from 6 to 10 wk of age. However, by 18 wk of age, the development of hypertension was more severe in the SSLepRmutant strain compared with SSWT rats (201 ± 10 vs. 155 ± 3 mmHg, respectively). Interestingly, proteinuria was substantially higher at 6 wk of age in the SSLepRmutant strain vs. SSWT rats (241 ± 27 vs. 24 ± 2 mg/day, respectively) and remained elevated until the end of the study. The kidneys from the SSLepRmutant strain displayed significant glomerular injury, including podocyte foot process effacement and lipid droplets compared with SSWT rats as early as 6 wk of age. By 18 wk of age, plasma creatinine levels were twofold higher in the SSLepRmutant strain vs. SSWT rats, suggesting the presence of chronic kidney disease (CKD). Overall, these results indicate that the SSLepRmutant strain develops podocyte injury and proteinuria independently of hyperglycemia and elevated arterial pressure that later progresses to CKD.
Keywords: obesity, leptin receptor, Dahl S rats, CKD, podocyte injury, lipid droplets
chronic kidney disease (CKD) is a major growing health problem and is one of the leading causes of death in the United States (30). While the two primary causes of CKD are hypertension and diabetes, the prevalence of obesity has increased dramatically within the last decade and is now considered an independent risk factor for CKD (14, 32). Moreover, a recent study revealed that 15% of nondiabetic patients with CKD suffer from morbid obesity (58). Furthermore, a report by Ejerblad et al. (32) suggested that obesity may play a significant role in the development of renal injury in patients without a history of hypertension, diabetes, and/or preexisting renal disease. These studies indicate that functional changes in the kidney occur in response to obesity that contribute to CKD.
The mechanisms by which obesity stimulates the development of renal injury in the absence of hypertension and diabetes remain unclear. The early stages of renal disease associated with obesity are renal vasodilation, hypertrophy, and increased renal blood flow (RBF) and glomerular filtration rate (GFR) (hyperfiltration). There are several components of obesity, including hyperinsulinemia (92), hyperglycemia (12), and elevations in arterial pressure (102, 103, 105), that are known to promote renal hyperfiltration. However, obesity in the absence of hypertension and diabetes has also been shown to cause elevations in RBF and GFR (14, 33, 68, 101). These early functional changes in renal hemodynamics associated with obesity may lead to increased transmission of systemic pressure to the glomerulus, causing damage to the glomerular filtration barrier and leading to proteinuria. In fact, previous studies have demonstrated that the development of proteinuria in obese patients is due, in part, to alterations in renal hemodynamics since lowering arterial pressure and GFR attenuates proteinuria (11, 14, 75).
Previous studies have demonstrated that patients suffering from obesity have a more than twofold greater risk of developing albuminuria and CKD than lean subjects (5, 16, 55, 60, 69, 98). However, a major complication in studying the obesity-CKD association is that some of the cardiovascular and metabolic disorders (i.e., hypertension, diabetes, dyslipidemia, and insulin resistance-metabolic syndrome; MetS) that are the result of obesity also contribute to the development and progression of CKD (16, 60). The consequences of obesity as an independent risk factor for the development of glomerular injury and proteinuria has received less attention due to a lack of an appropriate animal model. Currently, there are several obese rodent models that develop characteristics of MetS (1, 9, 21, 35–37, 43, 45, 47, 65, 71, 72, 89, 108, 111). Most of these models have a disruption in leptin receptor signaling that results in hyperphagia and leads to the development of obesity. Although these obese rodent strains have also served as models of renal disease, the onset and severity of renal injury vary widely in these models. The obese Zucker fatty (OZF) rat, which spontaneously developed a missense mutation (fatty, fa) in the leptin receptor gene (LepR) (112, 113), exhibits mild hypertension and hyperglycemia (≤200 mg/dl) along with characteristics of CKD including progressive proteinuria and a decline in renal function (21). A substrain of the OZF rat, the Zucker diabetic fatty (ZDF) rat, was created by breeding OZF rats that develop hyperglycemia (≥300 mg/dl) (20). The ZDF strain displays glomerular injury and proteinuria after the development of robust diabetes in the absence of hypertension (48). In another obese rodent model, the Koletsky rat carries a nonsense mutation in the leptin receptor (56, 57) and displays glomerular injury and proteinuria in the presence of hypertension along with impaired renal function, suggesting the presence of CKD (34, 35, 110). Similarly, the spontaneously hypertensive heart failure strain containing the corpulent mutation in the LepR gene (SHHF/Mcc-cp), which results from several backcrosses between the Koletsky and spontaneously hypertensive (SHR) rats, develops severe hypertension and renal disease that includes progressive proteinuria with elevated plasma creatinine (Pcr) levels (108). The obese, diabetic ZSF1 rats, which were developed by a F1 hybrid cross between a ZDF rat and lean SHHF rat, suffer from hypertension, glomerulosclerosis, and proteinuria with a decline in renal function by 20 wk of age (9, 43, 74). The diabetic obese db/db mouse model contains a mutation in the leptin receptor and develops little to no renal injury unless hypertension is induced by decreasing nitric oxide (NO) availability within its genetic background (65, 111). Taken together, while these obese rodent models develop some form of renal disease, the onset of renal injury occurs after the development of hypertension, diabetes, and/or age-related mechanisms. Hence, these models have limited use when one is investigating the direct impact of obesity on the development of renal injury in the absence of hypertension and diabetes.
The Dahl salt-sensitive (SS) rat is a well-established model of salt-dependent hypertension that is vastly susceptible to the development of renal injury (17, 26, 27, 70, 78, 81, 82, 90, 106). The kidneys of the SS rat are exposed to elevations in glomerular capillary hydrostatic pressure due to impaired renal autoregulation that ultimately leads to the development of glomerular and renal injury (104). However, the SS strain is not obese. In the current study, we created a 16-base pair frame-shift deletion in exon 11 of the leptin receptor gene (LepR) within the genetic background of the SS rat (SSLepRmutant strain) using zinc-finger nucleases that develops obesity without overt diabetes. We hypothesize that the development of obesity in the SSLepRmutant strain increases the early susceptibility to glomerular and renal injury in SS rats. In support of our hypothesis, SS rats fed a high-fat diet to induce a 15–25% increase in body weight display hypertension, proteinuria, and glomerular/tubular injury, suggesting that weight gain increases the susceptibility to renal disease in the SS strain (8, 52, 53, 67, 86). Similarly, Hattori and colleagues (46) observed progressive proteinuria, increased mesangial expansion, and reduced renal function in the obese DahlS.Z-Leprfa/Leprfa rat, a model created from a genetic cross between SS rats and heterozygous OZF rats, compared with their control counterparts. Moreover, these rats develop hypertension and hyperglycemia as early as 11 wk of age (46). However, the assessment of early changes in proteinuria and glomerular injury before 11 wk of age was not examined. Therefore, the aim of the current study was to assess the effects of obesity on the early temporal changes in metabolic and physiological parameters as well as the development of renal disease in our nondiabetic obese SSLepRmutant strain compared with their lean wild-type littermates, the SSWT strain.
METHODS
General.
Experiments were performed in 77 6–18 wk-old male SSWT and SSLepRmutant rats. SSWT and SSLepRmutant strains were obtained from our in-house colony of heterozygous SSLepRmutant (SSHETLepRmutant) rats that were originally obtained from Drs. Aron Geurts and Howard Jacob from the Medical College of Wisconsin. The rats were housed in the Laboratory Animal Facility at the University of Mississippi Medical Center, which is approved by the American Association for the Accreditation of Laboratory Animal Care. The rats had free access to food and water throughout the study. Rats were fed a 1% NaCl diet (TD8640; Harlan Laboratories, Madison, WI) to minimize the development of hypertension. All protocols were approved by the Animal Care Committee of the University of Mississippi Medical Center.
Creation and genotyping of the SSLepRmutant strain.
SSLepRmutant rats were created using zinc-finger technology at the Medical College of Wisconsin (MCW) using the PhysGen Knockout (http://rgd.mcw.edu/) as previously described (41, 42). Zinc-finger nuclease (ZFN) constructs (Sigma-Aldrich, St. Louis, MO) targeting the following sequence: AGCATCGTACTGCCCacaatgGGACATGGTCACAAG on chromosome 5 in exon 11 were used to cause a 16-base pair frame-shift deletion in the leptin receptor (LepR) gene within the genetic background of the SS rat (SS-Leprem2Mcwi strain). The pronucleus of fertilized SS/McwiHsd rat embryos was injected with mRNAs encoding the LepR ZFNs (10 ng/μl) and transferred to pseudopregnant females. To identify SSLepRmutant founders, genomic DNA extracted from ear punch tissues were screened with PCR and CEL-1 assay as previously described (64). The strain is designated as SS-Leprem2Mcwi (hereafter called SSLepmutant). Heterozygote animals (SSHETLepRmutant) from subsequent generations were used to generate homozygous mutants (SSLepRmutant strain) and wild-type littermates (SSWT strain). Genotyping and sequencing were performed by the Molecular and Genomic Facility at the University of Mississippi Medical Center. Briefly, genomic DNA collected from ear punch biopsies of pups at weaning was purified using the Wizard SV 96 Genomic DNA purification System (Promega, Madison, WI). For genotyping, the region of deletion targeted by the ZFNs within exon 11 of LepR was amplified via PCR using Platinum Taq DNA Polymerase High Fidelity Mix (Invitrogen, Grand Island, NY), dNTP mix (Invitrogen), and LepR-specific primers for the ZFN mutation site (forward 5′-CCACTGATGAAAAATGACTCAC-3′ and reverse 5′-ATGGGCCATGAGAACGTAAG-3′). The products were then loaded on a 3% agarose gel and visualized by ethidium bromide staining where the wild-type leptin receptor was 209 base pairs long and the mutated leptin receptor was 193 base pairs long. To validate the LepR disruption, PCR products were then purified using a PureLink Quick PCR Purification Kit (Invitrogen) to remove primers and excess reagents and prepared for Sanger sequencing using Dye Terminator Cycle Sequencing Mix (Beckman Coulter, Brea, CA) and the same primers used for genotyping described above. PCR products were then ethanol precipitated and resuspended in Sample Loading Solution (Beckman Coulter), then sequenced on the CEQ8000 Genetic Analysis System (Beckman Coulter). To confirm that the SSLepRmutant strain contained dysfunctional leptin receptors, blood samples from the tail vein were collected to measure plasma leptin levels at 6 wk of age (Quantikine Mouse/Rat Leptin ELISA, R&D Systems, Minneapolis, MN).
Protocol 1: comparison of time course of changes in metabolic parameters in SSWT rats and the SSLepRmutant strain.
Experiments were performed in 6-wk-old SSWT and SSLepRmutant rats. Body weight and blood glucose levels were monitored every 4 wk until the rats reached 18 wk of age. Blood samples were collected from the tail vein for measurement of blood glucose levels (glucometer, Bayer HealthCare, Mishawaka, IN). The samples were collected with ad libitum feeding 3 h into the lights-on cycle between 10 and 11 AM. Baseline and terminal plasma cholesterol and triglyceride concentrations were determined by ELISA (Cayman Chemical, Ann Arbor, MI).
Measurement of glucose tolerance and plasma insulin concentration levels.
SSWT and SSLepRmutant rats were subjected to an intraperitoneal glucose tolerance test (IPGTT) at 6 and 18 wk of age. Three days before the IPGTT, a blood sample (300–500 μl) was collected from the tail vein to measure plasma insulin concentrations (Rat Insulin ELISA, Mercodia, Uppsala, Sweden). On the day of the IPGTT, after the determination of fasting (2 h) blood glucose levels, the rats received an injection of 2 g/kg of glucose solution (ip). Blood samples (5–10 μl) were collected from the tail vein at 15, 30, 60, 90, and 120 min after the glucose load.
Protocol 2: comparison of the time course of changes in systolic arterial pressure, proteinuria, and podocalyxin excretion in SSWT rats and the obese SSLepRmutant strain.
Experiments were performed in 6-wk-old SSWT and SSLepRmutant rats. Systolic blood pressure (SAP) was measured in conscious animals via the tail-cuff method (MC4000 BP Analysis System, Hatteras Instruments, Cary, NC) at 6, 10, 14, and 18 wk of age. Before baseline measurements, the rats were trained and acclimated to restraint for 20–30 min for 4 consecutive days, and SAP was measured at the same time of day. At each time period, the rats were placed in metabolic cages to collect an overnight urine sample to study time course changes in the excretion of protein and podocalyxin. Urinary total protein concentration was determined colorimetrically using the Bradford method (Bio-Rad, Hercules, CA), and urinary podocalyxin concentration, an indicator of podocyte injury, was determined by ELISA (Exocell, Philadelphia, PA). Baseline and terminal blood samples were collected to determine Pcr (Wako Chemicals, Richmond, VA) to assess renal function.
Renal histopathology.
Kidneys were collected, weighed, and fixed in a 10% buffered formalin solution at 6 and 18 wk of age. Paraffin sections (3 μm) were prepared and stained with periodic acid-Schiff and Masson's trichrome to assess the degree of glomerular injury and renal fibrosis, respectively, in ∼30 images/section. Thirty glomeruli per section were scored in a blinded fashion on a 0–4 scale with 0 representing a normal glomerulus, 1 representing a 25% of loss, 2 representing a 50% loss, 3 representing a 75% loss, and 4 representing >75% loss of capillaries in the tuft. Additional analysis was performed to determine the degree of renal fibrosis. Images were captured using a Nikon Eclipse 55i microscope equipped with a Nikon DS-Fi1 color camera (Nikon, Melville, NY) and analyzed for the percentage of the image stained blue (primarily collagen) in the Masson's trichrome-stained sections using NIS-Elements D 3.0 software. Ten to 15 representative fields were analyzed per section.
Electron microscopy.
For transmission electron microscopy, kidneys from SSWT and SSLepRmutant rats at 6 and 18 wk of age were perfused with 2% glutaraldehyde, then ∼1 mm of cortex was obtained and placed in 10% formaldehyde buffered at pH 7.4 (Carson-Millonig Formulation, Ricca Chemical, Arlington, TX). Next, cortical samples were postfixed for 1 h with OsO4 followed by a graded series of chilled ethanol solutions (35–100%) ending with a final dehydration with 100% acetone. Samples were infiltrated overnight in Eponate 12 resin, and ultrathin sections (70–90 nm) were obtained and collected on copper support grids. Afterward, sections were poststained with uranyl acetate and lead citrate and examined using a JOEL 1400 plus transmission electron microscope (JOEL USA, Peabody, MA).
Statistical analysis.
Mean values ± SE are presented. The significance of difference in mean values for a single time point was determined by an unpaired t-test using SigmaPlot 12 software (Systat Software, San Jose, CA). The time course changes in metabolic and physiological parameters were compared between and within strains using a two-way repeated measures ANOVA followed by the Holm-Sidak test for preplanned comparisons with the same software mentioned above. A P value <0.05 was considered significantly different.
RESULTS
Validation of a dysfunctional leptin receptor in the SSLepRmutant strain.
To determine whether the leptin receptor was dysfunctional in the SSLepRmutant strain, we sequenced the targeted area and measured plasma leptin levels at 6 wk of age as well as temporal changes in body weight until the rats reached 18 wk age between SSWT and SSLepRmutant strains (Fig. 1). Sequencing revealed a 16-base pair frame-shift deletion in the LepR gene, resulting in a predicted stop codon 30 nonsense amino acids downstream (Fig. 1A). The genotypes of the SSWT (209 bp), SSHETLepRmutant (209 and 193 base pairs), and SSLepRmutant (193 base pairs) strains were verified using PCR as shown in Fig. 1B. The body weight at 6 wk of age was significantly higher in the SSLepRmutant strain compared with SSWT rats (238 ± 8 vs. 176 ± 6 g, respectively) and remained higher throughout the course of the study (528 ± 15 vs. 373 ± 4 g, respectively) (Fig. 1C). Moreover, plasma leptin levels were markedly elevated in the SSLepRmutant strain vs. SSWT rats (84.7 ± 4.5 vs. 2.1 ± 0.4 ng/ml, respectively) (Fig. 1D).
Fig. 1.

Sequencing (A), genotyping (B), and comparison of weight gain (C), and plasma leptin levels (D) of wild-type Dahl salt-sensitive (SSWT) rats, SS heterozygous leptin receptor mutant rats (SSHETLepRmutant strain), and the nondiabetic obese SS leptin receptor mutant (SSLepRmutant) strain. Numbers in parentheses indicate the number of rats studied per group. Values are means ± SE. *Significant difference from the corresponding value within the same strain at baseline (P < 0.05). †Significant difference from the corresponding value in SSWT rats (P < 0.05).
Comparison of metabolic parameters in SSWT and SSLepRmutant rats.
The time course of changes in blood glucose, plasma insulin levels, glucose tolerance, and plasma lipid levels are presented in Fig. 2. At baseline, nonfasting blood glucose levels were normal (≤120 mg/dl) for both strains and remained within the normal physiological range throughout the course of the study (Fig. 2A). However, as early as 6 wk of age, the plasma concentration of insulin was markedly elevated in the SSLepRmutant strain compared with the values measured in SSWT rats (4.7 ± 0.1 vs. 0.3 ± 0.03 ng/ml, respectively) and continued to be significantly elevated until the end of the study (Fig. 2B). Glucose tolerance tests were performed at 6 and 18 wk of age (Fig. 2, C and D). We observed a 50% increase in peak blood glucose levels at 6 wk of age in the SSLepRmutant strain vs. SSWT rats. Blood glucose levels returned to baseline 90 min after administration of the glucose load in the SSLepRmutant strain and 60 min in the SSWT rats. By 18 wk of age, the impairment in glucose tolerance was significantly greater in the SSLepRmutant strain compared with SSWT rats. Plasma cholesterol (Fig. 2E) and triglyceride (Fig. 2F) levels were measured at 6 and 18 wk of age. Plasma cholesterol was significantly higher in the SSLepRmutant strain vs. the SSWT rats as early as 6 wk of age (178 ± 15 vs. 112 ± 5 mg/dl, respectively). Additionally, at this same time point, plasma triglycerides were higher in the SSLepRmutant strain vs the SSWT rats (180 ± 34 vs. 41 ± 6 mg/dl). At 18 wk of age, plasma cholesterol levels were threefold higher in the SSLepRmutant strain vs. SSWT rats. Similarly, serum triglycerides were significantly higher in the SSLepRmutant strain compared with the values measured in their wild-type counterparts (1,550 ± 522 and 165 ± 27 mg/dl, respectively).
Fig. 2.

Comparison of time course measurements of blood glucose and plasma insulin levels (A and B), the progression of impaired glucose tolerance by IPGTT (C and D), and plasma cholesterol and triglyceride levels (E and F) in SSWT rats and the SSLepRmutant strain. Numbers in parentheses indicate the number of rats studied per group. Values are means ± SE. *Significant difference from the corresponding value within the same strain at baseline (P < 0.05). †Significant difference from the corresponding value in SSWT rats (P < 0.05).
Time course of changes in SAP, protein excretion, and podocalyxin excretion in SSWT rats and the SSLepRmutant strain.
Comparisons of the development of hypertension, proteinuria, and podocyte injury between SSWT rats and the SSLepRmutant strain are presented in Fig. 3. While SAP was not significantly different at 6 and 10 wk of age in the SSWT and SSLepRmutant strains, SSLepRmutant rats subsequently developed severe systolic hypertension compared with the values measured in SSWT rats by the end of the study (201 ± 10 vs. 155 ± 3 mmHg, respectively) (Fig. 3A). Interestingly, protein excretion was significantly higher at 6 and 10 wk of age in the SSLepRmutant strain vs. SSWT rats before elevations in arterial pressure. Proteinuria increased to 569 ± 60 mg/day in the SSLepRmutant strain while it only increased to 88 ± 5 mg/day in SSWT rats by 18 wk of age (Fig. 3B). Similar to protein excretion, podocalyxin excretion was significantly elevated by more than threefold in the SSLepRmutant strain vs. SSWT rats by 6 wk of age (64 ± 14 vs. 17 ± 2 μg/day, respectively) without any differences in SAP and continued to increase to 247 ± 62 vs. 47 ± 7 μg/day, respectively, by 18 wk of age (Fig. 3C).
Fig. 3.
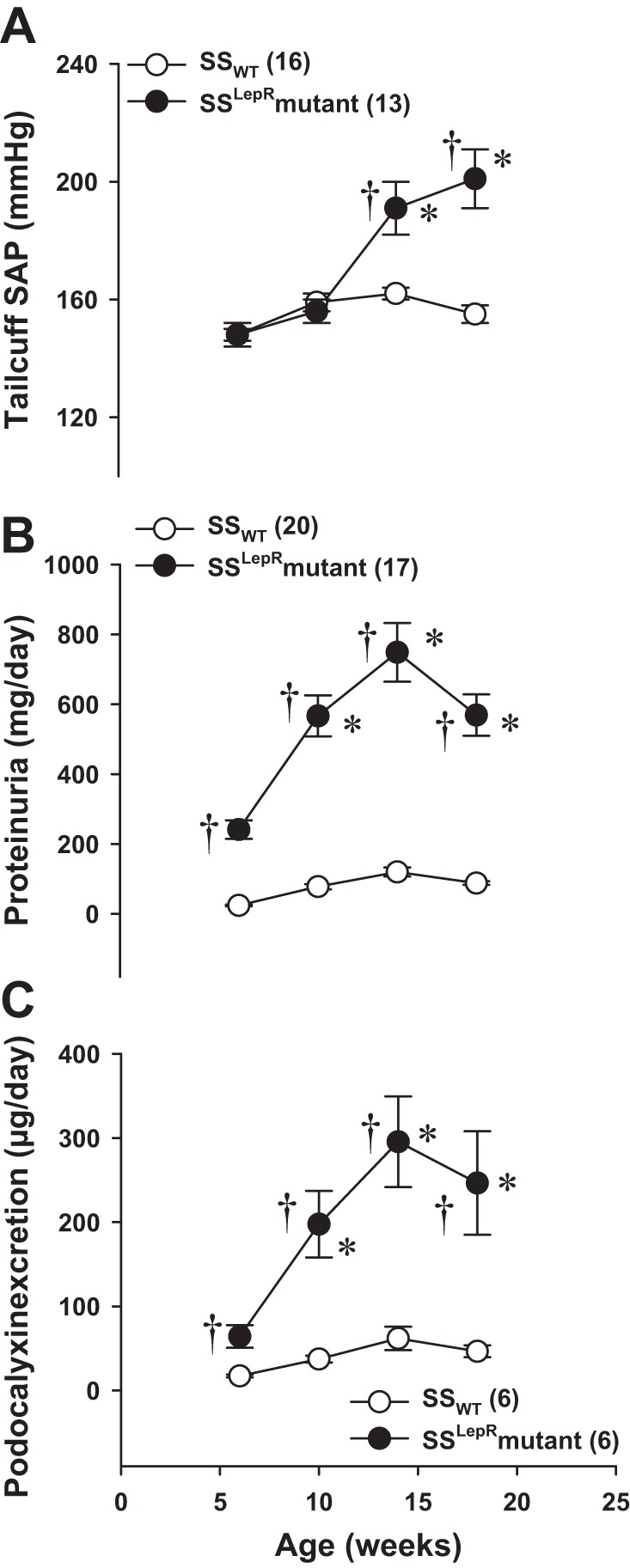
Time course measurements of systolic arterial pressure (SAP; A), protein excretion (B), and podocalyxin excretion (C), a marker of podocyte injury, in SSWT rats and the SSLepRmutant strain. Numbers in parentheses indicate the number of rats studied per group. Values are means ± SE. *Significant difference from the corresponding value within the same strain at baseline (P < 0.05). †Significant difference from the corresponding value in SSWT rats (P < 0.05).
Renal pathology.
Images of renal histopathology in SSWT rats and the SSLepRmutant strain are represented in Fig. 4. Compared with SSWT rats, the kidneys from the SSLepRmutant strain exhibited severe glomerulosclerosis and mesangial expansion (PAS staining) as early as 6 wk of age (Fig. 4, A and B) and continued to progress until the end of the study (18 wk of age) (Fig. 4, C and D). At 6 wk of age, we did not observe any differences in renal fibrosis between SSWT and SSLepRmutant strains (Fig. 4, E and F). However, by 18 wk of age, kidneys from the SSLepRmutant strain showed severe indications of interstitial fibrosis, protein casts, tubular necrosis, and dilated tubules while the SSWT did not (Fig. 4, G and H).
Fig. 4.

Comparison of renal histology. Periodic acid-schiff (PAS) staining (A–D) and Masson's trichrome staining (E–H) in SSWT rats and the SSLepRmutant strain are shown.
Multianalyses of the degree of renal injury in SSWT rats and the SSLepRmutant strain are represented in Fig. 5. The glomerular injury score was significantly greater in the SSLepRmutant strain at 6 wk of age and further increased by 18 wk of age compared with the values measured in SSWT rats (Fig. 5A). While renal fibrosis (blue staining) in the SSLepRmutant strain vs. SSWT rats at 6 wk of age did not reach statistical significance (P = 0.053), there was a tendency for it to be elevated (Fig. 5B). However, renal fibrosis increased by threefold in the SSLepRmutant strain compared with SSWT rats by 18 wk of age. Additionally, we observed a significant increase in kidney weight (renal hypertrophy) in the SSLepRmutant strain vs. the values seen in SSWT rats (1.9 ± 0.089 vs. 1.41 ± 0.003 g, respectively). When evaluating renal function, we observed that Pcr levels were similar between both strains at 6 wk of age (Fig. 5C). However, Pcr levels more than doubled in the SSLepRmutant strain vs. control SSWT rats by 18 wk of age (1.31 ± 0.29 vs. 0.52 ± 0.03 mg/dl, respectively).
Fig. 5.
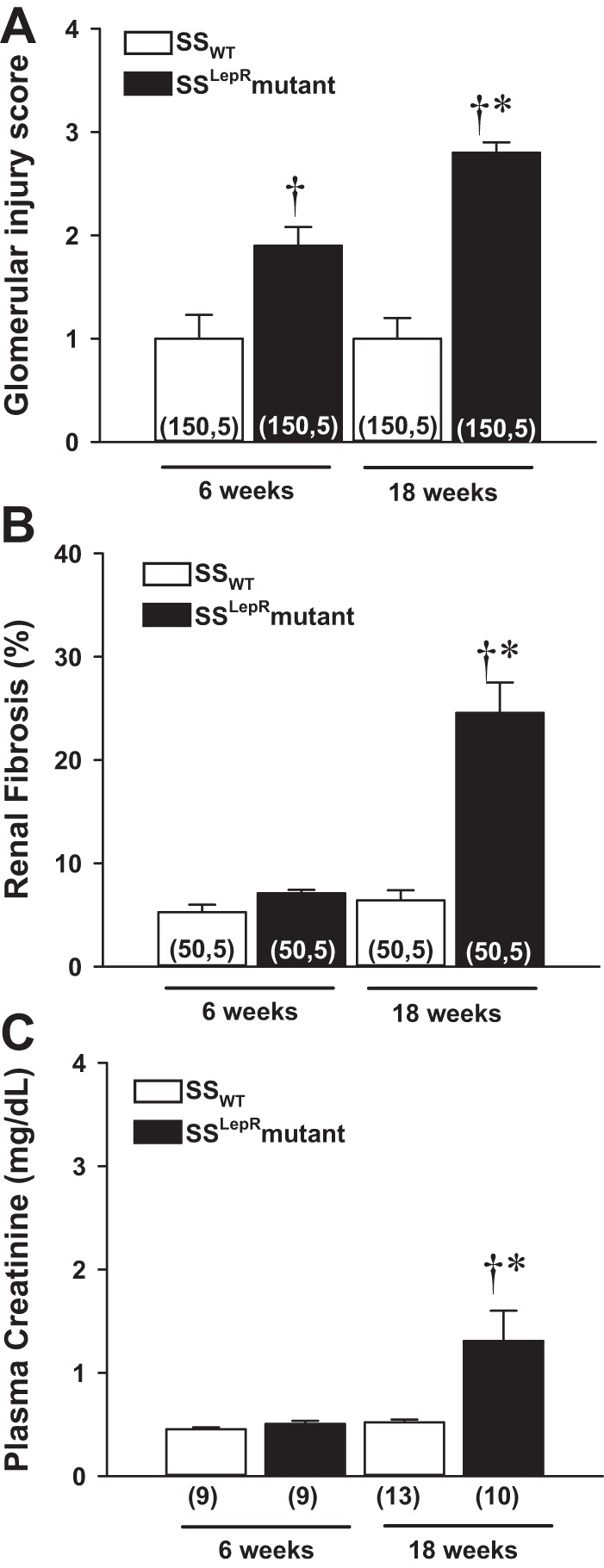
Comparison of glomerular injury score (A), renal fibrosis (B), and plasma creatinine (Pcr) levels (C) in SSWT rats and the SSLepRmutant strain. Numbers in parentheses indicate either the number of glomeruli/images and rats (A and B) or only the number of rats (C) studied per group. Values are means ± SE. *Significant difference from the corresponding value within the same strain at baseline (P < 0.05). †Significant difference from the corresponding value in SSWT rats (P < 0.05).
Electron microscopy.
Representative electron micrographs of the ultrastructure of glomerular capillaries in SSWT and SSLepRmutant rats are presented in Fig. 6. At 6 wk of age, SSWT rats exhibited a normal appearance of the glomerular filtration barrier (Fig. 6A). However, glomerular capillaries from the SSLepRmutant strain displayed minor podocyte foot process effacement (black filled arrows) and lipid droplets (black open arrows) at the same time period (Fig. 6B). By 18 wk of age, we observed severe foot process fusion and lipid accumulation in the SSLepRmutant strain compared with SSWT rats (Fig. 6, C and D).
Fig. 6.

Representative electron micrographs of the ultrastructure of the glomerular filtration barrier (A–D) in SSWT and SSLepRmutant strain. The filled black arrows represent podocyte foot process effacement, and the open black arrows represent lipid droplets.
DISCUSSION
Several rodent models of obesity have been developed. However, most of these models have limited use in the investigation of mechanisms responsible for the early development of renal injury associated with obesity due to the development of diabetes and hypertension. For example, the OZF rat develops mild hyperglycemia around 16 wk of age with proteinuria occurring at 40 wk of age, but Pcr levels remain within the normal range until it increases by more than twofold after 60 wk of age. Thus the slow progression of renal disease in the OZF rat cannot be separated from age-related mechanisms (21, 63). ZDF rats display severe hyperglycemia as early as 12 wk of age followed by the development of renal injury including severe glomerulosclerosis and albuminuria with a decline in GFR after 28 wk of age (48, 96). The obese Koletsky, SHHF/Mcc-cp, and ZSF1 strains, which contain the hypertensive genetic background of the SHR rat, present with hypertension as early as 6–8 wk of age but do not develop progressive renal injury until after 20 wk of age (9, 34, 35, 43, 74, 108, 110). Db/db mice only display albuminuria and glomerular abnormalities with a decline in GFR when mild to moderate hypertension is introduced by decreasing NO availability within its genetic background (48, 65). Therefore, it is difficult to identify any initial mechanisms specifically related to obesity in the absence of diabetes and hypertension. The current study determined the temporal changes in metabolic and cardiovascular parameters in a novel model of nondiabetic obesity which was created by mutating the leptin receptor in the SS rat genetic background using zinc-finger nuclease technology. The SS genetic background was used since SS rats exhibit impaired autoregulation of RBF and GFR and are more susceptible to the development of glomerular disease. The SSLepRmutant strain developed obesity and insulin resistance in the absence of hyperglycemia. Proteinuria and podocalyxin excretion were markedly elevated before any significant increase in SAP in the SSLepRmutant strain vs. SSWT rats at 6 wk of age, suggesting early characteristics of glomerular injury. However, by the end of the study, the SSLepRmutant strain developed severe elevated SAP and progressive proteinuria compared with their control counterparts. The kidneys from the SSLepRmutant strain displayed renal abnormalities, including glomerulosclerosis, renal fibrosis, and tubular necrosis while kidneys from SSWT rats did not. We also observed a twofold increase in Pcr levels in the SSLepRmutant strain compared with SSWT rats, suggesting the occurrence of CKD.
A large percentage of obese animal models exhibit type II diabetes. However, in the current study, the SSLepRmutant strain developed obesity, insulin resistance, and hyperinsulinemia by 6 wk of age, which did not stimulate overt diabetes or hyperglycemia. Moreover, this nonhyperglycemic trend continued throughout the course of the study. Yet, we do not find this result quite surprising since the obese DahlS.Z-Leprfa/Leprfa rat only develops mild hyperglycemia with hyperinsulinemia by 11 wk of age (46). However, there are conflicting reports in the OZF rat model developing hyperglycemia (6, 13, 51). Rohner-Jeanrenaud et al. (79, 80) observed impaired glucose tolerance and elevated plasma insulin levels without hyperglycemia. In contrast, several studies have provided evidence that OZF rats display hyperglycemia after 16 wk of age (≥170 mg/dl) (21, 63). SHHF/Mcc-cp rats are hyperinsulinemic at an early age with hyperglycemia due to marked hyperplasia of pancreatic β cells (3, 97, 108). The ZDF strain exhibits hyperglycemia (≥300 mg/dl) between 10 and 12 wk age due to an autosomal recessive defect in the islet cells of the pancreas independently of the leptin receptor mutation (20, 23, 107). Plasma insulin levels increase significantly from 6 to 8 wk of age and fall dramatically after 10–15 wk of age, which contributes to the elevations in blood glucose levels in the ZDF rat (73, 95). The Koletsky and ZSF1 strains present with mild hyperglycemia and hyperinsulinemia that progresses to severe diabetes (≥300 mg/dl) after 20 wk of age (9, 34, 35, 43, 74, 110). Similarly, the db/db mouse develops severe hyperglycemia by 10 wk of age (≥400 mg/dl) (1, 45) with plasma insulin levels peaking at 12 wk of age and rapidly declining after 24 wk of age (7, 22, 45). These data suggest the varying differences in the levels of glucose in these obese rat and mouse strains may involve the degree of insulin sensitivity and resistance and/or when the decline of insulin secretion from the β cells of the pancreas occurs.
The SS rat is a commonly used animal model to study salt-sensitive hypertension-induced renal disease (17, 26, 27, 70, 78, 81, 82, 90, 106). Moreover, the development of renal injury in the SS rat is associated with a lack of autoregulation of RBF, which contributes to elevations in glomerular capillary pressure that eventually lead to glomerular injury independently of hypertension (104). Previous work from our laboratory recently demonstrated that inducing diabetes in the SS strain with streptozotocin is associated with renal hyperfiltration and progressive proteinuria without significant changes in arterial pressure (85). Similar results were observed in the current study in which proteinuria and podocalyxin excretion were significantly higher in the obese SSLepRmutant strain compared with SSWT rats at 6 wk of age in the absence of elevations in systolic arterial pressure and blood glucose levels. The results from the current study suggest that the SS rat is more susceptible to the development of renal injury associated with nondiabetic obesity. However, unlike the hypertension and diabetic studies in SS rats, the initial mechanism that triggers renal disease in the nondiabetic obese SSLepRmutant strain remains to be determined but more than likely involves the lack of leptin signaling, causing obesity-related effects including changes in renal hemodynamics and insulin resistance. Obesity has been associated with increased albuminuria and renal injury independently of diabetes (11, 14, 25, 31, 76). Moreover, previous studies have demonstrated that the development of obesity-related proteinuria is due, in part, to alterations in renal hemodynamics since lowering arterial pressure and GFR attenuates proteinuria (11, 14, 75). Welsh et al. (100) observed that insulin resistance specifically at the podocyte level stimulates glomerular injury and albuminuria in the absence of hyperglycemia. These studies indicate that the early functional changes in the kidney that occur in response to obesity in the SSLepRmutant strain may be due to elevations in GFR and insulin resistance.
Interestingly, we observed the severe development of hypertension in the SSLepRmutant strain during the course of the study, which is not typical in similar models of obesity that are deficient in leptin receptor signaling. Db/db mice have similar or marked reductions in arterial pressure compared with their lean control littermates (10, 84, 109). The obese Zucker rat model develops moderate hypertension late in life (61) while their substrain counterpart, the Zucker diabetic fatty rat, does not (24, 29, 96). The lack of elevations in arterial pressure in these models is primarily due to the loss of leptin's mediated activation of the sympathetic nervous system. In contrast, there are a few models that develop hypertension in the wake of leptin receptor signaling. For instance, despite the LepR mutations on the SHR genetic background, the obese Koletsky, SHHF/Mcc-cp, and ZSF1 strains develop hypertension by 9 wk of age independently of leptin signaling (9, 34, 35, 43, 74, 108, 110). Similarly, deficiency of leptin receptor function in the DahlS.Z-Leprfa/Leprfa and SSLepRmutant strains causes hyperphagia-induced increases in food/sodium intake that may lead to elevations in arterial pressure due to the genetic susceptibility to salt-sensitive hypertension. However, in the current study, baseline SAP measurements were in the hypertensive range (>140 mmHg) but were similar between both strains until 14 wk of age, suggesting the difference in sodium intake due to hyperphagia does not play a significant role in causing early elevations in arterial pressure in the SSLepRmutant strain. We hypothesize that the severe development of hypertension after 10 wk of age observed in the SSLepRmutant strain may be attributed to reduced renal function. Overall, these studies suggest that some rodent models of obesity that lack leptin receptor signaling and are genetically predisposed to hypertension may develop elevations in arterial pressure.
The major finding in the current study is that the renal disease in the nondiabetic obese SSLepRmutant strain rapidly progresses to CKD by 18 wk of age. Pcr, a marker of renal function, was elevated by more than twofold in the SSLepRmutant strain and averaged 1.3 mg/dl compared with the normal value of 0.5 mg/dl observed in the SSWT strain, suggesting the presence of CKD. The reduction in renal function was associated with the severe development of SAP elevations and renal structural abnormalities, including podocyte foot process effacement, glomerulosclerosis, mesangial expansion, and renal fibrosis in the SSLepRmutant strain. In contrast, development of renal disease in the other obese animal models with leptin receptor signaling deficiency progresses slowly or not all. Db/db mice and ZDF rats develop mild renal disease, which may be due to the lack of increased SAP (1, 2, 28, 48). While the Koletsky, SHHF-Mcc-cp, and ZSF1 strains develop severe hypertension as well as diabetes, these rats rarely present with any significant renal abnormalities before 20 wk of age (9, 34, 35, 43, 74, 108, 110) or unless fed a high-carbohydrate diet (93, 94). In OZF rats, the transition from renal injury to CKD occurs slowly and does not develop until these rats are >40 wk of age (21, 63). The DahlS.Z-Leprfa/Leprfa strain is very similar to our SSLepRmutant strain, but the renal histological abnormalities are less severe. One possible reason for the progression of CKD in some of these models of obesity that lack leptin receptor signaling with insulin resistance and mild or no diabetes (i.e., DahlS.Z-Leprfa/Leprfa and OZF strains) is dyslipidemia (38, 46, 87). Dyslipidemia is a risk factor for the development of proteinuria and promotes the progression of CKD (62, 66, 83) by stimulating podocyte apoptosis and mesangial sclerosis (54). In the current study, the SSLepRmutant strain exhibited significant hypertriglyceridemia at the end of the study that was associated with impaired renal function. However, further studies are needed to determine whether long-term lipid-lowering therapy (i.e., statins and fibrates) prevents the progression of renal injury in these unique rodent obese models that develop CKD.
Like the other models of obesity that are of limited use in examining the direct impact of obesity on renal injury, the SSLepRmutant strain has limitations as well. As early as 6 wk of age, the SSLepRmutant strain displays characteristics of MetS, including insulin resistance, elevated SAP, and increased plasma triglyceride levels. Insulin resistance is associated with a higher prevalence of renal injury (15, 18). In the current study, the SSLepRmutant strain displayed impaired glucose tolerance and hyperinsulinemia along with progressive proteinuria at 6 wk of age. To support the relationship between insulin resistance and renal disease, Welsh and colleagues (100) demonstrated that insulin resistance specifically at the podocyte level contributes to podocyte injury and albuminuria as early as 5 wk of age. While we did not observe any early differences in arterial pressure between the SSWT and SSLepRmutant strains, SAP was in the hypertensive range. We and others have previously demonstrated that the SS strain is genetically susceptible to the development of hypertension-induced glomerular injury due to impaired renal autoregulation (4, 39, 40, 88, 103, 104). However, the level of podocyte injury and proteinuria was more severe in the SSLepRmutant strain compared with SSWT rats, suggesting that hypertension alone does not play a significant role in the early development of renal injury in the SSLepRmutant strain. Hypertriglyceridemia is a risk factor for developing proteinuria (91). Interestingly, we observed increases in plasma triglyceride levels and lipid droplets in the podocytes at 6 wk of age that was associated with glomerular injury and proteinuria in the SSLepRmutant strain. Wang et al. (99) recently observed treatment with simvastatin had no effect on plasma triglyceride levels but prevented renal lipidosis and renal disease, suggesting that lipid accumulation contributes to renal injury. Overall, the early development of these MetS features may contribute to the early podocyte and renal abnormalities in the SSLepRmutant strain. However, the SSLepRmutant strain is the first reported obese rodent model that develops progressive renal disease at an early age in the absence of hyperglycemia and elevated arterial pressure compared with its lean littermates.
Recent studies suggest that patients with obesity in the absence of diabetes have an increased risk for the development of CKD, but the mechanisms remain unclear (19, 44, 49, 50, 98). Despite this major health concern, little is known about the pathogenesis of renal disease associated with nondiabetic obesity due to the lack of an appropriate animal model, which has inhibited our understanding of the causes and progression of disease symptoms that could lead to development of new therapeutic interventions. However, in the current study, we observed that the SSLepRmutant strain develops many of the cardiovascular and metabolic characteristics of MetS along with CKD. Furthermore, the development of renal injury occurs at an early age independently of hyperglycemia and elevations in arterial pressure. Thus the SSLepRmutant strain may be useful in investigating the potential mechanisms involved in both the early and late stages of development of CKD associated with nondiabetic obesity. Another area of research that may benefit from using the SSLepRmutant strain is studying the effects of obesity on the progression of renal injury in a rodent strain that is susceptible to renal disease. This may be of use in high-risk minorities, including African Americans and Hispanics that are at greater risk for developing obesity and CKD than Caucasians (59, 77).
GRANTS
J. M. Williams is supported by funds from the National Institutes of General Medical Sciences of the National Institutes of Health (NIGMS/NIH) Obesity, Cardiorenal and Metabolic Diseases-COBRE (P20GM104357) and the American Heart Association (12SDG9440034). S. P. Didion is supported by the National Heart, Lung and Blood Institute of the National Institutes of Health (NHLBI/NIH) Grant HL107632. M. R. Garrett is supported by NHLBI/NIH Grant HL094446. The work performed through the UMMC Molecular and Genomics Facility is supported, in part, by funds from the NIGMS/NIH including Mississippi INBRE (P20GM103476), Center for Psychiatric Neuroscience (CPN)-COBRE (P30GM103328), and Obesity, Cardiorenal and Metabolic Diseases-COBRE (P20GM104357). The SSLepRmutant strain was developed as part of funds from the NIH (DP2-OD008396 to A. M. Guerts).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
K.C.M., L.T., A.C.J., S.P.D., A.M.G., M.R.G., and J.M.W. performed experiments; K.C.M., L.T., A.C.J., A.M.G., M.R.G., and J.M.W. analyzed data; K.C.M., S.P.D., M.R.G., and J.M.W. interpreted results of experiments; K.C.M., M.R.G., and J.M.W. prepared figures; K.C.M. and J.M.W. drafted manuscript; K.C.M., M.R.G., and J.M.W. edited and revised manuscript; K.C.M., L.T., A.C.J., A.M.G., M.R.G., and J.M.W. approved final version of manuscript; J.M.W. provided conception and design of research.
ACKNOWLEDGMENTS
We thank DesTenee Green, Brianca Fizer, Joshua Jefferson, and Denisha Spires for excellent technical assistance. We also thank Dr. Howard Jacob for valuable input and role in creating the SSLepRmutant strain.
REFERENCES
- 1.Allen TJ, Cooper ME, Lan HY. Use of genetic mouse models in the study of diabetic nephropathy. Curr Diab Rep 4: 435–440, 2004. [DOI] [PubMed] [Google Scholar]
- 2.Alonso-Galicia M, Brands MW, Zappe DH, Hall JE. Hypertension in obese Zucker rats. Role of angiotensin II and adrenergic activity. Hypertension 28: 1047–1054, 1996. [DOI] [PubMed] [Google Scholar]
- 3.Atgie C, Hadj-Sassi A, Bukowiecki L, Mauriege P. High lipolytic activity and dyslipidemia in a spontaneous hypertensive/NIH corpulent (SHR/N-cp) rat: a genetic model of obesity and type 2 diabetes mellitus. J Physiol Biochem 65: 33–41, 2009. [DOI] [PubMed] [Google Scholar]
- 4.Azar SLC, Iwai J, Weller D. Single nephron dynamics during high Na+ intake and early hypertension in Dahl rats. Jpn Heart J 20: 138–140, 1979. [Google Scholar]
- 5.Bagby SP. Obesity-initiated metabolic syndrome and the kidney: a recipe for chronic kidney disease? J Am Soc Nephrol 15: 2775–2791, 2004. [DOI] [PubMed] [Google Scholar]
- 6.Banday AA, Fazili FR, Marwaha A, Lokhandwala MF. Mitogen-activated protein kinase upregulation reduces renal D1 receptor affinity and G-protein coupling in obese rats. Kidney Int 71: 397–406, 2007. [DOI] [PubMed] [Google Scholar]
- 7.Berglund O, Frankel BJ, Hellman B. Development of the insulin secretory defect in genetically diabetic (db/db) mouse. Acta Endocrinol (Copenh) 87: 543–551, 1978. [DOI] [PubMed] [Google Scholar]
- 8.Beyer AM, Raffai G, Weinberg B, Fredrich K, Lombard JH. Dahl salt-sensitive rats are protected against vascular defects related to diet-induced obesity. Hypertension 60: 404–410, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bilan VP, Salah EM, Bastacky S, Jones HB, Mayers RM, Zinker B, Poucher SM, Tofovic SP. Diabetic nephropathy and long-term treatment effects of rosiglitazone and enalapril in obese ZSF1 rats. J Endocrinol 210: 293–308, 2011. [DOI] [PubMed] [Google Scholar]
- 10.Bodary PF, Shen Y, Ohman M, Bahrou KL, Vargas FB, Cudney SS, Wickenheiser KJ, Myers MG Jr, Eitzman DT. Leptin regulates neointima formation after arterial injury through mechanisms independent of blood pressure and the leptin receptor/STAT3 signaling pathways involved in energy balance. Arterioscler Thromb Vasc Biol 27: 70–76, 2007. [DOI] [PubMed] [Google Scholar]
- 11.Bosma RJ, van der Heide JJ, Oosterop EJ, de Jong PE, Navis G. Body mass index is associated with altered renal hemodynamics in non-obese healthy subjects. Kidney Int 65: 259–265, 2004. [DOI] [PubMed] [Google Scholar]
- 12.Brands MW, Bell TD, Rodriquez NA, Polavarapu P, Panteleyev D. Chronic glucose infusion causes sustained increases in tubular sodium reabsorption and renal blood flow in dogs. Am J Physiol Regul Integr Comp Physiol 296: R265–R271, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cai XJ, Lister CA, Buckingham RE, Pickavance L, Wilding J, Arch JR, Wilson S, Williams G. Down-regulation of orexin gene expression by severe obesity in the rats: studies in Zucker fatty and Zucker diabetic fatty rats and effects of rosiglitazone. Brain Res Mol Brain Res 77: 131–137, 2000. [DOI] [PubMed] [Google Scholar]
- 14.Chagnac A, Weinstein T, Korzets A, Ramadan E, Hirsch J, Gafter U. Glomerular hemodynamics in severe obesity. Am J Physiol Renal Physiol 278: F817–F822, 2000. [DOI] [PubMed] [Google Scholar]
- 15.Chen J, Muntner P, Hamm LL, Fonseca V, Batuman V, Whelton PK, He J. Insulin resistance and risk of chronic kidney disease in nondiabetic US adults. J Am Soc Nephrol 14: 469–477, 2003. [DOI] [PubMed] [Google Scholar]
- 16.Chen J, Muntner P, Hamm LL, Jones DW, Batuman V, Fonseca V, Whelton PK, He J. The metabolic syndrome and chronic kidney disease in US adults. Ann Intern Med 140: 167–174, 2004. [DOI] [PubMed] [Google Scholar]
- 17.Chen PY, St John PL, Kirk KA, Abrahamson DR, Sanders PW. Hypertensive nephrosclerosis in the Dahl/Rapp rat. Initial sites of injury and effect of dietary l-arginine supplementation. Lab Invest 68: 174–184, 1993. [PubMed] [Google Scholar]
- 18.Cheng HT, Huang JW, Chiang CK, Yen CJ, Hung KY, Wu KD. Metabolic syndrome and insulin resistance as risk factors for development of chronic kidney disease and rapid decline in renal function in elderly. J Clin Endocrinol Metab 97: 1268–1276, 2012. [DOI] [PubMed] [Google Scholar]
- 19.Chertow GM, Hsu CY, Johansen KL. The enlarging body of evidence: obesity and chronic kidney disease. J Am Soc Nephrol 17: 1501–1502, 2006. [DOI] [PubMed] [Google Scholar]
- 20.Clark JB, Palmer CJ, Shaw WN. The diabetic Zucker fatty rat. Proc Soc Exp Biol Med 173: 68–75, 1983. [DOI] [PubMed] [Google Scholar]
- 21.Coimbra TM, Janssen U, Grone HJ, Ostendorf T, Kunter U, Schmidt H, Brabant G, Floege J. Early events leading to renal injury in obese Zucker (fatty) rats with type II diabetes. Kidney Int 57: 167–182, 2000. [DOI] [PubMed] [Google Scholar]
- 22.Coleman DL, Hummel KP. Hyperinsulinemia in pre-weaning diabetes (db) mice. Diabetologia 10, Suppl: 607–610, 1974. [DOI] [PubMed] [Google Scholar]
- 23.Corsetti JP, Sparks JD, Peterson RG, Smith RL, Sparks CE. Effect of dietary fat on the development of non-insulin dependent diabetes mellitus in obese Zucker diabetic fatty male and female rats. Atherosclerosis 148: 231–241, 2000. [DOI] [PubMed] [Google Scholar]
- 24.Cosson E, Valensi P, Laude D, Mesangeau D, Dabire H. Arterial stiffness and the autonomic nervous system during the development of Zucker diabetic fatty rats. Diabetes Metab 35: 364–370, 2009. [DOI] [PubMed] [Google Scholar]
- 25.Csernus K, Lanyi E, Erhardt E, Molnar D. Effect of childhood obesity and obesity-related cardiovascular risk factors on glomerular and tubular protein excretion. Eur J Pediatr 164: 44–49, 2005. [DOI] [PubMed] [Google Scholar]
- 28.D'Angelo G, Mintz JD, Tidwell JE, Schreihofer AM, Pollock DM, Stepp DW. Exaggerated cardiovascular stress responses and impaired beta-adrenergic-mediated pressor recovery in obese Zucker rats. Hypertension 48: 1109–1115, 2006. [DOI] [PubMed] [Google Scholar]
- 29.Daniels A, Linz D, van Bilsen M, Rutten H, Sadowski T, Ruf S, Juretschke HP, Neumann-Haefelin C, Munts C, van der Vusse GJ, van Nieuwenhoven FA. Long-term severe diabetes only leads to mild cardiac diastolic dysfunction in Zucker diabetic fatty rats. Eur J Heart Fail 14: 193–201, 2012. [DOI] [PubMed] [Google Scholar]
- 30.Di Lullo L, Gorini A, Russo D, Santoboni A, Ronco C. Left ventricular hypertrophy in chronic kidney disease patients: from pathophysiology to treatment. Cardiorenal Med 5: 254–266, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ding W, Mak RH. Early markers of obesity-related renal injury in childhood. Pediatr Nephrol 30: 1–4, 2015. [DOI] [PubMed] [Google Scholar]
- 32.Ejerblad E, Fored CM, Lindblad P, Fryzek J, McLaughlin JK, Nyren O. Obesity and risk for chronic renal failure. J Am Soc Nephrol 17: 1695–1702, 2006. [DOI] [PubMed] [Google Scholar]
- 33.Eknoyan G. Obesity and chronic kidney disease. Nefrologia 31: 397–403, 2011. [DOI] [PubMed] [Google Scholar]
- 34.Ernsberger P, Johnson JL, Rosenthal T, Mirelman D, Koletsky RJ. Therapeutic actions of allylmercaptocaptopril and captopril in a rat model of metabolic syndrome. Am J Hypertens 20: 866–874, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ernsberger P, Koletsky RJ, Friedman JE. Molecular pathology in the obese spontaneous hypertensive Koletsky rat: a model of syndrome X. Ann NY Acad Sci 892: 272–288, 1999. [DOI] [PubMed] [Google Scholar]
- 36.Friedman JE, Ishizuka T, Liu S, Farrell CJ, Bedol D, Koletsky RJ, Kaung HL, Ernsberger P. Reduced insulin receptor signaling in the obese spontaneously hypertensive Koletsky rat. Am J Physiol Endocrinol Metab 273: E1014–E1023, 1997. [DOI] [PubMed] [Google Scholar]
- 37.Frisbee JC. Hypertension-independent microvascular rarefaction in the obese Zucker rat model of the metabolic syndrome. Microcirculation 12: 383–392, 2005. [DOI] [PubMed] [Google Scholar]
- 38.Gades MD, Van Goor H, Kaysen GA, Johnson PR, Horwitz BA, Stern JS. Brief periods of hyperphagia cause renal injury in the obese Zucker rat. Kidney Int 56: 1779–1787, 1999. [DOI] [PubMed] [Google Scholar]
- 39.Ge Y, Murphy SR, Fan F, Williams JM, Falck JR, Liu R, Roman RJ. Role of 20-HETE in the impaired myogenic and TGF responses of the Af-Art of Dahl salt-sensitive rats. Am J Physiol Renal Physiol 307: F509–F515, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ge Y, Murphy SR, Lu Y, Falck J, Liu R, Roman RJ. Endogenously produced 20-HETE modulates myogenic and TGF response in microperfused afferent arterioles. Prostaglandins Other Lipid Mediat 102: 42–48, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Geurts AM, Cost GJ, Freyvert Y, Zeitler B, Miller JC, Choi VM, Jenkins SS, Wood A, Cui X, Meng X, Vincent A, Lam S, Michalkiewicz M, Schilling R, Foeckler J, Kalloway S, Weiler H, Menoret S, Anegon I, Davis GD, Zhang L, Rebar EJ, Gregory PD, Urnov FD, Jacob HJ, Buelow R. Knockout rats via embryo microinjection of zinc-finger nucleases. Science 325: 433, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Geurts AM, Cost GJ, Remy S, Cui X, Tesson L, Usal C, Menoret S, Jacob HJ, Anegon I, Buelow R. Generation of gene-specific mutated rats using zinc-finger nucleases. Methods Mol Biol 597: 211–225, 2010. [DOI] [PubMed] [Google Scholar]
- 43.Griffin KA, Abu-Naser M, Abu-Amarah I, Picken M, Williamson GA, Bidani AK. Dynamic blood pressure load and nephropathy in the ZSF1 (fa/facp) model of type 2 diabetes. Am J Physiol Renal Physiol 293: F1605–F1613, 2007. [DOI] [PubMed] [Google Scholar]
- 44.Griffin KA, Kramer H, Bidani AK. Adverse renal consequences of obesity. Am J Physiol Renal Physiol 294: F685–F696, 2008. [DOI] [PubMed] [Google Scholar]
- 45.Han KL, Choi JS, Lee JY, Song J, Joe MK, Jung MH, Hwang JK. Therapeutic potential of peroxisome proliferator-activated receptor-alpha/gamma dual agonist with alleviation of endoplasmic reticulum stress for the treatment of diabetes. Diabetes 57: 737–745, 2008. [DOI] [PubMed] [Google Scholar]
- 46.Hattori T, Murase T, Ohtake M, Inoue T, Tsukamoto H, Takatsu M, Kato Y, Hashimoto K, Murohara T, Nagata K. Characterization of a new animal model of metabolic syndrome: the DahlS Z-Lepr(fa)/Lepr(fa) rat. Nutr Diabetes 1: e1, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hennes MM, McCune SA, Shrago E, Kissebah AH. Synergistic effects of male sex and obesity on hepatic insulin dynamics in SHR/Mcc-cp rat. Diabetes 39: 789–795, 1990. [DOI] [PubMed] [Google Scholar]
- 48.Hoshi S, Shu Y, Yoshida F, Inagaki T, Sonoda J, Watanabe T, Nomoto K, Nagata M. Podocyte injury promotes progressive nephropathy in Zucker diabetic fatty rats. Lab Invest 82: 25–35, 2002. [DOI] [PubMed] [Google Scholar]
- 49.Hsu CY, McCulloch CE, Iribarren C, Darbinian J, Go AS. Body mass index and risk for end-stage renal disease. Ann Intern Med 144: 21–28, 2006. [DOI] [PubMed] [Google Scholar]
- 50.Iseki K, Ikemiya Y, Kinjo K, Inoue T, Iseki C, Takishita S. Body mass index and the risk of development of end-stage renal disease in a screened cohort. Kidney Int 65: 1870–1876, 2004. [DOI] [PubMed] [Google Scholar]
- 51.Janssen U, Phillips AO, Floege J. Rodent models of nephropathy associated with type II diabetes. J Nephrol 12: 159–172, 1999. [PubMed] [Google Scholar]
- 52.Jin C, Jeon Y, Kleven DT, Pollock JS, White JJ, Pollock DM. Combined endothelin a blockade and chlorthalidone treatment in a rat model of metabolic syndrome. J Pharmacol Exp Ther 351: 467–473, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jin C, O'Boyle S, Kleven DT, Pollock JS, Pollock DM, White JJ. Antihypertensive and anti-inflammatory actions of combined azilsartan and chlorthalidone in Dahl salt-sensitive rats on a high-fat, high-salt diet. Clin Exp Pharmacol Physiol 41: 579–588, 2014. [DOI] [PubMed] [Google Scholar]
- 54.Joles JA, Kunter U, Janssen U, Kriz W, Rabelink TJ, Koomans HA, Floege J. Early mechanisms of renal injury in hypercholesterolemic or hypertriglyceridemic rats. J Am Soc Nephrol 11: 669–683, 2000. [DOI] [PubMed] [Google Scholar]
- 55.Kanauchi M, Kanauchi K, Kimura K, Inoue T, Saito Y. Associations of chronic kidney disease with the metabolic syndrome in non-diabetic elderly. Nephrol Dial Transplant 21: 3608–3609, 2006. [DOI] [PubMed] [Google Scholar]
- 56.Koletsky S. Animal model: obese hypertensive rat. Am J Pathol 81: 463–466, 1975. [PMC free article] [PubMed] [Google Scholar]
- 57.Koletsky S. Pathologic findings and laboratory data in a new strain of obese hypertensive rats. Am J Pathol 80: 129–142, 1975. [PMC free article] [PubMed] [Google Scholar]
- 58.Kramer HJ, Saranathan A, Luke A, Durazo-Arvizu RA, Guichan C, Hou S, Cooper R. Increasing body mass index and obesity in the incident ESRD population. J Am Soc Nephrol 17: 1453–1459, 2006. [DOI] [PubMed] [Google Scholar]
- 59.Kumanyika S, Grier S. Targeting interventions for ethnic minority and low-income populations. Future Child 16: 187–207, 2006. [DOI] [PubMed] [Google Scholar]
- 60.Kurella M, Lo JC, Chertow GM. Metabolic syndrome and the risk for chronic kidney disease among nondiabetic adults. J Am Soc Nephrol 16: 2134–2140, 2005. [DOI] [PubMed] [Google Scholar]
- 61.Kurtz TW, Morris RC, Pershadsingh HA. The Zucker fatty rat as a genetic model of obesity and hypertension. Hypertension 13: 896–901, 1989. [DOI] [PubMed] [Google Scholar]
- 62.Manttari M, Tiula E, Alikoski T, Manninen V. Effects of hypertension and dyslipidemia on the decline in renal function. Hypertension 26: 670–675, 1995. [DOI] [PubMed] [Google Scholar]
- 63.McCaleb ML, Sredy J. Metabolic abnormalities of the hyperglycemic obese Zucker rat. Metabolism 41: 522–525, 1992. [DOI] [PubMed] [Google Scholar]
- 64.Miller JC, Holmes MC, Wang J, Guschin DY, Lee YL, Rupniewski I, Beausejour CM, Waite AJ, Wang NS, Kim KA, Gregory PD, Pabo CO, Rebar EJ. An improved zinc-finger nuclease architecture for highly specific genome editing. Nat Biotechnol 25: 778–785, 2007. [DOI] [PubMed] [Google Scholar]
- 65.Mohan S, Reddick RL, Musi N, Horn DA, Yan B, Prihoda TJ, Natarajan M, Abboud-Werner SL. Diabetic eNOS knockout mice develop distinct macro- and microvascular complications. Lab Invest 88: 515–528, 2008. [DOI] [PubMed] [Google Scholar]
- 66.Muntner P, Coresh J, Smith JC, Eckfeldt J, Klag MJ. Plasma lipids and risk of developing renal dysfunction: the atherosclerosis risk in communities study. Kidney Int 58: 293–301, 2000. [DOI] [PubMed] [Google Scholar]
- 67.Nagae A, Fujita M, Kawarazaki H, Matsui H, Ando K, Fujita T. Effect of high fat loading in Dahl salt-sensitive rats. Clin Exp Hypertens 31: 451–461, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Naumnik B, Mysliwiec M. Renal consequences of obesity. Med Sci Monit 16: RA163–RA170, 2010. [PubMed] [Google Scholar]
- 69.Ninomiya T, Kiyohara Y. Albuminuria and chronic kidney disease in association with the metabolic syndrome. J Cardiometab Syndr 2: 104–107, 2007. [DOI] [PubMed] [Google Scholar]
- 70.O'Donnell MP, Kasiske BL, Katz SA, Schmitz PG, Keane WF. Lovastatin but not enalapril reduces glomerular injury in Dahl salt-sensitive rats. Hypertension 20: 651–658, 1992. [DOI] [PubMed] [Google Scholar]
- 71.Ogura M, Urabe M, Akimoto T, Onishi A, Ito C, Ito T, Tsukahara T, Mizukami H, Kume A, Muto S, Kusano E, Ozawa K. Interleukin-10 expression induced by adeno-associated virus vector suppresses proteinuria in Zucker obese rats. Gene Ther 19: 476–482, 2012. [DOI] [PubMed] [Google Scholar]
- 72.Osmond JM, Mintz JD, Stepp DW. Preventing increased blood pressure in the obese Zucker rat improves severity of stroke. Am J Physiol Heart Circ Physiol 299: H55–H61, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Paulsen SJ, Vrang N, Larsen LK, Larsen PJ, Jelsing J. Stereological assessment of pancreatic beta-cell mass development in male Zucker Diabetic Fatty (ZDF) rats: correlation with pancreatic beta-cell function. J Anat 217: 624–630, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Prabhakar S, Starnes J, Shi S, Lonis B, Tran R. Diabetic nephropathy is associated with oxidative stress and decreased renal nitric oxide production. J Am Soc Nephrol 18: 2945–2952, 2007. [DOI] [PubMed] [Google Scholar]
- 75.Praga M, Hernandez E, Andres A, Leon M, Ruilope LM, Rodicio JL. Effects of body-weight loss and captopril treatment on proteinuria associated with obesity. Nephron 70: 35–41, 1995. [DOI] [PubMed] [Google Scholar]
- 76.Praga M, Morales E, Herrero JC, Perez Campos A, Dominguez-Gil B, Alegre R, Vara J, Martinez MA. Absence of hypoalbuminemia despite massive proteinuria in focal segmental glomerulosclerosis secondary to hyperfiltration. Am J Kidney Dis 33: 52–58, 1999. [DOI] [PubMed] [Google Scholar]
- 77.Pulgaron ER, Delamater AM. Obesity and type 2 diabetes in children: epidemiology and treatment. Curr Diab Rep 14: 508, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Raij L, Azar S, Keane W. Mesangial immune injury, hypertension, and progressive glomerular damage in Dahl rats. Kidney Int 26: 137–143, 1984. [DOI] [PubMed] [Google Scholar]
- 79.Rohner-Jeanrenaud F, Hochstrasser AC, Jeanrenaud B. Hyperinsulinemia of preobese and obese fa/fa rats is partly vagus nerve mediated. Am J Physiol Endocrinol Metab 244: E317–E322, 1983. [DOI] [PubMed] [Google Scholar]
- 80.Rohner-Jeanrenaud F, Proietto J, Ionescu E, Jeanrenaud B. Mechanism of abnormal oral glucose tolerance of genetically obese fa/fa rats. Diabetes 35: 1350–1355, 1986. [DOI] [PubMed] [Google Scholar]
- 81.Roman RJ, Alonso-Galicia M, Wilson TW. Renal P450 metabolites of arachidonic acid and the development of hypertension in Dahl salt-sensitive rats. Am J Hypertens 10: 63S–67S, 1997. [PubMed] [Google Scholar]
- 82.Roman RJ, Ma YH, Frohlich B, Markham B. Clofibrate prevents the development of hypertension in Dahl salt-sensitive rats. Hypertension 21: 985–988, 1993. [DOI] [PubMed] [Google Scholar]
- 83.Schaeffner ES, Kurth T, Curhan GC, Glynn RJ, Rexrode KM, Baigent C, Buring JE, Gaziano JM. Cholesterol and the risk of renal dysfunction in apparently healthy men. J Am Soc Nephrol 14: 2084–2091, 2003. [DOI] [PubMed] [Google Scholar]
- 84.Senador D, Kanakamedala K, Irigoyen MC, Morris M, Elased KM. Cardiovascular and autonomic phenotype of db/db diabetic mice. Exp Physiol 94: 648–658, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Slaughter TN, Paige A, Spires D, Kojima N, Kyle PB, Garrett MR, Roman RJ, Williams JM. Characterization of the development of renal injury in type-1 diabetic Dahl salt-sensitive rats. Am J Physiol Regul Integr Comp Physiol 305: R727–R734, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Spradley FT, De Miguel C, Hobbs J, Pollock DM, Pollock JS. Mycophenolate mofetil prevents high-fat diet-induced hypertension and renal glomerular injury in Dahl SS rats. Physiol Rep 1: e00137, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Stevenson FT, Wheeldon CM, Gades MD, van Goor H, Stern JS. Hyperphagia as a mediator of renal disease initiation in obese Zucker rats. Obes Res 9: 492–499, 2001. [DOI] [PubMed] [Google Scholar]
- 88.Takenaka T, Forster H, De Micheli A, Epstein M. Impaired myogenic responsiveness of renal microvessels in Dahl salt-sensitive rats. Circ Res 71: 471–480, 1992. [DOI] [PubMed] [Google Scholar]
- 89.Tofovic SP, Kusaka H, Kost CK Jr, Bastacky S. Renal function and structure in diabetic, hypertensive, obese ZDFxSHHF-hybrid rats. Ren Fail 22: 387–406, 2000. [DOI] [PubMed] [Google Scholar]
- 90.Tolins JP, Raij L. Comparison of converting enzyme inhibitor and calcium channel blocker in hypertensive glomerular injury. Hypertension 16: 452–461, 1990. [DOI] [PubMed] [Google Scholar]
- 91.Tozawa M, Iseki K, Iseki C, Oshiro S, Ikemiya Y, Takishita S. Triglyceride, but not total cholesterol or low-density lipoprotein cholesterol levels, predict development of proteinuria. Kidney Int 62: 1743–1749, 2002. [DOI] [PubMed] [Google Scholar]
- 92.Tucker BJ, Anderson CM, Thies RS, Collins RC, Blantz RC. Glomerular hemodynamic alterations during acute hyperinsulinemia in normal and diabetic rats. Kidney Int 42: 1160–1168, 1992. [DOI] [PubMed] [Google Scholar]
- 93.Velasquez MT, Abraham AA, Kimmel PL, Farkas-Szallasi T, Michaelis OEt. Diabetic glomerulopathy in the SHR/N-corpulent rat: role of dietary carbohydrate in a model of NIDDM. Diabetologia 38: 31–38, 1995. [DOI] [PubMed] [Google Scholar]
- 94.Velasquez MT, Kimmel PL, Michaelis OEt Carswell N, Abraham A, Bosch JP. Effect of carbohydrate intake on kidney function and structure in SHR/N-cp rats. A new model of NIDDM. Diabetes 38: 679–685, 1989. [DOI] [PubMed] [Google Scholar]
- 95.Veniant MM, LeBel CP. Leptin: from animals to humans. Curr Pharm Des 9: 811–818, 2003. [DOI] [PubMed] [Google Scholar]
- 96.Vora JP, Zimsen SM, Houghton DC, Anderson S. Evolution of metabolic and renal changes in the ZDF/Drt-fa rat model of type II diabetes. J Am Soc Nephrol 7: 113–117, 1996. [DOI] [PubMed] [Google Scholar]
- 97.Voyles NR, Powell AM, Timmers KI, Wilkins SD, Bhathena SJ, Hansen C, Michaelis OEt, Recant L. Reversible impairment of glucose-induced insulin secretion in SHR/N-cp rats. Genetic model of type II diabetes. Diabetes 37: 398–404, 1988. [DOI] [PubMed] [Google Scholar]
- 98.Wahba IM, Mak RH. Obesity and obesity-initiated metabolic syndrome: mechanistic links to chronic kidney disease. Clin J Am Soc Nephrol 2: 550–562, 2007. [DOI] [PubMed] [Google Scholar]
- 99.Wang C, Li Q, Zhen J, Xu Y, Sun S. Simvastatin ameliorates renal lipidosis through the suppression of renal CXCL16 expression in mice with adriamycin-induced nephropathy. Int J Clin Exp Pathol 8: 15696–15707, 2015. [PMC free article] [PubMed] [Google Scholar]
- 100.Welsh GI, Hale LJ, Eremina V, Jeansson M, Maezawa Y, Lennon R, Pons DA, Owen RJ, Satchell SC, Miles MJ, Caunt CJ, McArdle CA, Pavenstadt H, Tavare JM, Herzenberg AM, Kahn CR, Mathieson PW, Quaggin SE, Saleem MA, Coward RJ. Insulin signaling to the glomerular podocyte is critical for normal kidney function. Cell Metab 12: 329–340, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wickman C, Kramer H. Obesity and kidney disease: potential mechanisms. Semin Nephrol 33: 14–22, 2013. [DOI] [PubMed] [Google Scholar]
- 102.Williams JM, Burke M, Lazar J, Jacob HJ, Roman RJ. Temporal characterization of the development of renal injury in FHH rats and FHH.1BN congenic strains. Am J Physiol Renal Physiol 300: F330–F338, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Williams JM, Fan F, Murphy S, Schreck C, Lazar J, Jacob HJ, Roman RJ. Role of 20-HETE in the antihypertensive effect of transfer of chromosome 5 from Brown Norway to Dahl salt-sensitive rats. Am J Physiol Regul Integr Comp Physiol 302: R1209–R1218, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Williams JM, Sarkis A, Hoagland KM, Fredrich K, Ryan RP, Moreno C, Lopez B, Lazar J, Fenoy FJ, Sharma M, Garrett MR, Jacob HJ, Roman RJ. Transfer of the CYP4A region of chromosome 5 from Lewis to Dahl S rats attenuates renal injury. Am J Physiol Renal Physiol 295: F1764–F1777, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Williams JM, Sarkis A, Lopez B, Ryan RP, Flasch AK, Roman RJ. Elevations in renal interstitial hydrostatic pressure and 20-hydroxyeicosatetraenoic acid contribute to pressure natriuresis. Hypertension 49: 687–694, 2007. [DOI] [PubMed] [Google Scholar]
- 106.Wilson TW, Alonso-Galicia M, Roman RJ. Effects of lipid-lowering agents in the Dahl salt-sensitive rat. Hypertension 31: 225–231, 1998. [DOI] [PubMed] [Google Scholar]
- 107.Yokoi N, Hoshino M, Hidaka S, Yoshida E, Beppu M, Hoshikawa R, Sudo K, Kawada A, Takagi S, Seino S. A novel rat model of type 2 diabetes: the Zucker Fatty Diabetes Mellitus ZFDM rat. J Diab Res 2013: 103731, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Youcef G, Olivier A, L'Huillier CP, Labat C, Fay R, Tabcheh L, Toupance S, Rodriguez-Gueant RM, Bergerot D, Jaisser F, Lacolley P, Zannad F, Laurent V, Pizard A. Simultaneous characterization of metabolic, cardiac, vascular and renal phenotypes of lean and obese SHHF rats. PLoS One 9: e96452, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zhang C, Park Y, Picchi A, Potter BJ. Maturation-induces endothelial dysfunction via vascular inflammation in diabetic mice. Basic Res Cardiol 103: 407–416, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zhang Q, Davis KJ, Hoffmann D, Vaidya VS, Brown RP, Goering PL. Urinary biomarkers track the progression of nephropathy in hypertensive and obese rats. Biomark Med 8: 85–94, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zhao HJ, Wang S, Cheng H, Zhang MZ, Takahashi T, Fogo AB, Breyer MD, Harris RC. Endothelial nitric oxide synthase deficiency produces accelerated nephropathy in diabetic mice. J Am Soc Nephrol 17: 2664–2669, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zucker LM. Two-way selection for body size in rats, with observations on simultaneous changes in coat color pattern and hood size. Genetics 45: 467–483, 1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zucker LM, Antoniades HN. Insulin and obesity in the Zucker genetically obese rat “fatty.” Endocrinology 90: 1320–1330, 1972. [DOI] [PubMed] [Google Scholar]