

Abstract
Here we tested impact of Tris-DBA on CLL B-cell survival. Indeed treatment of CLL B-cells with Tris-DBA induced apoptosis in a dose-dependent manner irrespective of IgVH mutational status. Further analyses suggest that Tris-DBA-induced apoptosis involves reduced expression of the anti-apoptotic proteins Bcl-xL, and XIAP with an upregulation of the pro-apoptotic protein BIM in CLL B-cells. Our findings also indicate that Tris-DBA targets the ribosomal protein (rp)-S6, an essential component of the Akt/mTOR signaling axis in CLL B-cells. Of interest, CLL bone marrow stromal cells were unable to protect the leukemic B-cells from Tris-DBA-induced apoptosis in an in vitro co-culture system. Finally, co-administration of Tris-DBA and the purine nucleoside analog fludarabine (F-ara-A) augmented CLL B-cell apoptosis levels in vitro showing synergistic effects. In total, Tris-DBA is effective at inducing apoptosis in CLL B-cells even in the presence of stromal cells likely by targeting directly the signal mediator, rpS6.
Keywords: CLL, Tris-DBA, fludarabine
Introduction
The effective treatment of B-cell chronic lymphocytic leukemia (CLL) is an important research goal as it still largely incurable despite the effectiveness of Chemoimmunotherapy (CIT) and an increasing number of effective agents targeting the B-cell receptor (BCR) signaling pathway. CIT regimens combing purine analogue-based chemotherapy in tandem with monoclonal antibodies have also improved response rates, progression free survival (PFS) and overall survival (OS) particularly for certain subsets of CLL patients. However it is generally agreed that the current upfront treatment CIT regimens for patients with CLL are not curative for most patients and for relapsed/refractory CLL patients post-CIT the options for effective therapy can still be improved.
To this end, the inhibition of signal pathways by novel oral bioavailable agents can induce high overall responses in relapsed/refractory CLL patients. For example clinical trials using Ibrutinib which is a BCR inhibitor via targeting of BTK, uncovered that approximately 90% of patients had a clinical benefit with PFS and OS after 26 months of continuous therapy of ~75% and 83% respectively[1]. When relapsed/refractory CLL patients were treated with the PI3K delta inhibitor (Idelalisib) responses were found in over 70% of patients and the PFS was 15.8 months while the overall survival had not yet been reached[2]. However within these regimens, which tend to be more durable, the current estimate is that 20–30% of all patients treated with these novel drugs will ultimately relapse. Thus novel drugs with unique mechanisms of action are still needed for CLL patients.
We have been interested in conducting preclinical testing in CLL on the unique organometallic complex called Tris (dibenzylideneacetone) dipalladium (Tris-DBA). This novel agent is a small-molecule palladium complex shown to have antiproliferative activity against melanoma tumor cells[3] in both the in vivo and in vitro settings. In addition it has been found to have activity in myeloma with or without other drug combinations[4]. Of interest, this compound has the ability to inhibit the activity of multiple signaling pathways including the activation of mitogen-activated protein kinase, Akt, STAT3, and S6 kinase some of which have been shown to be activated in CLL B-cells[5–9]. Indeed Akt and STAT3 are known to play key roles in the enhancement of survival for CLL B-cells[10,11].
Given the need for novel drugs in CLL and since Tris-DBA was known to be able to target key signaling molecules which remain constitutively active in CLL B-cells influencing cell survival and resistance to apoptosis, we directed our efforts to conduct an in vitro pre-clinical study testing the impact, if any, of Tris-DBA on CLL B-cell survival.
Materials and Methods[12]
Patient selection and CLL sample processing
Blood was obtained from CLL patients who had provided written informed consent under a protocol approved by the Mayo Clinic Institutional Review Board according to the regulations of the Declaration of Helsinki or from healthy volunteers. All CLL patients had a confirmed diagnosis using the IWCLL 2008 definition[13]. Patients in this cohort were from all Rai stages and had not been treated at the time of blood collection. CLL cells were isolated from heparinized venous blood by density gradient centrifugation. To ensure purity of the CLL B-cells we used flow cytometry (FACScan, Becton Dickinson) and the isolated cells were predominantly CLL B-cells (>90% CD5+/CD19+). Lymphocytes from healthy volunteers were also separated by density gradient centrifugation. Freshly isolated CLL B-cells or peripheral blood mononuclear cells (PBMCs) from normal healthy individuals were cultured in serum-free AIM-V medium at 37°C in an atmosphere containing 95% air and 5% CO2. In all experiments described below we used only CLL PBMCs that consisted of at least 92% CD5+/CD19+ CLL B-cells.
Reagents
The following antibodies were used for western blotting and were purchased from the indicated suppliers: Antibodies to, phospho-Akt, phospho-STAT3, phospho-S6ribosomal protein (ser235/236), phospho-p44/42 MAPK (ERK1/2), phosphor-PI3Kp85, Akt, STAT3, S6 ribosomal protein, p44/42 MAPK (ERK1/2), PI3Kp85, Mcl-1, XIAP, Bcl-xL and PARP were purchased from Cell Signaling Technology. β-Actin and BIM were obtained from Santa Cruz Biotechnology. Bcl-2 was from BD Pharmingen. The following reagents were purchased from the indicated suppliers: Ficoll-Paque Plus (GE Healthcare); AIM-V medium, PBS and MEM Alpha medium (Gibco, Life Technologies); DMSO and propidium iodide (PI) (Sigma Aldrich); CD3 APC, CD16 FITC, and CD19 APC (BD Biosciences); annexin-V FITC (Life Technologies); and pan-caspase inhibitor Z-VAD-fmk (BD Pharmingen, BD Biosciences). Tris (dibenzylideneacetone) dipalladium (Tris-DBA) was a gift from Dr. Jack L. Arbiser, MD, Dept. of Dermatology, Emory University, School of Medicine, Winship Cancer Institute, Atlanta, GA.
Apoptosis assay
Primary CLL B-cells (1.0 × 106/mL) were treated with either vehicle (DMSO) or Tris-DBA for 24, 48 and 72 h at increasing doses (1.0–10 μM) in serum-free AIM-V medium. Cells were washed with PBS, stained with annexin V-FITC/PI and analyzed for apoptosis induction by flow cytometry (FACScan). Similarly, freshly isolated normal PBMCs (1.0 × 106 cells/mL) were treated with Tris-DBA (2.5–10 μM) for 24, 48 and 72 h. Cells were harvested and analyzed for the induction of cell death after staining the cells with Allophycocyanin (APC)-conjugated anti-CD19, anti-CD3 or anti-CD16 antibody, annexin-FITC and PIon a FACSCalibur Instrument (Becton Dickinson) using Cell Quest software. The use of reagents specific for B-cells (anti-CD19), T-cells (anti-CD3) or NK cells (anti-CD16) allowed us to discriminate between these 3 cell types in the PMBC preparations and their individual survival pattern. Cells staining with annexin V-FITC and/or PI were considered positive for cell death.
Treatment of CLL B-cells with Tris-DBA in the presence of stromal cells
For stromal experiments, the human bone marrow mesenchymal stromal cells (BMSCs) were grown and maintained in confluent monolayers as described previously[14]. BMSCs were cultured in 12-well tissue-culture plates at a cell density of 1.0 × 105/well overnight, washed twice with serum-free AIM-V medium, and then incubated with primary CLL B-cells at a cell density of 1.0 × 106/mL cells/well. CLL B-cells were cultured in direct contact with BMSCs with serum-free AIM-V medium for 24 h followed by exposure of the cells to increasing doses of Tris-DBA (1–10 μM) or left untreated (DMSO control). For comparison, CLL B-cells cultured without stromal cells were treated similarly with Tris-DBA or DMSO. After 24 h, CLL B-cells were harvested, washed in PBS, and stained with APC conjugated antibody to CD19 and annexin V-FITC/PI, followed by analysis on a FACSCalibur for apoptosis induction.
Combined treatment of CLL B-cells with Tris-DBA and fludarabine or chlorambucil
CLL B-cells were treated with various doses of Tris-DBA in combination with F-ara-A or chlorambucil at a 1:1 or 1: 5 ratio respectively, or single agent alone for 24 h. Cells were harvested, stained with annexin/PI, and viability analyzed by flow cytometry. After concentration-effect curves were generated for each agent, data were analyzed using the CalcuSyn software program (Biosoft), which uses the method of Chou and Talalay[15], to determine whether combination treatment yields synergistic, additive or antagonistic effects on cell viability. A combination index (CI) of 1 indicates an additive effect, a CI above 1 indicates an antagonistic effect, and a CI below 1 indicates a synergistic effect[16].
Immunoblot
Purified CLL B-cells (4.0 × 106/mL) were treated with DMSO or Tris-DBA (4 μM) with or without pre-treatment of the cells with the pan-caspase inhibitor Z-VAD-fmk and then cells were lysed in NP40-lysis buffer, and whole-cell extract was prepared as described previously.[17,18] Protein content was determined, and equal amounts of proteins were loaded on SDS-polyacrylamide gels after digesting in Laemmli SDS-sample buffer. Separated proteins were transferred onto 0.45-μm nitrocellulose papers (BioRad) and immunostained with specific antibodies. Protein bands were detected using an enhanced chemiluminescence detection kit (Pierce).
Statistical analysis
The percent kill and percentage viable cells were evaluated across CLL patients and were summarized graphically and quantitatively. These percentages were analyzed graphically for each dose level independently, as well as across dose levels graphically. Means and SEs were calculated using Excel (Microsoft Corporation). Graphical analyses were done using PRIZM plot software.
Results
Tris-DBA induces apoptosis in CLL B-cells
To explore the impact of Tris-DBA on CLL B-cell survival, primary CLL B-cells were treated with increasing doses of Tris-DBA for 24 h. At the indicated time, cells were harvested, stained with annexin V/PI, and analyzed by flow cytometry for viability. Tris-DBA treatment induced apoptosis in CLL B-cells in a dose-dependent manner (Fig. 1A) over 24 h. While the sensitivity of CLL B-cells to Tris-DBA was not dependent on the IgVH mutational status, we detected that Tris-DBA treatment produced a lower LD50 value for the mutated CLL B-cell cohort when compared to the unmutated IgVH cohort (Fig. 1A). Under the same experimental conditions, the average LD50 dose for normal CD19+ B lymphocytes was noted at approximately 5 μM. Normal blood T-cells were less sensitive to Tris-DBA with an LD50 of 6.5 μM while CD16+ NK cells were insensitive to Tris-DBA induced apoptosis (Fig. 1B).
Figure 1A and B. Tris-DBA induces apoptosis in CLL B-cells.

Primary CLL B-cells characterized as either mutated IgVH (n=7) or unmutated IgVH (n=6) in serum-free AIM-V medium or normal PBMC in RPMI were treated with increasing doses of Tris-DBA (2.5–10 μM) for 24 h. At the indicated time, cells were harvested, stained with annexin V/PI, and analyzed by flow cytometry for viability. The apoptosis levels for PBMC were subdivided based on two color flow for annexin V and reactivity for either CD3 (T-cells), CD19 (B-cells) or CD16 (NK cells).
To investigate whether Tris-DBA induced apoptosis in CLL B-cells involves poly (ADP ribose) polymerase (PARP) cleavage, purified CLL B-cells were treated with Tris-DBA at sub-lethal dose. Cell lysates were analyzed for PARP cleavage in Western blot using a specific antibody which recognizes both the native (116 kDa) and cleaved (86 kDa) forms of PARP. Indeed, our findings suggest that Tris-DBA induces PARP-cleavage in CLL B-cells to induce apoptosis (Fig. 2A). The DNA repair enzyme PARP can be cleaved by executioner caspases (caspases 3, 7) upon activation or translocation of apoptosis-inducing factor (AIF) from mitochondria to the nucleus, independent of caspase-activation[19]. To examine whether Tris-DBA induced PARP-cleavage was caspase-dependent, CLL B-cells were treated with Tris-DBA as described above with or without pre-treatment of the cells with the pan-caspase inhibitorZ-VAD-fmk. Cell lysates were analyzed for the PARP-cleavage in Western blot. We found that Z-VAD-fmk blocked Tris-DBA induced PARP-cleavage in CLL B-cells (Fig. 2B) suggesting that Tris-DBA induced PARP-cleavage is caspase-dependent.
Figure 2A and B. Tris-DBA induced apoptosis in CLL B-cells involves poly (ADP ribose) polymerase (PARP) cleavage and is caspase dependent.

A. Purified CLL B-cells (4.0 × 106/ml) were treated with Tris-DBA at sub-lethal dose of 4μM. Cell lysates were then analyzed for PARP cleavage in Western blot using a specific antibody which recognizes both the native (116 kDa) and cleaved (86 kDa) forms of PARP. B. CLL B-cells were treated with Tris-DBA at a dose of 4μM with or without pre-treatment of the cells with the pan-caspase inhibitor Z-VAD-fmk. Cell lysates were then analyzed for the PARP-cleavage via western blot. Actin was used as a loading control. CLL patients are indicated by arbitrary numbers (P1 and P2).
Tris-DBA targets the ribosomal protein S6 and modulates expression of the Bcl-2 family proteins in CLL B-cells
We and others have shown that several survival pathways are operative in CLL B-cells including activation of constituent molecules such as Akt, and STAT3[20–25]. Therefore, to examine the effect of Tris-DBA on Akt and STAT3 activation, we analyzed lysates from primary CLL B-cells treated with Tris-DBA. However, we did not find any significant and consistent levels of inhibition of PI3Kp85/Akt/STAT3 activation by Tris-DBA in CLL B-cells (Fig. 3A). In addition, Tris-DBA did not inhibit the constitutive phosphorylation levels on mTOR or p70S6K in CLL B-cells (data not shown). In an attempt to further identify the signaling mediator(s) impacted by the Tris-DBA treatment, the same cell lysates were analyzed for modulation of the further downstream signal mediator of the mTOR/p70S6K or ERK/p90RSK axis, rpS6. Here CLL B-cells were treated with 4μM of Tris-DBA for 8 and 16 h, followed by Western blot analyses (Fig. 3A). At both 8 and 16 h of Tris-DBA treatment, a significant inhibition of rpS6 phosphorylation (S235/236 sites) was observed. However, 8 h of Tris-DBA treatment had no effect on ERK1/2 phosphorylation in 2 out of 4 patients. Only at 16 h of Tris-DBA exposure, we were able to detect a significant reduction of P-ERK1/2 levels in CLL B-cells. Additionally, Tris-DBA had no effect on P-p90 RSK at the constitutive serine/threonine phosphorylation sites, S380/Thr359 (data not shown). Thus, it is likely that Tris-DBA directly targets the rpS6 in CLL B-cells, as it did not show any inhibitory effects on the upstream regulators of rpS6 (Fig. 3A).
Figure 3A and B. Tris-DBA targets the ribosomal protein S6 kinase and has variable ability to modulate expression of Bcl-2 family proteins in CLL B-cells.

A. CLL B-cells from previously untreated CLL patients were treated with a sub-lethal dose of Tris-DBA (4μM) or left untreated (DMSO) for 8 and 16 h, were analyzed for the activation status of P-rpS6 (S235/236), P-ERK (42/44), P-PI3Kp85,P-Akt(S473), and P-STAT3 (S727) by Western blots using specific antibodies. The blots were then stripped and reprobed to detect total rpS6, ERK (42/44), PI3Kp85, Akt and STAT3 respectively. NIH ImageJ quantitation data for the phosphorylated over total protein values are shown with bar graphs. B. Tris-DBA modulates expression of the Bcl-2 family proteins. Tris-DBA treated B-Cell lysates were also examined for the expression of the pro-apoptotic protein BIM and anti-apoptotic proteins Bcl-xL, and XIAP by Western blot analyses using specific antibodies. Actin was used as a loading control. Levels of anti-apoptotic proteins Bcl-xL and XIAP with or without Tris-DBA treatments are also quantitated using ImageJ software and presented after normalization with respective values of Actin by bar diagram. Data from 4 representative patients is shown. CLL patients are indicated by arbitrary numbers (P1 – P4).
Given the robust apoptosis we then attempted to determine if there were recurrent alterations of Bcl-2 family proteins when CLL B-cells were exposed to Tris-DBA. We therefore determined the impact of Tris-DBA on these Bcl-2 family member proteins in CLL B-cells after exposing the cells to a sub-lethal dose of Tris-DBA for 8 or 16 h. While we did not find any significant alteration in Mcl-1 or Bcl-2 levels (data not shown), there was a variable impact of Tris-DBA on XIAP and Bcl-xL anti-apoptotic protein levels in CLL B-cells as shown in Fig. 3B. In addition, Tris-DBA treatment also increased the expression of pro-apoptotic protein BIM (small and extra-large fragments) in CLL B-cells from one patient (P1) and inhibited the expression levels of Bcl-xL (P2–P4) and XIAP in CLL B-cells from P2–P4 (Fig. 3B).
Tris-DBA overcomes stromal protection of CLL B-cells
Previously, we and others have shown that stromal cells protect CLL B-cells from spontaneous and drug-induced apoptosis through soluble and contact-mediated interactions[26,27]. To further assess the ability of Tris-DBA to induce apoptosis in CLL B-cells we evaluated its impact on CLL B-cell apoptosis when co-cultured with stromal cells. For this set of experiments, CLL B-cells were cultured with human primary bone marrow stroma for 24 h, where Tris-DBA was added at increasing concentrations (0, 1, 2.5, 7.5 and 10 μM), and then CLL B-cells were harvested and analyzed for apoptosis induction by staining with annexin/PI. In these experiments it was evident that the presence of primary stromal cells could partially abrogate the impact of Tris-DBA in inducing apoptosis of CLL B-cells at lower doses but this abrogation was markedly reduced at the higher Tris-DBA doses [7.5 and 10 μM] (Fig. 4).
Figure 4. Tris-DBA overcomes stromal protection of CLL B-cells.
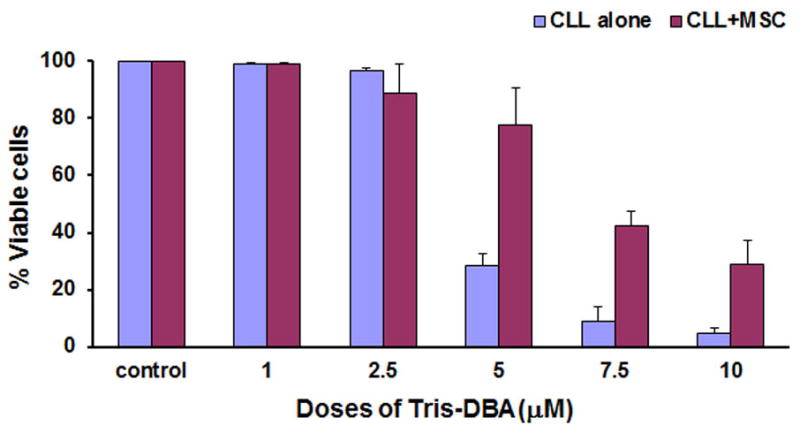
CLL B-cells (1.0 × 106/ml) were cultured with human primary bone marrow stroma for 24 h as described by us before, where Tris-DBA was then added at increasing concentrations (0, 1, 2.5, 7.5 and 10 μM), and then CLL B-cells were harvested and analyzed for apoptosis by staining with annexin/PI.
Combination treatment with Tris-DBA and chemotherapy increases apoptosis levels in CLL B-cells
To evaluate the combined effect of Tris-DBA with two commonly used chemotherapeutic agents in CLL (fludarabine and chlorambucil), we treated primary CLL B-cells (n = 6) with Tris-DBA (0.11–2.73 μM) in combination with either F-ara-A or chlorambucil at a ratio 1:1 or 1:5 respectively, or single agent alone. Following a 24-h treatment, cells were harvested and induction of apoptosis was assessed using annexin/PI staining. To more specifically evaluate for synergy and/or additive vs. antagonistic impact of Tris-DBA on CLL B-cells with either chemotherapeutic drug we then utilized a CalcuSyn software program as previously described[12,28]. CLL B-cells from the majority of CLL patients (5 of 6) tested for apoptosis sensitivity to the combination of F-ara-A and Tris-DBA had additive or synergistic combination index (CI) values (Fig. 5A), while the combination of chlorambucil and Tris-DBA resulted in around one third of CLL B-cell samples tested showing an antagonistic CI value (Fig. 5B)[15].
Figure 5A and B. Combination treatment with Tris-DBA and chemotherapy increases apoptosis in CLL B-cells.

To evaluate the combined effect of Tris-DBA with two commonly used chemotherapeutic agents in CLL (fludarabine and chlorambucil), we treated primary CLL B-cells (n = 6) with either Tris-DBA (0.11–2.73 μM) or F-ara-A (0.11–2.73 μM) or chlorambucil (0.5–10.0 μM) alone or in combination. Following 24-h treatment, cells were harvested and induction of cell death was assessed using annexin/PI staining. The data is presented for both figures as points above or below the line designated as 1.
Discussion
Our study had found that an organo-palladium, Tris-DBA, was very effective at inducing apoptosis in CLL B-cells as measured by positivity of the leukemic B-cells for annexin-V incorporation regardless of their IgVH mutation status. This finding for the first time shows that this drug can induce meaningful apoptosis in CLL B-cells and extends the spectrum of malignant cells known to be sensitive to Tris-DBA which now include both CLL and melanoma. Of interest when Tris-DBA was combined with a chemotherapeutic drug, fludarabine, used previously in the treatment of CLL we found that it could be synergistic or additive in the induction of CLL B-cell apoptosis. Finally, in vitro studies suggest that Tris-DBA targets the ribosomal protein S6 (rpS6) by reducing its phosphorylation levels. rpS6 is a component of the small 40S ribosomal subunit and has been associated with multiple physiological and pathophysiological functions[29–31]. It is a critical ribosomal protein subject to phosphorylation via a number of activation stimuli [32–34] and this latter event can play a critical regulatory role in multiple cellular processes mediated by rpS6.
Tris-DBA-mediated apoptosis induction in the leukemic B-cells was found to be robust with an LD50 less than 4.0 μM for CLL B-cells with a mutated IgVH status and less than 4.5 μM for CLL B-cells from individuals with an unmutated IgVH status. Of interest, we found that Tris-DBA mediated apoptosis induction in CLL B-cells involved caspase-dependent PARP cleavage. However, we were not successful in finding a consistent change in the Bcl-2 family of anti-apoptotic proteins in CLL B-cells exposed to Tris-DBA. Several anti-apoptotic proteins including Mcl-1, XIAP, and Bcl-2 are known to be critical to apoptotic resistance in CLL B-cells[35–37]. Tris-DBA induced apoptosis in CLL B-cells did not show any obvious changes in either Mcl-1 or Bcl-2, the most commonly affected anti-apoptotic proteins when CLL B-cells are exposed to known therapeutic agents. In addition, while we did find down regulation of both XIAP and Bcl-xL these were not consistent and one patient exhibited an increase in the pro-apoptotic protein BIM. This set of findings suggested to us an alternative mode of action for Tris-DBA on CLL B-cells that results in apoptosis.
To establish if alternate pathways were modulated in CLL B-cells by Tris-DBA we chose to examine the status of multiple signaling pathways in CLL B-cells exposed to the agent in vitro. However, our findings were unable to document any significant alteration of the Akt/mTOR/p70S6K, STAT3 signaling pathways in Tris-DBA treated CLL B-cells. This is in contrast with the prior in vitro findings in melanoma cell lines where Tris-DBA inhibited MAPK, Akt, STAT3 and phospho-S6 kinase[3]. Interestingly when we evaluated further downstream of the Akt/mTOR/p70S6K signaling axis, we did find a consistent inhibition of the P-rpS6 in the leukemic B-cells exposed to Tris-DBA. Activation of rpS6 can be induced as a downstream effect from either the Akt/mTOR/p70S6K pathway[29,38] or the MAPK pathway[39]. However it appears that inhibition of P-rpS6 in CLL B-cells by Tris-DBA is likely a direct effect on rpS6 as there was no consistent modulation of the upstream signaling effectors like mTOR or p70S6K and there was no impact of Tris-DBA on P-ERK1/2 at the time points when rpS6 was found to be inhibited. MAPK is known to activate 40S ribosomal protein S6 kinase (RSK), which in turn phosphorylates rpS6 however its phosphorylation can occur via other stimuli including tumor promoting factors and growth factors[38,39].
The exact function of rpS6 in CLL B-cells is not known but prior studies in other systems have shown multiple effects of this molecule on mRNA translation, cell proliferation, and glucose metabolism. We have found in an earlier study that Tris-DBA can modify VEGF secretion in melanoma cells[3]. In this study we did not evaluate this aspect in CLL B-cells however VEGF is known to be important in enhancing CLL B-cell survival. Our earlier studies found that Tris-DBA inhibited N-myristoyltransferase-1 in melanoma cells[3] but we were unable to find any significant inhibition of that enzyme or related members in CLL B-cells (data not shown)[29,40]. It is our intent to continue to study and define the exact mechanism(s) for leukemic B-cell apoptosis induction by this drug as it can pinpoint unique targets for therapy.
In summary, our finding that Tris-DBA induces killing in CLL B-cells is of clinical interest for a variety of reasons. First, we found brisk induction of apoptosis with a steep dose response curve for both mutated and the unmutated IgVH type B-cell clones. Secondly, we noted that Tris-DBA mediated apoptosis was only partially modified by co-culture with stromal cells and finally when combined with fludarabine we found significant and consistent enhancement of CLL B-cell apoptosis over the use of either drug alone. This report provides important information supporting the concept that Tris-DBA has potential for use in CLL therapy and that its use alone or in combination therapy should be tested in clinical trials.
References
- 1.Byrd JC, Furman RR, Coutre SE, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. The New England journal of medicine. 2013;369(1):32–42. doi: 10.1056/NEJMoa1215637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brown JR, Byrd JC, Coutre SE, et al. Idelalisib, an inhibitor of phosphatidylinositol 3-kinase p110delta, for relapsed/refractory chronic lymphocytic leukemia. Blood. 2014;123(22):3390–3397. doi: 10.1182/blood-2013-11-535047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bhandarkar SS, Bromberg J, Carrillo C, et al. Tris (dibenzylideneacetone) dipalladium, a N-myristoyltransferase-1 inhibitor, is effective against melanoma growth in vitro and in vivo. Clinical cancer research : an official journal of the American Association for Cancer Research. 2008;14(18):5743–5748. doi: 10.1158/1078-0432.CCR-08-0405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de la Puente P, Azab F, Muz B, Luderer M, Arbiser J, Azab AK. Tris DBA palladium overcomes hypoxia-mediated drug resistance in multiple myeloma. Leukemia & lymphoma. 2015:1–10. doi: 10.3109/10428194.2015.1099645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Palacios F, Abreu C, Prieto D, et al. Activation of the PI3K/AKT pathway by microRNA-22 results in CLL B-cell proliferation. Leukemia. 2015;29(1):115–125. doi: 10.1038/leu.2014.158. [DOI] [PubMed] [Google Scholar]
- 6.Negro R, Gobessi S, Longo PG, et al. Overexpression of the autoimmunity-associated phosphatase PTPN22 promotes survival of antigen-stimulated CLL cells by selectively activating AKT. Blood. 2012;119(26):6278–6287. doi: 10.1182/blood-2012-01-403162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee YK, Shanafelt TD, Bone ND, Strege AK, Jelinek DF, Kay NE. VEGF receptors on chronic lymphocytic leukemia (CLL) B cells interact with STAT 1 and 3: implication for apoptosis resistance. Leukemia. 2005;19(4):513–523. doi: 10.1038/sj.leu.2403667. [DOI] [PubMed] [Google Scholar]
- 8.Hazan-Halevy I, Harris D, Liu Z, et al. STAT3 is constitutively phosphorylated on serine 727 residues, binds DNA, and activates transcription in CLL cells. Blood. 2010;115(14):2852–2863. doi: 10.1182/blood-2009-10-230060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Frank DA, Mahajan S, Ritz J. B lymphocytes from patients with chronic lymphocytic leukemia contain signal transducer and activator of transcription (STAT) 1 and STAT3 constitutively phosphorylated on serine residues. The Journal of clinical investigation. 1997;100(12):3140–3148. doi: 10.1172/JCI119869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yoon JY, Ishdorj G, Graham BA, Johnston JB, Gibson SB. Valproic acid enhances fludarabine-induced apoptosis mediated by ROS and involving decreased AKT and ATM activation in B-cell-lymphoid neoplastic cells. Apoptosis : an international journal on programmed cell death. 2014;19(1):191–200. doi: 10.1007/s10495-013-0906-7. [DOI] [PubMed] [Google Scholar]
- 11.Bodo J, Zhao X, Sharma A, et al. The phosphatidylinositol 3-kinases (PI3K) inhibitor GS-1101 synergistically potentiates histone deacetylase inhibitor-induced proliferation inhibition and apoptosis through the inactivation of PI3K and extracellular signal-regulated kinase pathways. British journal of haematology. 2013;163(1):72–80. doi: 10.1111/bjh.12498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ghosh AK, Kay NE, Secreto CR, Shanafelt TD. Curcumin inhibits prosurvival pathways in chronic lymphocytic leukemia B cells and may overcome their stromal protection in combination with EGCG. Clinical cancer research : an official journal of the American Association for Cancer Research. 2009;15(4):1250–1258. doi: 10.1158/1078-0432.CCR-08-1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hallek M, Cheson BD, Catovsky D, et al. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood. 2008;111(12):5446–5456. doi: 10.1182/blood-2007-06-093906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ding W, Knox TR, Tschumper RC, et al. Platelet-derived growth factor (PDGF)-PDGF receptor interaction activates bone marrow-derived mesenchymal stromal cells derived from chronic lymphocytic leukemia: implications for an angiogenic switch. Blood. 2010;116(16):2984–2993. doi: 10.1182/blood-2010-02-269894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Advances in enzyme regulation. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 16.Chang TT, Chou TC. Rational approach to the clinical protocol design for drug combinations: a review. Acta paediatrica Taiwanica = Taiwan er ke yi xue hui za zhi. 2000;41(6):294–302. [PubMed] [Google Scholar]
- 17.Ghosh AK, Secreto C, Boysen J, et al. The novel receptor tyrosine kinase Axl is constitutively active in B-cell chronic lymphocytic leukemia and acts as a docking site of nonreceptor kinases: implications for therapy. Blood. 2011;117(6):1928–1937. doi: 10.1182/blood-2010-09-305649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boysen J, Sinha S, Price-Troska T, et al. The tumor suppressor axis p53/miR-34a regulates Axl expression in B-cell chronic lymphocytic leukemia: implications for therapy in p53-defective CLL patients. Leukemia. 2014;28(2):451–455. doi: 10.1038/leu.2013.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu SW, Andrabi SA, Wang H, et al. Apoptosis-inducing factor mediates poly(ADP-ribose) (PAR) polymer-induced cell death. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(48):18314–18319. doi: 10.1073/pnas.0606528103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ringshausen I, Schneller F, Bogner C, et al. Constitutively activated phosphatidylinositol-3 kinase (PI-3K) is involved in the defect of apoptosis in B-CLL: association with protein kinase Cdelta. Blood. 2002;100(10):3741–3748. doi: 10.1182/blood-2002-02-0539. [DOI] [PubMed] [Google Scholar]
- 21.Li P, Harris D, Liu Z, et al. STAT3-activated GM-CSFRalpha translocates to the nucleus and protects CLL cells from apoptosis. Molecular cancer research : MCR. 2014;12(9):1267–1282. doi: 10.1158/1541-7786.MCR-13-0652-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rozovski U, Wu JY, Harris DM, et al. Stimulation of the B-cell receptor activates the JAK2/STAT3 signaling pathway in chronic lymphocytic leukemia cells. Blood. 2014;123(24):3797–3802. doi: 10.1182/blood-2013-10-534073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ghosh AK, Shanafelt TD, Cimmino A, et al. Aberrant regulation of pVHL levels by microRNA promotes the HIF/VEGF axis in CLL B cells. Blood. 2009;113(22):5568–5574. doi: 10.1182/blood-2008-10-185686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cheng S, Ma J, Guo A, et al. BTK inhibition targets in vivo CLL proliferation through its effects on B-cell receptor signaling activity. Leukemia. 2014;28(3):649–657. doi: 10.1038/leu.2013.358. [DOI] [PubMed] [Google Scholar]
- 25.Ding W, Shanafelt TD, Lesnick CE, et al. Akt inhibitor MK2206 selectively targets CLL B-cell receptor induced cytokines, mobilizes lymphocytes and synergizes with bendamustine to induce CLL apoptosis. British journal of haematology. 2014;164(1):146–150. doi: 10.1111/bjh.12564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kay NE, Shanafelt TD, Strege AK, Lee YK, Bone ND, Raza A. Bone biopsy derived marrow stromal elements rescue chronic lymphocytic leukemia B-cells from spontaneous and drug induced cell death and facilitates an “angiogenic switch”. Leukemia research. 2007;31(7):899–906. doi: 10.1016/j.leukres.2006.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fiorcari S, Brown WS, McIntyre BW, et al. The PI3-kinase delta inhibitor idelalisib (GS-1101) targets integrin-mediated adhesion of chronic lymphocytic leukemia (CLL) cell to endothelial and marrow stromal cells. PloS one. 2013;8(12):e83830. doi: 10.1371/journal.pone.0083830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sinha S, Boysen J, Nelson M, et al. Targeted Axl Inhibition Primes Chronic Lymphocytic Leukemia B Cells to Apoptosis and Shows Synergistic/Additive Effects in Combination with BTK Inhibitors. Clinical cancer research : an official journal of the American Association for Cancer Research. 2015;21(9):2115–2126. doi: 10.1158/1078-0432.CCR-14-1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Magnuson B, Ekim B, Fingar DC. Regulation and function of ribosomal protein S6 kinase (S6K) within mTOR signalling networks. The Biochemical journal. 2012;441(1):1–21. doi: 10.1042/BJ20110892. [DOI] [PubMed] [Google Scholar]
- 30.Kim SH, Jang YH, Chau GC, Pyo S, Um SH. Prognostic significance and function of phosphorylated ribosomal protein S6 in esophageal squamous cell carcinoma. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc. 2013;26(3):327–335. doi: 10.1038/modpathol.2012.161. [DOI] [PubMed] [Google Scholar]
- 31.Hu LY, Sun ZG, Wen YM, et al. ATP-mediated protein kinase B Akt/mammalian target of rapamycin mTOR/p70 ribosomal S6 protein p70S6 kinase signaling pathway activation promotes improvement of locomotor function after spinal cord injury in rats. Neuroscience. 2010;169(3):1046–1062. doi: 10.1016/j.neuroscience.2010.05.046. [DOI] [PubMed] [Google Scholar]
- 32.Kim D, Akcakanat A, Singh G, Sharma C, Meric-Bernstam F. Regulation and localization of ribosomal protein S6 kinase 1 isoforms. Growth factors. 2009;27(1):12–21. doi: 10.1080/08977190802556986. [DOI] [PubMed] [Google Scholar]
- 33.Wang ML, Panasyuk G, Gwalter J, et al. Regulation of ribosomal protein S6 kinases by ubiquitination. Biochemical and biophysical research communications. 2008;369(2):382–387. doi: 10.1016/j.bbrc.2008.02.032. [DOI] [PubMed] [Google Scholar]
- 34.Jastrzebski K, Hannan KM, Tchoubrieva EB, Hannan RD, Pearson RB. Coordinate regulation of ribosome biogenesis and function by the ribosomal protein S6 kinase, a key mediator of mTOR function. Growth factors. 2007;25(4):209–226. doi: 10.1080/08977190701779101. [DOI] [PubMed] [Google Scholar]
- 35.Cervantes-Gomez F, Lamothe B, Woyach JA, et al. Pharmacological and Protein Profiling Suggests Venetoclax (ABT-199) as Optimal Partner with Ibrutinib in Chronic Lymphocytic Leukemia. Clinical cancer research : an official journal of the American Association for Cancer Research. 2015;21(16):3705–3715. doi: 10.1158/1078-0432.CCR-14-2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Choudhary GS, Al-Harbi S, Mazumder S, et al. MCL-1 and BCL-xL-dependent resistance to the BCL-2 inhibitor ABT-199 can be overcome by preventing PI3K/AKT/mTOR activation in lymphoid malignancies. Cell death & disease. 2015;6:e1593. doi: 10.1038/cddis.2014.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhuang J, Laing N, Oates M, Lin K, Johnson G, Pettitt AR. Selective IAP inhibition results in sensitization of unstimulated but not CD40-stimulated chronic lymphocytic leukaemia cells to TRAIL-induced apoptosis. Pharmacology research & perspectives. 2014;2(6):e00081. doi: 10.1002/prp2.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Soliman GA. The mammalian target of rapamycin signaling network and gene regulation. Current opinion in lipidology. 2005;16(3):317–323. doi: 10.1097/01.mol.0000169352.35642.06. [DOI] [PubMed] [Google Scholar]
- 39.Roux PP, Shahbazian D, Vu H, et al. RAS/ERK signaling promotes site-specific ribosomal protein S6 phosphorylation via RSK and stimulates cap-dependent translation. The Journal of biological chemistry. 2007;282(19):14056–14064. doi: 10.1074/jbc.M700906200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim YK, Kim S, Shin YJ, et al. Ribosomal protein S6, a target of rapamycin, is involved in the regulation of rRNA genes by possible epigenetic changes in Arabidopsis. The Journal of biological chemistry. 2014;289(7):3901–3912. doi: 10.1074/jbc.M113.515015. [DOI] [PMC free article] [PubMed] [Google Scholar]