

Intermittent hypoxia (IH)-induced inflammation increases the risk of atherosclerosis in obstructive sleep apnea (OSA) patients. However, the effect of IH on macrophage foam cell formation, a key player in atherosclerosis, has not been elucidated. We demonstrate for the first time that IH-induced foam cell formation is diminished by IKK-β deletion. Our findings highlight the importance of the IKK-β- dependent NF-κB pathway and the potential of this pathway as a therapeutic target.
Keywords: NF-κB, foam cell, macrophage, intermittent hypoxia, IKK-β
Abstract
Obstructive sleep apnea (OSA) is a common sleep disorder characterized by intermittent hypoxia (IH). Clinical studies have previously shown that OSA is an independent risk factor for atherosclerosis. Atherogenicity in OSA patients has been assumed to be associated with the NF-κB pathways. Although foam cells are considered to be a hallmark of atherosclerosis, how IH as in OSA affects their development has not been fully understood. Therefore, we hypothesized that IH induces macrophage foam cell formation through NF-κB pathway activation. To test this hypothesis, peritoneal macrophages collected from myeloid-restricted IKK-β-deleted mice were incubated with native LDL and exposed to either IH or normoxia. After exposure, NF-κB pathway activity and intracellular cholesterol were measured. In control macrophages, IH significantly increased NF-κB pathway activity by 93% compared with normoxia (P < 0.05). However, such response to IH was diminished by IKK-β deletion (increased by +31% compared with normoxia; P = 0.64), suggesting that IKK-β is critical for IH-induced NF-κB pathway activation. Likewise, in control macrophages, total cholesterol was increased in IH compared with normoxia (65.7 ± 3.8 μg/mg cellular protein and 53.2 ± 1.2, respectively; P < 0.05). However, this IH-induced foam cell formation was disappeared when IKK-β was deleted (52.2 ± 1.2 μg/mg cellular protein for IH and 46.3 ± 1.7 for normoxia; P = 0.55). This IH-mediated effect still existed in macrophages without LDL receptor. Taken together, our findings show that IH activates the IKK-β-dependent NF-κB pathway and that this, in turn, induces foam cell formation in murine macrophages.
NEW & NOTEWORTHY
Intermittent hypoxia (IH)-induced inflammation increases the risk of atherosclerosis in obstructive sleep apnea (OSA) patients. However, the effect of IH on macrophage foam cell formation, a key player in atherosclerosis, has not been elucidated. We demonstrate for the first time that IH-induced foam cell formation is diminished by IKK-β deletion. Our findings highlight the importance of the IKK-β- dependent NF-κB pathway and the potential of this pathway as a therapeutic target.
obstructive sleep apnea (OSA) is a common sleep disorder predominant in developed countries, including the United States. The prevalence of OSA in the adult population in the U.S. was reported to be more than 10% (31, 43). Interestingly, OSA patients not only experience sleep fragmentation and daytime sleepiness but also have increased risks of a variety of clinical conditions, such as dyslipidemia, diabetes mellitus, and cardiovascular events (11, 35, 56). Furthermore, OSA has been identified as an independent risk factor for atherosclerosis that results in cardiovascular events (4, 12, 13).
One of the hallmark characteristics of OSA is intermittent hypoxia (IH). From large-scale clinical studies, the risk of atherosclerosis seems to be correlated to the severity of OSA (20, 30). This link between IH and atherosclerosis was confirmed in animal experiments in which mice exposed to IH developed aortic atherosclerotic plaques (3, 14, 15, 23, 32, 49). The mechanism of such phenomenon is not yet fully understood. However, one clinical study has already demonstrated that the severity of the OSA is correlated with the serum level of TNF-α, which is dependent on NF-κB activity (47). Furthermore, from observations on human subjects, it has been documented that NF-κB activation contributes to atherosclerosis development (8). Taken together, it has been proposed that IH induces NF-κB activation, which in turn may contribute to atherogenicity in OSA patients (34, 38). This is further supported by rodent experiments in which IH-exposed mice on high-fat diet (HFD) developed less atherosclerosis when NF-κB p50 was deleted (15, 51).
To elucidate some of the complex mechanisms of atherosclerosis development, it is essential to focus on cell-specific contribution. Macrophages seem to have an essential role in atherosclerosis development at all phases, from early fatty streaks to advanced lesions with necrotic cores (31). The finding that reduction of macrophages by CD11b-diphthelia toxin receptor or deletion of macrophage colony-stimulating factor ameliorates HFD-induced atherosclerosis in mice (50, 53) emphasizes the importance of macrophages in atherosclerosis.
Lipid-laden macrophages (foam cells) in the lesions are one of the earliest histological changes in atherosclerosis (52). Foam cells play a critical role in atherosclerosis development through activation of inflammatory pathways by secreting proinflammatory mediators including IL-6 and TNF-α and simultaneously recruiting immune cells and smooth muscle cells (17, 31). Therefore, it is essential to elucidate the effect of IH on foam cell formation to address the atherogenicity in OSA patients.
To date, a few researchers have documented IH-induced foam cell formation in macrophages and one implied the possible involvement of inflammation (29, 32). Evidence indicates that IH activates the NF-κB pathways in HeLa cells (48). In addition, inhibition of these pathways by overexpression of IκBα results in a reduction of foam cell formation in murine peritoneal macrophages (16). Thus we speculate that IH induces foam cell formation by NF-κB activation in macrophages.
IKK-β-dependent NF-κB activation has been especially implicated in human atherosclerosis (37). However, in spite of the importance of IH in atherogenicity, to our knowledge, no one has studied the role of IKK-β on IH-induced macrophage foam cell formation. Therefore, we hypothesized that IH exposure elicits IKK-β-dependent NF-κB activation, which in turn induces macrophage foam cell formation. To test this hypothesis, we studied THP-1 cell line and IKK-β knockout mice and proved that deletion of IKK-β reduces IH-induced foam cell formation in murine peritoneal macrophages.
METHODS
Cell culture.
THP-1 cells were originally obtained from American Tissue Type Cell Collection (ATCC, Manassas, VA) and were maintained in RPMI 1640 medium supplemented with 40 μg/ml gentamicin (Invitrogen, Carlsbad, CA) and 10% fetal bovine serum (FBS; Omega Scientific, Tarzana, CA). THP-1 cells were seeded at a density of 2 × 106 cells/ml and incubated with 50 μM of phorbol 12-myristate 13-acetate (PMA) for 72 h to induce differentiation into macrophages. After induction, the media were changed to low glucose (1 g/l) DMEM medium free of phenol red (11054-020; GIBCO, Grand Island, NY) supplemented with gentamicin and 10% FBS. We avoided phenol red because it is reported to have estrogenic activity (7) and estrogen inhibits foam cell formation in human monocyte-derived macrophages (36). The cells were incubated in a normoxia incubator for 24 h before IH exposure.
Animals.
Ldlr−/− mice on a C57BL/6J background were purchased (stock no. 002207; The Jackson Laboratory, Bar Harbor, ME). Ikk-βF/F mice were kindly provided by Dr. Michael Karin at the University of California, San Diego, and myeloid-specific IKK-β knockout mice (Ikk-βΔMye) were generated as previously described (19). We crossed Ikk-βΔMye mice with Ldlr−/− mice to generate Ikk-βΔMye-Ldlr−/− double knockout mice. Ikk-βF/F and Ikk-βF/F-Ldlr−/− mice were used as control animals. All animals used in this study were 2- to 3-mo-old male mice fed with a regular chow diet.
This study was conducted in conformity with the Guiding Principles for Research Involving Animals and Human Beings and was approved by the University of California, San Diego, Institutional Animal Care and Use Committee (Protocol No. S-05534).
Isolation of peritoneal macrophages.
Peritoneal macrophages were collected by peritoneal lavage with pyrogen-free PBS 3 days following peritoneal injection of 1 ml of 3% sterile thioglycolate (BD, Sparks, MD) to increase the yield of macrophages. The collected cells were incubated for 3 h before the plates were washed twice with PBS to remove nonadherent cells. Macrophage monolayers were then cultured overnight in phenol red-free low-glucose DMEM containing 1% FBS for lipid starvation. Before IH or normoxia exposure, media were changed to phenol red-free low glucose DMEM supplemented with different concentrations of native human LDL (BT-903; Alfa Aesar, Ward Hill, MA).
IH exposure.
The incubator was specifically designed to periodically regulate oxygen concentration by controlling gas outlets of O2, CO2, and N2 using a computerized system (LabVIEW; National Instruments, Austin, TX). The macrophages were exposed to IH in the incubator (25% O2 for 8 min, 0% for 12 min, CO2 was maintained at 5% throughout the exposure). One major pitfall of in vitro IH exposure is the difficulty of generating the fluctuations of dissolved oxygen in media because of the relatively high resistance of the transport of oxygen from the air to cells. (6) Therefore, we validated the system by measuring the dissolved oxygen in media and confirmed its cyclic oscillation from 20.4 ± 0.12 to 2.76 ± 0.05% before starting (mean ± SD).
Lipid staining.
Macrophages were plated onto Lab-Tek chambered glass slides (Nalge Nunc, Naparville, IL), fixed with 4% paraformaldehyde for 15 min, and stained with Oil red O and hematoxylin. Intracellular neutral lipid was measured by Nile red staining using the AdipoRed Assay Reagent (Lonza, Walkersville, MD). The intensity of fluorescence was measured with excitation at 485 nm and emission at 530 nm.
Cholesterol loading and quantification of intracellular cholesterol.
After differentiated THP-1 cells or peritoneal macrophages were loaded with cholesterol by incubation with 200 μg/ml of human native LDL for 24 h to induce foam cell formation, the macrophages were washed twice with ice-cold PBS. The cellular lipids were extracted with hexane/isopropanol (3:2, v/v), as previously noted (22). Total cholesterol (TC) and free cholesterol (FC) were determined by the Amplex Red Cholesterol assay kit (Thermo Fisher Scientific, Waltham, MA). Cholesteryl ester (CE) was calculated by subtracting FC from TC. After lipid extraction, cellular protein was dissolved in 0.2 N sodium hydroxide and 1% SDS, and protein concentration was measured using the BCA assay (Sigma-Aldrich, St. Louis, MO).
Western blot analysis.
Phosphorylated p-65, an indicator of activated NF-κB pathways, was performed using thioglycolate-elicited peritoneal macrophages. Macrophages were washed twice with ice-cold PBS, and nuclear protein was extracted by using NE-PER Nuclear and Cytoplasmic Extraction Reagents and Halt Protease Inhibitor Cocktail (Thermo Fisher Scientific, Waltham, MA). The nuclear protein content was quantified and 25 μg was used for analysis. The membrane was incubated with the phospholylated-p65 antibody (no. 3033; Cell Signaling Technology, Danvers, MA). The results were normalized with HDAC1 (sc-7872; Santa Cruz Biotechnology, Dallas, TX) to correct for well-to-well variability.
Statistics.
Data are reported as means ± SE. Differences between groups were analyzed using the Student's unpaired t-test or two-way ANOVA with post hoc Tukey's multiple comparison test (GraphPad Prism; GraphPad Software, San Diego, CA) as appropriate. Differences in the means were considered statistically significant when P < 0.05.
RESULTS
IH induces foam cell formation in THP-1 cells.
THP-1 human monocyte/macrophage-like cells were incubated in media with native LDL at various concentrations in either normoxia or IH for 24 h (Fig. 1A). IH exposure induced a significant increase of lipid particles (Nile red staining) when 400 or 800 μg/ml of native LDL were added to the cultures (114 ± 4% in normoxia and 128 ± 3% in IH for 400 μg/ml and 127 ± 3% in normoxia and 140 ± 3% in IH for 800 μg/ml; values are percentages of the relative fluorescence units of normoxia-exposed cells incubated without LDL; P < 0.05 for both comparisons; Fig. 1B). Remarkably, IH also significantly increased intracellular TC when cells were incubated with 200 μg/ml of native LDL (34.7 ± 1.2 μg/mg of cellular protein in normoxia and 40.1 ± 0.8 in IH; P < 0.05; Fig. 2A). Additionally, there was an IH-induced increase of CE (5.9 ± 0.4 μg/mg of cellular protein in normoxia and 8.9 ± 0.5 in IH for CE; P < 0.05; Fig. 2C). IH did not increase FC (28.8 ± 2.8 μg/mg of cellular protein in normoxia and 31.3 ± 2.0 in IH; P = 0.34; Fig. 2B).
Fig. 1.

Effects of intermittent hypoxia (IH) on intracellular lipids in THP-1 cells. A: microscopy images of Oil red O stained THP-1 cells incubated with different concentrations of native LDL. The cells were exposed to either normoxia or IH for 24 h. B: quantification of intracellular neutral lipids by Nile red staining. RFU, relative fluorescence units. Values are expressed as means ± SE; n = 6. *P < 0.05 vs. normoxia.
Fig. 2.

Effects of IH on intracellular cholesterol in THP-1 cells. A: total cholesterol. B: free cholesterol. C: cholesteryl ester. Values are expressed as means ± SE; n = 10. *P < 0.05, compared with IH-exposed cells incubated with 200 μg/dl native LDL.
IH activates the NF-κB pathway in peritoneal macrophages.
To assess the effect of IH on the NF-κB pathways, peritoneal macrophages from Ikk-βF/F and Ikk-βΔMye mice were incubated under either normoxia or IH. In the IH-exposed macrophages from Ikk-βF/F mice, phosphorylated p-65 protein expression in the nucleus was increased by 93% compared with the normoxia-exposed Ikk-βF/F macrophages (P = 0.05, Fig. 3). Interestingly, we observed no significant increase of NF-κB pathway activity in response to IH when IKK-β was deleted (+31% activity increase in IH compared with normoxia; P = 0.64; Fig. 3). Therefore, we conclude that IH increases the NF-κB pathway activity and is dependent on pathways where IKK-β is a key component.
Fig. 3.
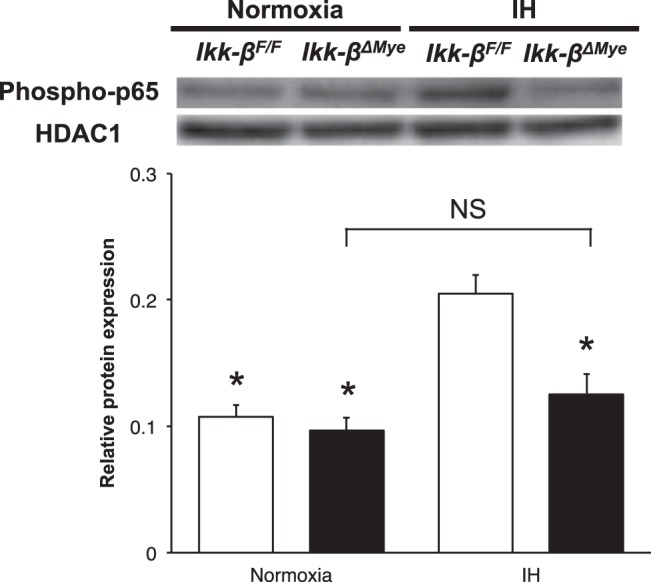
IKK-β-dependent NF-κB activation by IH in peritoneal macrophages. The activity of the NF-κB pathway was measured as phosphorylated p65 in nuclear protein. Representative data of Western blot (top) and summary data (bottom). Values are expressed as means ± SE; n = 5. *P < 0.05, compared with IH-exposed Ikk-βF/F macrophages.
IH-induced foam cell formation depends on the IKK-β-dependent NF-κB pathway.
To investigate the role of IKK-β-dependent NF-κB activation in IH-induced foam cell formation, macrophages from Ikk-βF/F and Ikk-βΔMye mice were incubated with native LDL (Fig. 4) and assayed to quantify intracellular cholesterol after 24-h in normoxia or IH (Fig. 5, A–C). TC of IH-exposed macrophages from Ikk-βF/F was significantly increased compared with normoxia when they were incubated with 200 μg/ml of native LDL (53.2 ± 1.2 μg/mg cellular protein in normoxia and 65.7 ± 3.8 in IH; P < 0.05; Fig. 5A). However, IH did not increase TC when IKK-β was deleted (46.3 ± 1.7 μg/mg cellular protein in normoxia vs. 52.2 ± 1.2 in IH; P = 0.55; Fig. 5A). We also observed a significant increase of FC in Ikk-βF/F macrophages exposed to IH compared with normoxia (48.0 ± 1.2 μg/mg cellular protein in normoxia and 59.2 ± 4.3 in IH; P < 0.05; Fig. 5B), and there was no IH-induced FC increase in Ikk-βΔMye macrophages (40.6 ± 2.4 μg/mg cellular protein in normoxia and 46.4 ± 1.3 in IH; P = 0.52; Fig. 5B). CE was increased when the cells were incubated with LDL but not increased by IH-exposure in both Ikk-βF/F and Ikk-βΔMye macrophages. In summary, IH increased intracellular TC in macrophages incubated with native LDL and showed a modest effect on FC, which was diminished by IKK-β deletion.
Fig. 4.

Microscopy images of Oil red O stained peritoneal macrophages from Ikk-βF/F and Ikk-βΔMye mice incubated with different concentrations of native LDL. The cells were exposed to either normoxia or IH for 24 h.
Fig. 5.

Effects of IH and deletion of IKK-β on intracellular cholesterol contents in peritoneal macrophages from Ikk-βΔMye and Ikk-βF/F mice where A: total cholesterol. B: free cholesterol. C: cholesteryl ester. Values are expressed as means ± SE; n = 7. *P < 0.05 vs. IH-exposed Ikk-βF/F macrophages incubated with 200 μg/dl native LDL.
Cholesterol from nonmodified native LDL is not taken up by LDL receptor-mediated internalization.
To elucidate the role of uptake through LDL receptor in IH-induced foam cell formation, peritoneal macrophages from Ikk-βF/F-Ldlr−/− and Ikk-βΔMye-Ldlr−/− mice were used. Intriguingly, we still observed the IH-induced TC increase that was diminished by IKK-β deletion (52.4 ± 1.7 μg/mg cellular protein in normoxia vs. 63.9 ± 4.8 in IH for Ikk-βF/F-Ldlr−/− and 45.8 ± 1.9 in normoxia vs. 47.9 ± 1.9 in IH for Ikk-βΔMye-Ldlr−/−; P < 0.05 and P > 0.99, respectively, Fig. 6A), and a modest effect of IH on FC was observed again (45.5 ± 1.9 μg/mg cellular protein in normoxia vs. 53.2 ± 2.8 in IH for Ikk-βF/F-Ldlr−/− and 38.8 ± 2.1 in normoxia vs. 40.9 ± 1.5 in IH for Ikk-βΔMye-Ldlr−/−; P = 0.09 and P > 0.99, respectively; Fig. 6B). CE was not increased by IH in both Ikk-βF/F-Ldlr−/− and Ikk-βΔMye-Ldlr−/− macrophages. These findings indicate that the role of LDLR on IH-induced foam cell formation is not major and imply the presence of another mechanism, including increased cholesterol intake from native LDL by pinocytosis and inhibition of cholesterol efflux (18).
Fig. 6.

Effects of IH and deletion of IKK-β on intracellular cholesterol contents in peritoneal macrophages from Ikk-βΔMye-Ldlr−/− and Ikk-βF/F-Ldlr−/− mice where A: total cholesterol. B: free cholesterol. C: cholesteryl ester. Values are expressed as means ± SE; n = 7. *P < 0.05 vs. IH-exposed Ikk-βF/F-Ldlr−/− macrophages incubated with 200 μg/dl native LDL.
DISCUSSION
The results of this research show that IH activates the NF-κB pathways and induces foam cell formation. To the best of our knowledge, this is the first report to directly show that IKK-β-dependent NF-κB activation causes IH-induced macrophage foam cell formation. Our result is consistent with previously published evidence, where researchers showed that IH exposure caused macrophage foam cell formation in vitro (28, 31). Furthermore, it has been shown that IH activates the NF-κB pathways in various types of cells (1, 21, 32, 41, 46, 48, 55). Other data demonstrated that activation of the NF-κB pathways contributed to foam cell formation where they employed PMA-treated THP-1 cells with inactive NF-κB/IκBα complex (16) and lipopolysaccharide-treated bone marrow-derived macrophages from NF-κB1-deficient mouse (24). Therefore, we speculated that IH causes NF-κB pathway activation, and in turn induces foam cell formation. In spite of the importance of OSA in atherogenicity, the IH-induced foam cell formation by NF-κB pathway activation was not fully understood. In this study, we showed that the IKK-β-dependent NF-κB activation is the pathway that causes IH-induced foam cell formation by using IKK-β-deleted mouse macrophages. Our results add essential information to our understanding of the increased risk of cardiovascular disease in OSA patients, which is now a big national health burden.
Evidence is growing to support the hypothesis that IH induces atherosclerosis in mice on HFD (3, 13, 15, 23, 32, 49). First, we have indeed shown that IH exposure induces foam cell formation in the THP-1 human macrophage cell line as well as mouse peritoneal macrophages. Although the significance of macrophage foam cells, the characteristic of atherosclerotic lesions, has been well established (31), only a few researchers have documented the induction of foam cell formation by IH exposure (29, 32). One of the important differences between our work and the work of others is that we supplemented the culture medium with native LDL, which is a weaker inducer of foam cell formation compared with chemically modified LDL, such as acetylated LDL, that was used by other investigators (29, 32). Since the use of chemically modified LDL for cholesterol-loading experiments has been previously criticized (26, 27), we used 200 to 800 μg/mg of native LDL. This range of LDL concentrations is equivalent to 10 to 40 mg/dl in plasma, a range that is considered normal in humans. It is very interesting that we were still able to observe the IH-induced effect on intracellular cholesterol metabolism in spite of using relatively low concentrations of nonmodified LDL.
One of the questions that can be raised is how cholesterol enters macrophages to produce foam cells. Of interest is that previous research has shown that activated macrophages by PMA or macrophage-colony-stimulating factor (M-CSF) increase their native LDL uptake through fluid-phase pinocytosis (2, 27, 28, 59). Thus we believe that IH likely works to mediate LDL uptake by this receptor-independent mechanism since IH continues to induce cholesterol accumulation in macrophages even when LDLR, which is the major receptor for LDL internalization, is absent.
The NF-κB pathways have two major activation signaling processes, canonical and noncanonical. IKK-β is the prevalent catalytic subunit of the IKK complex that is necessary for the canonical pathway of NF-κB activation (25). IKK-β-dependent NF-κB activation has been implicated in human atherosclerosis (37) and the importance of IKK-β in the pathogenesis of atherosclerosis has been well documented (5, 39). An important finding in our work is that we show that IH activates IKK-β-dependent NF-κB pathways in murine peritoneal macrophages because our IKK-β-deleted macrophages in IH did not activate the NF-κB pathways and did not accumulate more cholesterol compared with normoxia. This indicates that the IKK-β-dependent pathway is essential for IH-induced NF-κB activation and foam cell formation.
One very important question to answer is related to the mechanisms of how IKK-β-dependent NF-κB activation regulates intracellular cholesterol metabolism. Cholesterol homeostasis in macrophages is regulated by many biochemical steps (31). Briefly, native LDL is taken up by LDLR or via pinocytosis (17, 26). Whereas, modified LDL, such as acetylated or oxidized LDL, is chiefly taken up by scavenger receptors (38). Then, internalized cholesterol is stored as CE (9, 18). As an initial step in the efflux, CE is hydrolyzed to form FC (18, 40). Lastly, ABCA-1 and ABCG-1 mediate cholesterol efflux, which is facilitated by SR-BI (45). Since FC can induce inflammation and cause cytotoxicity (33, 54, 60), macrophages are normally protected from accumulation of excess FC because it can be injurious to them. Remarkably, IH significantly increased intracellular FC in the peritoneal macrophages from mice, an effect that was not observed when IKK-β was deleted. We observed a significant increase of CE but not FC in THP-1 cells. The reason for this difference could be explained by the difference in intracellular cholesterol metabolism between human and mouse (10) or possibly between cell lines and primary cells (41).
Lipid assays of lesions from various stages of human and animal atherosclerosis reveal a progressive intracellular FC increase in macrophages as lesions become more advanced (44). Since it has been reported that constant hypoxia also increases intracellular FC in murine macrophage cell lines and bone marrow-derived macrophages (42), further investigations are required to address whether IH and constant hypoxia share similar mechanisms to increase FC and, furthermore, the role of IKK-β-dependent NF-κB pathways on this.
There are a few limitations to our study. Firstly, we used thioglycolate-elicited macrophages. Although many researchers in this field have used these types of macrophages, it is known that this agent itself can induce inflammation (58). However, there was no difference in NF-κB pathway activity between Ikk-βF/F and Ikk-βΔMye macrophages in normoxia. Bone marrow-derived macrophages could be an alternative method, but M-CSF that is used to differentiate cells into to macrophages is also known to augment macrophage pinocytosis and induce foam cell formation (59). Secondly, IKK-β deletion in this model was reported not to cause complete deletion (∼75% reduction of the expression; Ref. 19). Nonetheless, we were still able to observe a significant reduction of NF-κB pathway activation in the IH-exposed macrophages when IKK-β was deleted. Thirdly, we did not address detailed mechanisms of this IH-induced foam cell formation by IKK-β-dependent NF-κB activation in this study. Fourthly, the uptake of native LDL is through LDLR or micropinocytosis, but modified LDL is taken up through scavenger receptors, such as SRA, CD36, and LOX-1 (38). Our aim in this paper was not how to assess whether oxidized LDL was taken up through these scavenger receptors. Lastly, we did not assess in this study the mechanisms of uptake or the correlation between IH-induced foam cell formation and atherogenicity in OSA patients, and in vivo animal experiments will be necessary to address this.
In summary, we have demonstrated that IH induces IKK-β-dependent NF-κB activation in macrophages and that inflammation is important for foam cell formation. Our findings have implications for the relation between OSA and macrophage foam cell formation through NF-κB pathway activation. Our results could potentially provide therapeutic targets to reduce the risk of cardiovascular disease in OSA patients.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grant 1P01-HL-098053.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
T.I. and G.G.H. conception and design of research; T.I. performed experiments; T.I., O.P., and G.G.H. analyzed data; T.I. and G.G.H. interpreted results of experiments; T.I., O.P., and G.G.H. prepared figures; T.I. and O.P. drafted manuscript; O.P. and G.G.H. edited and revised manuscript; G.G.H. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Michael Karin at University of California, San Diego for providing Ikk-βF/F mice.
REFERENCES
- 1.Angelo MF, Aguirre A, Aviles Reyes RX, Villarreal A, Lukin J, Melendez M, Vanasco V, Barker P, Alvarez S, Epstein A, Jerusalinsky D, Ramos AJ. The proinflammatory RAGE/NF-kappaB pathway is involved in neuronal damage and reactive gliosis in a model of sleep apnea by intermittent hypoxia. PLoS One 9: e107901, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anzinger JJ, Chang J, Xu Q, Buono C, Li Y, Leyva FJ, Park BC, Greene LE, Kruth HS. Native low-density lipoprotein uptake by macrophage colony-stimulating factor-differentiated human macrophages is mediated by macropinocytosis and micropinocytosis. Arterioscler Thromb Vasc Biol 30: 2022–2031, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arnaud C, Poulain L, Levy P, Dematteis M. Inflammation contributes to the atherogenic role of intermittent hypoxia in apolipoprotein-E knock out mice. Atherosclerosis 219: 425–431, 2011. [DOI] [PubMed] [Google Scholar]
- 4.Baguet JP, Barone-Rochette G, Tamisier R, Levy P, Pepin JL. Mechanisms of cardiac dysfunction in obstructive sleep apnea. Nat Rev Cardiol 9: 679–688, 2012. [DOI] [PubMed] [Google Scholar]
- 5.Baker RG, Hayden MS, Ghosh S. NF-kappaB, inflammation, and metabolic disease. Cell Metab 13: 11–22, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baumgardner JE, Otto CM. In vitro intermittent hypoxia: challenges for creating hypoxia in cell culture. Respir Physiol Neurobiol 136: 131–139, 2003. [DOI] [PubMed] [Google Scholar]
- 7.Berthois Y, Katzenellenbogen JA, Katzenellenbogen BS. Phenol red in tissue culture media is a weak estrogen: implications concerning the study of estrogen-responsive cells in culture. Proc Natl Acad Sci USA 83: 2496–2500, 1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brand K, Page S, Rogler G, Bartsch A, Brandl R, Knuechel R, Page M, Kaltschmidt C, Baeuerle PA, Neumeier D. Activated transcription factor nuclear factor-kappa B is present in the atherosclerotic lesion. J Clin Invest 97: 1715–1722, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chang TY, Chang CC, Lin S, Yu C, Li BL, Miyazaki A. Roles of acyl-coenzyme A:cholesterol acyltransferase-1 and -2. Curr Opin Lipidol 12: 289–296, 2001. [DOI] [PubMed] [Google Scholar]
- 10.Dietschy JM, Turley SD. Control of choletserol turnover in the mouse. J Biol Chem 277: 3801–3804, 2002. [DOI] [PubMed] [Google Scholar]
- 11.Dong JY, Zhang YH, Qin LQ. Obstructive sleep apnea and cardiovascular risk: meta-analysis of prospective cohort studies. Atherosclerosis 229: 489–495, 2013. [DOI] [PubMed] [Google Scholar]
- 12.Drager LF, Polotsky VY, Lorenzi-Filho G. Obstructive sleep apnea: an emerging risk factor for atherosclerosis. Chest 140: 534–542, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Drager LF, Togeiro SM, Polotsky VY, Lorenzi-Filho G. Obstructive sleep apnea: a cardiometabolic risk in obesity and the metabolic syndrome. J Am Coll Cardiol 62: 569–576, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Drager LF, Yao Q, Hernandez KL, Shin MK, Bevans-Fonti S, Gay J, Sussan TE, Jun JC, Myers AC, Olivecrona G, Schwartz AR, Halberg N, Scherer PE, Semenza GL, Powell DR, Polotsky VY. Chronic intermittent hypoxia induces atherosclerosis via activation of adipose angiopoietin-like 4. Am J Respir Crit Care Med 188: 240–248, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fang G, Song D, Ye X, Mao SZ, Liu G, Liu SF. Chronic intermittent hypoxia exposure induces atherosclerosis in ApoE knockout mice: role of NF-kappaB p50. Am J Pathol 181: 1530–1539, 2012. [DOI] [PubMed] [Google Scholar]
- 16.Ferreira V, van Dijk KW, Groen AK, Vos RM, van der Kaa J, Gijbels MJ, Havekes LM, Pannekoek H. Macrophage-specific inhibition of NF-kappaB activation reduces foam-cell formation. Atherosclerosis 192: 283–290, 2007. [DOI] [PubMed] [Google Scholar]
- 17.Glass CK, Witztum JL. Atherosclerosis the road ahead. Cell 104: 503–516, 2001. [DOI] [PubMed] [Google Scholar]
- 18.Ghosh S. Macrophage cholesterol homeostasis and metabolic diseases: critical role of cholesteryl ester mobilization. Expert Rev Cardiovasc Ther 9: 329–340, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Greten FR, Eckmann L, Greten TF, Park JM, Li ZW, Egan LJ, Kagnoff MF, Karin M. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 118: 285–296, 2004. [DOI] [PubMed] [Google Scholar]
- 20.Gunnarsson SI, Peppard PE, Korcarz CE, Barnet JH, Aeschlimann SE, Hagen EW, Young T, Hla KM, Stein JH. Obstructive sleep apnea is associated with future subclinical carotid artery disease: thirteen-year follow-up from the Wisconsin sleep cohort. Arterioscler Thromb Vasc Biol 34: 2338–2342, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Han Q, Yeung SC, Ip MS, Mak JC. Intermittent hypoxia-induced NF-kappaB and HO-1 regulation in human endothelial EA.hy926 cells. Cell Biochem Biophys 66: 431–441, 2013. [DOI] [PubMed] [Google Scholar]
- 22.Hara A, Radin NS. Lipid extraction of tissues with a low-toxicity solvent. Anal Biochem 90: 420–426, 1978. [DOI] [PubMed] [Google Scholar]
- 23.Jun J, Reinke C, Bedja D, Berkowitz D, Bevans-Fonti S, Li J, Barouch LA, Gabrielson K, Polotsky VY. Effect of intermittent hypoxia on atherosclerosis in apolipoprotein E-deficient mice. Atherosclerosis 209: 381–386, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kanters E, Gijbels MJ, van der Made I, Vergouwe MN, Heeringa P, Kraal G, Hofker MH, de Winther MP. Hematopoietic NF-kappaB1 deficiency results in small atherosclerotic lesions with an inflammatory phenotype. Blood 103: 934–940, 2004. [DOI] [PubMed] [Google Scholar]
- 25.Karin M. Nuclear factor-kappaB in cancer development and progression. Nature 441: 431–436, 2006. [DOI] [PubMed] [Google Scholar]
- 26.Kruth HS. Receptor-independent fluid-phase pinocytosis mechanisms for induction of foam cell formation with native low-density lipoprotein particles. Curr Opin Lipidol 22: 386–393, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kruth HS, Huang W, Ishii I, Zhang WY. Macrophage foam cell formation with native low density lipoprotein. J Biol Chem 277: 34573–34580, 2002. [DOI] [PubMed] [Google Scholar]
- 28.Kruth HS, Jones NL, Huang W, Zhao B, Ishii I, Chang J, Combs CA, Malide D, Zhang WY. Macropinocytosis is the endocytic pathway that mediates macrophage foam cell formation with native low density lipoprotein. J Biol Chem 280: 2352–2360, 2005. [DOI] [PubMed] [Google Scholar]
- 29.Lattimore JD, Wilcox I, Nakhla S, Langenfeld M, Jessup W, Celermajer DS. Repetitive hypoxia increases lipid loading in human macrophages-a potentially atherogenic effect. Atherosclerosis 179: 255–259, 2005. [DOI] [PubMed] [Google Scholar]
- 30.Lee W, Nagubadi S, Kryger MH, Mokhlesi B. Epidemiology of obstructive sleep apnea: a population-based perspective. Expert Rev Respir Med 2: 349–364, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li AC, Glass CK. The macrophage foam cell as a target for therapeutic intervention. Nat Med 8: 1235–1242, 2002. [DOI] [PubMed] [Google Scholar]
- 32.Li RC, Haribabu B, Mathis SP, Kim J, Gozal D. Leukotriene B4 receptor-1 mediates intermittent hypoxia-induced atherogenesis. Am J Respir Crit Care Med 184: 124–131, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li Y, Schwabe RF, DeVries-Seimon T, Yao PM, Gerbod-Giannone MC, Tall AR, Davis RJ, Flavell R, Brenner DA, Tabas I. Free cholesterol-loaded macrophages are an abundant source of tumor necrosis factor-alpha and interleukin-6: model of NF-kappaB- and map kinase-dependent inflammation in advanced atherosclerosis. J Biol Chem 280: 21763–21772, 2005. [DOI] [PubMed] [Google Scholar]
- 34.Libby P. Inflammation in atherosclerosis. Nature 420: 868–874, 2002. [DOI] [PubMed] [Google Scholar]
- 35.Marin JM, Carrizo SJ, Vicente E, Agusti AG. Long-term cardiovascular outcomes in men with obstructive sleep apnoea-hypopnoea with or without treatment with continuous positive airway pressure: an observational study. Lancet 365: 1046–1053, 2005. [DOI] [PubMed] [Google Scholar]
- 36.McCrohon JA, Nakhla S, Jessup W, Stanley KK, Celermajer DS. Estrogen and progesterone reduce lipid accumulation in human monocyte-derived macrophages: a sex-specific effect. Circulation 100: 2319–2325, 1999. [DOI] [PubMed] [Google Scholar]
- 37.Monaco C, Andreakos E, Kiriakidis S, Mauri C, Bicknell C, Foxwell B, Cheshire N, Paleolog E, Feldmann M. Canonical pathway of nuclear factor kappa B activation selectively regulates proinflammatory and prothrombotic responses in human atherosclerosis. Proc Natl Acad Sci USA 101: 5634–5639, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moore KJ, Freeman MW. Scavenger receptors in atherosclerosis: beyond lipid uptake. Arterioscler Thromb Vasc Biol 26: 1702–1711, 2006. [DOI] [PubMed] [Google Scholar]
- 39.Moore KJ, Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell 145: 341–355, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Okazaki H, Igarashi M, Nishi M, Sekiya M, Tajima M, Takase S, Takanashi M, Ohta K, Tamura Y, Okazaki S, Yahagi N, Ohashi K, Amemiya-Kudo M, Nakagawa Y, Nagai R, Kadowaki T, Osuga J, Ishibashi S. Identification of neutral cholesterol ester hydrolase, a key enzyme removing cholesterol from macrophages. J Biol Chem 283: 33357–33364, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Oliver KM, Garvey JF, Ng CT, Veale DJ, Fearon U, Cummins EP, Taylor CT. Hypoxia activates NF-kappaB-dependent gene expression through the canonical signaling pathway. Antioxid Redox Signal 11: 2057–2064, 2009. [DOI] [PubMed] [Google Scholar]
- 42.Parathath S, Mick SL, Feig JE, Joaquin V, Grauer L, Habiel DM, Gassmann M, Gardner LB, Fisher EA. Hypoxia is present in murine atherosclerotic plaques and has multiple adverse effects on macrophage lipid metabolism. Circ Res 109: 1141–1152, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Peppard PE, Young T, Palta M, Dempsey J, Skatrud J. Longitudinal study of moderate weight change and sleep-disordered breathing. JAMA 284; 3015–3021, 2000. [DOI] [PubMed] [Google Scholar]
- 44.Rapp JH, Connor WE, Lin DS, Inahara T, Porter JM. Lipids of human atherosclerotic plaques and xanthomas: clues to the mechanism of plaque progression. J Lipid Res 24: 1329–1335, 1983. [PubMed] [Google Scholar]
- 45.Rosenson RS, Brewer HB Jr, Davidson WS, Fayad ZA, Fuster V, Goldstein J, Hellerstein M, Jiang XC, Phillips MC, Rader DJ, Remaley AT, Rothblat GH, Tall AR, Yvan-Charvet L. Cholesterol efflux and atheroprotection: advancing the concept of reverse cholesterol transport. Circulation 125: 1905–1919, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ryan S, McNicholas WT, Taylor CT. A critical role for p38 map kinase in NF-kappaB signaling during intermittent hypoxia/reoxygenation. Biochem Biophys Res Commun 355: 728–733, 2007. [DOI] [PubMed] [Google Scholar]
- 47.Ryan S, Taylor CT, McNicholas WT. Predictors of elevated nuclear factor-kappaB-dependent genes in obstructive sleep apnea syndrome. Am J Respir Crit Care Med 174: 824–830, 2006. [DOI] [PubMed] [Google Scholar]
- 48.Ryan S, Taylor CT, McNicholas WT. Selective activation of inflammatory pathways by intermittent hypoxia in obstructive sleep apnea syndrome. Circulation 112: 2660–2667, 2005. [DOI] [PubMed] [Google Scholar]
- 49.Savransky V, Nanayakkara A, Li J, Bevans S, Smith PL, Rodriguez A, Polotsky VY. Chronic intermittent hypoxia induces atherosclerosis. Am J Respir Crit Care Med 175: 1290–1297, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Smith JD, Trogan E, Ginsberg M, Grigaux C, Tian J, Miyata M. Decreased atherosclerosis in mice deficient in both macrophage colony-stimulating factor (op) and apolipoprotein E. Proc Natl Acad Sci USA 92: 8264–8268, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Song D, Fang G, Mao SZ, Ye X, Liu G, Gong Y, Liu SF. Chronic intermittent hypoxia induces atherosclerosis by NF-kappaB-dependent mechanisms. Biochim Biophys Acta 1822: 1650–1659, 2012. [DOI] [PubMed] [Google Scholar]
- 52.Stary HC, Chandler AB, Glagov S, Guyton JR, Insull W Jr, Rosenfeld ME, Schaffer SA, Schwartz CJ, Wagner WD, Wissler RW. A definition of initial, fatty streak, and intermediate lesions of atherosclerosis A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation 89: 2462–2478, 1994. [DOI] [PubMed] [Google Scholar]
- 53.Stoneman V, Braganza D, Figg N, Mercer J, Lang R, Goddard M, Bennett M. Monocyte/macrophage suppression in CD11b diphtheria toxin receptor transgenic mice differentially affects atherogenesis and established plaques. Circ Res 100: 884–893, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sun Y, Ishibashi M, Seimon T, Lee M, Sharma SM, Fitzgerald KA, Samokhin AO, Wang Y, Sayers S, Aikawa M, Jerome WG, Ostrowski MC, Bromme D, Libby P, Tabas IA, Welch CL, Tall AR. Free cholesterol accumulation in macrophage membranes activates Toll-like receptors and p38 mitogen-activated protein kinase and induces cathepsin K. Circ Res 104: 455–465, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Taylor CT, Kent BD, Crinion SJ, McNicholas WT, Ryan S. Human adipocytes are highly sensitive to intermittent hypoxia induced NF-kappaB activity and subsequent inflammatory gene expression. Biochem Biophys Res Commun 447: 660–665, 2014. [DOI] [PubMed] [Google Scholar]
- 56.Young T, Finn L, Peppard PE, Szklo-Coxe M, Austin D, Nieto FJ, Stubbs R, Hla KM. Sleep disordered breathing and mortality: eighteen-year follow-up of the Wisconsin sleep cohort. Sleep 31: 1071–1078, 2008. [PMC free article] [PubMed] [Google Scholar]
- 57.Young T, Palta M, Dempsey J, Skatrud J, Weber S, Badr S. The occurrence of sleep-disordered breathing among middle-aged adults. N Engl J Med 328: 1230–1235, 1993. [DOI] [PubMed] [Google Scholar]
- 58.Zhang X, Goncalves R, Mosser DM. The isolation and characterization of murine macrophages. Curr Protoc Immunol 14: Unit 14.11, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhao B, Li Y, Buono C, Waldo SW, Jones NL, Mori M, Kruth HS. Constitutive receptor-independent low density lipoprotein uptake and cholesterol accumulation by macrophages differentiated from human monocytes with macrophage-colony-stimulating factor (M-CSF). J Biol Chem 281: 15757–15762, 2006. [DOI] [PubMed] [Google Scholar]
- 60.Zhu X, Lee JY, Timmins JM, Brown JM, Boudyguina E, Mulya A, Gebre AK, Willingham MC, Hiltbold EM, Mishra N, Maeda N, Parks JS. Increased cellular free cholesterol in macrophage-specific Abca1 knock-out mice enhances pro-inflammatory response of macrophages. J Biol Chem 283: 22930–22941, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]