

Abstract
Objective
Chronic therapy with synthetic glucocorticoids has been associated with cardiovascular side effects, although differential interindividual susceptibility to glucocorticoids has been observed. The objective of this study was to identify the molecular mechanisms leading to differential glucocorticoid responses in endothelial cells.
Approach and Results
We tested the sensitivity of 42 human umbilical vein endothelial cells (HUVECs) to dexamethasone as determined by changes in gene expression, promoter transactivation, and procoagulant activity. We identified that 16 HUVECs were sensitive in every test, 14 HUVECs were sensitive in at least 1 test and 12 HUVECs were resistant in every test to dexamethasone. Nuclear translocation assays revealed that Dex-sensitive HUVECs have higher basal and Dex-stimulated levels of nuclear glucocorticoid receptor compared with Dex-resistant HUVECs. Cycloheximide assays revealed that Dex-resistant HUVECs have significantly shorter glucocorticoid receptor protein half-lives than Dex-sensitive HUVECs. Dex-resistant HUVECs have a stronger interaction of glucocorticoid receptor with the proteasomal recruiting protein, BCL2-associated athanogene 1 (BAG1), as shown by immunoprecipitation assays. Silencing BAG1 expression increased Dex-sensitivity in resistant HUVECs, whereas BAG1 overexpression decreased Dex-sensitivity in sensitive HUVECs. Finally, Dex-resistant HUVECs presented higher BAG1 expression than Dex-sensitive HUVECs.
Conclusions
In vitro endothelial sensitivity to Dex varies within individuals and is inversely proportional to BAG1 protein expression and glucocorticoid receptor protein turnover.
Keywords: coagulation, endothelium, glucocorticoids, sensitivity
Glucocorticoids are potent therapeutic agents used for reducing inflammation by targeting inflammatory gene expression in leukocytes.1,2 However, adverse side effects of GCs limit the use of these agents and often require further treatment. GCs induce short-term side effects in the cardiovascular system of fetuses, neonates, and adults that include hypertension, dyslipidemia, and thrombosis.3–8 Chronic synthetic GC treatment has been correlated with increased cardiovascular risk of heart failure and myocardial infarction.3–7 In addition, chronic GC therapy induces similar effects as observed in Cushing’s syndrome and the metabolic syndrome, including hyperglycemia, decreased insulin sensitivity, hypertension, and obesity.3–8 Nevertheless, the mechanisms responsible for GC induction of cardiovascular side effects have not been fully elucidated and conflicting results suggest significant human variability in response to GCs. Furthermore, GC biological effects vary according to the site of action (vascular smooth muscle, endothelium, myocardium, and macrophages). For instance, GCs induce strong anti-inflammatory profiles in immune cells while inducing a proatherogenic and contractile phenotype in vascular smooth muscle and endothelial cells.3–8 Various in vivo studies on GC-induced hypertension have shown an association with decreased levels of the vasodilatory nitric oxide partly attributable to decreased expression of endothelial nitric oxide synthase (eNOS).9,10 GCs also decrease vasodilatory prostacyclin levels and increase vasocontractile endothelin-1 levels.5–8 Although GCs decrease nicotinamide adenine dinucleotide phosphate oxidase expression in neutrophils,1,11 they increase it in endothelial cells resulting in oxidative stress, quenching of NO, and further endothelial dysfunction.5–8,12 Microarray studies have indicated that GCs upregulate genes involved in atherogenesis and hemostasis such as the cell adhesion molecules, prothrombotic genes such as factor VIII, and antifibrinolytic genes such as the plasminogen activator inhibitor-1 (PAI1).13 More importantly, GCs synergize with other inflammatory mediators in upregulating various cardiovascular risk markers, such as PAI1.14,15
GCs mediate their biological effects by the ubiquitously expressed glucocorticoid receptor (GR).1,2,16–22 Alternative splicing produces 2 main isoforms (α and β), where GRβ is localized in the nucleus and has a dominant negative effect on GRα through the formation of GRα/GRβ heterodimers.18–21 GRα is the biologically relevant isoform capable of binding ligand.19–21 GRα protein synthesis, signal transduction, and degradation steps are guided by a variety of molecular chaperones that regulate its activity.22,23 Bound to HSP70, GRα will undergo conformational changes that result in a folded conformation with low affinity for the substrate. The immature ADP-HSP70-GR complex can bind to the BCL2-associated athanogene 1 (BAG1), repressing GRα transactivation.24–26 HSP90 binding to the GR complex leads to the opening of the GR hormone–binding cleft, which allows access to GC ligands. After GC binds to the mature GR form, active GR complexes dissociate from the HSP70–HSP90 chaperone complex and translocate to the nucleus.16–21 Once inside the nucleus, the active GRα regulates transcription of numerous genes and is itself regulated by the nuclear chaperone system.21–23 For instance, nuclear BAG1 can compete with GR for DNA binding or interact directly with GR to stimulate proteasomal degradation.24–26
Various modes of transcription regulation by GR complexes have been described.27,28 First, positive regulation of target genes, or transactivation, is mediated by the binding of the GR dimers to the glucocorticoid response element (GRE) domains of enhancer–promoters and thereby upregulating transcription of target genes such as the antifibrinolytic gene PAI1.29 Negative GREs have also been described for some genes where GR dimers bind and decrease transcription.27,28 However, most of the anti-inflammatory effects of GCs have been proposed to be mediated via transrepression.16–21,30 Transrepression does not require GR dimer binding to DNA, and it has been proposed to occur by interaction with other transcription factors, as reported for glucocorticoid-mediated downregulation of eNOS.9,10,30
It is well known that different tissues respond to cortisol and synthetic GCs with different sensitivity.17,18,31–35 In addition to differential tissue sensitivity, GC sensitivity, measured by a dexamethasone suppression test of hypothalamic control of cortisol secretion, varies greatly between individuals.4–6 However, within individuals, GC sensitivity is rather stable. This suggests that, in humans, a threshold for glucocorticoid sensitivity might be genetically determined. Endogenous tissue sensitivity to cortisol has been reported to vary according to GR polymorphisms and isoform expression, differential cortisol metabolism, expression of downstream signaling effectors, and regulation of GR chaperones.17,18,31–37 To date, the molecular mechanisms responsible for interindividual variability in response to GCs remain incompletely understood.
The objective of this study was to elucidate the molecular mechanisms responsible for human variability in endothelial sensitivity to GCs. Our approach was to examine the in vitro sensitivity of human umbilical vein endothelial cells (HUVECs) to Dex, as determined by gene expression regulation and procoagulant activity. We investigated the molecular regulation of endothelial GR, including GR isoform expression, nuclear translocation, and interaction with the chaperone system. We found significant differences in endothelial sensitivity to Dex that are attributable to differential regulation of the GR protein.
Materials and Methods
Materials and Methods are available in the online-only Supplement.
Results
Differential Sensitivity of HUVECs to Dex-Mediated Gene Regulation and Procoagulant Activity
We hypothesized that endothelial sensitivity to glucocorticoids is set by genetic and epigenetic mechanisms before birth. To test this hypothesis, we first examined the response of 42 HUVECs to Dex-mediated PAI1 upregulation and eNOS downregulation. These 2 genes were chosen because of their relevance to cardiovascular disease and to glucocorticoid regulation by various transcriptional and post-transcriptional mechanisms.38–43 Sensitivity was defined by Dex-mediated gene expression regulation in a dose-dependent manner by ≥50% of basal levels. As shown in Figure 1, of the 42 HUVECs tested, 16 HUVECs responded to Dex-mediated regulation of both PAI1 and eNOS, 7 HUVECs responded to Dex-mediated PAI1 upregulation only, 7 HUVECs responded to Dex-mediated eNOS downregulation only, and 12 HUVECs did not respond to either test. The regulation of PAI1 and eNOS by Dex showed a strong correlation between mRNA and protein levels indicating the likelihood of Dex-mediated transcriptional mechanisms of action. Of interest, although all HUVECs originated from healthy pregnancies, Dex-sensitivity was associated with higher maternal body mass index and blood pressure (Table II in the online-only Data Supplement).
Figure 1.
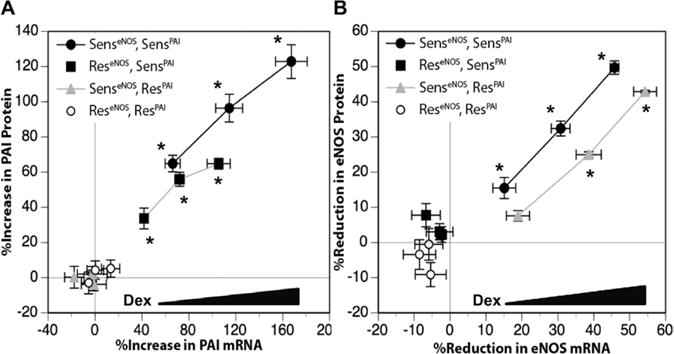
Differential dexamethasone (Dex)-sensitivity in human umbilical vein endothelial cells (HUVECs) as determined by gene expression regulation. Confluent and quiescent HUVECs were treated with various doses of Dex (0.1, 0.33, and 1 μmol/L) for 24 h and then analyzed for plasminogen activator inhibitor1 (PAI1) and endothelial nitric oxide synthase (eNOS) mRNA and protein as described under Methods in the online-only Data Supplement. A, Differences in Dex-mediated PAI1 upregulation. B, Differences in Dex-mediated eNOS downregulation. HUVECs were assigned to 4 groups, Dex-sensitive for both eNOS and PAI-1 (SenseNOSSensPAI1, n=16), Dex-sensitive for PAI1 only (ReseNOSSensPAI1, n=7), Dex-sensitive for eNOS only (SenseNOSResPAI1, n=7), and Dex-resistant for both PAI1 and eNOS (ReseNOSResPAI-1, n=12). Data points represent the correlation of mRNA and protein for each dose of Dex. Data are shown as the mean±SEM for each group. *P<0.05, basal vs Dex for both mRNA and protein.
Dex-sensitivity differences were confirmed by additional in vitro assays, which are shown in Tables III through V in the online-only Data Supplement. First, we observed similar differences in Dex-mediated regulation of other genes, including upregulation of Factor VIII, von Willebrand factor, and intercellular adhesion molecule-1; and downregulation of vascular endothelial growth factor A. We also evaluated the ability of Dex to stimulate a minimal GRE-luciferase construct. As expected, Dex-sensitive cells stimulated GRE-mediated transcriptional activation in a stronger manner than Dex-resistant cells. Finally, we performed an in vitro procoagulant activity assay and determined that Dex decreased the activated partial thromboplastin time of HUVECs in Dex-sensitive, but not in Dex-resistant HUVECs. Altogether, we were able to characterize our 42 HUVECs in terms of in vitro sensitivity to Dex to investigate the molecular basis of differential sensitivity to glucocorticoids.
Differences in Dex-Sensitivity Are Attributable to Increased GR Protein Turnover
Although many factors can affect tissue sensitivity to glucocorticoids, it is well accepted that a key factor is the availability and activation of the GRα protein.16–28 We analyzed the expression levels of GR isoforms α and β. Real-time reverse transcription-polymerase chain reaction assays revealed that GRα is the main isoform expressed in HUVECs (≈99% of total GR), and that there were no significant differences in either GRα or GRβ mRNA expression between Dex-sensitive and Dex-resistant HUVECs (Table VI in the online-only Data Supplement). Immunoblotting assays revealed that the major isoforms of GRα in HUVECs are the A (94 kDa), B (90 kDa), and C (82 kDa) isoforms; the GRαD isoform and GRβ isoforms were undetectable. There were significant differences between Dex-sensitive and Dex-resistant HUVECs in GRα protein expression (Figure 2A). Although Dex decreased GRα protein levels in both groups, basal and Dex-stimulated GRα protein levels were significantly higher in Dex-sensitive HUVECs as compared with Dex-resistant HUVECs (Figure 2A). Next, we studied the degradation rates of GRα protein. As reported for other cell types, Dex decreased the t1/2 of GRα protein in both Dex-sensitive and Dex-resistant HUVECs (Figure 2B). However, Dex-resistant HUVECs have a shorter basal and Dex-stimulated GRα protein t1/2 compared with Dex-sensitive HUVECs (Figure 2B). The basal t1/2 of GRα protein was 61±18.3 hours in Dex-sensitive HUVECs and 37.6±17.1 hours in Dex-resistant HUVECs (P<0.05). The Dex-stimulated t1/2 of GRα protein was 14.5±3.0 hours in Dex-sensitive cells and 8.2±1.6 hours in Dex-resistant cells (P<0.05).
Figure 2.
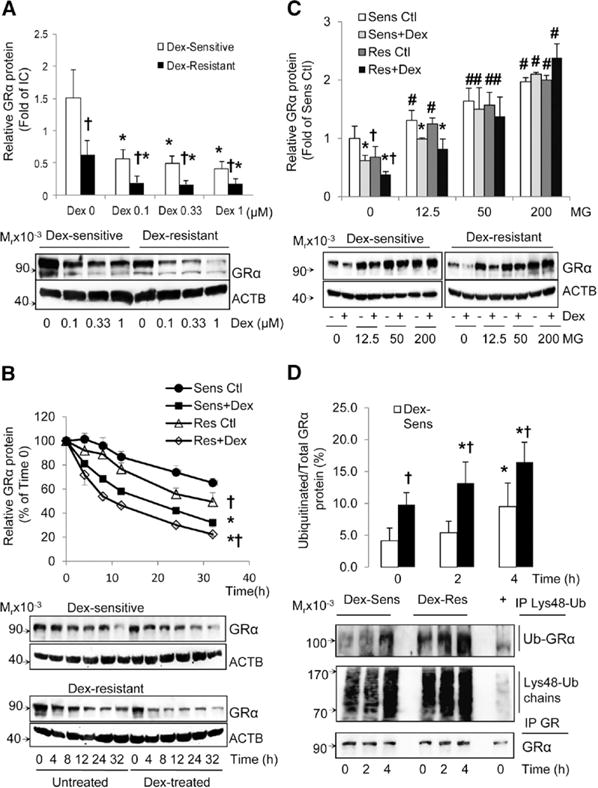
Dexamethasone (Dex)-sensitivity is linked to decreased glucocorticoid receptor α (GRα) protein turnover by the proteasomal system. A, Dex dose-dependent decreases of GRα protein levels. Dex-sensitive (n=16) and Dex-resistant (n=12) HUVECs were treated or not with Dex for 24 h and GRα and β-Actin (ACTB) protein analyzed by immunoblot. B, Time-dependent degradation of GRα protein. Dex-sensitive HUVECs (n=10) and Dex-resistant HUVECs (n=10) were treated or not with Dex (1 μmol/L) and then exposed to cycloheximide (10 μg/mL) and GRα protein analyzed at various time points via immunoblot. C, Role of the proteasomal system in GRα protein degradation. Dex-sensitive HUVECs (n=5) and Dex-resistant HUVECs (n=5) were pretreated with the proteasome inhibitor MG132 at various doses for 1 h before treating with Dex (1 μmol/L) for 18 h and GRα protein levels were analyzed by immunoblot. D, Analysis of ubiquitinated GRα protein levels. Dex-sensitive and Dex-resistant HUVECs (n=5 each) were pretreated with MG132 (200 nmol/L) for 1 h, then treated with Dex (1 μmol/L) for 0, 2, and 4 h and lysine-48 polyubiquitin chains immunoprecipitated as described in Methods in the online-only Data Supplement. All data represent mean±SD. *P<0.05, basal vs Dex-treated; †P<0.05, Dex-sensitive vs Dex-resistant cells; and #P<0.05, basal vs MG132-treated.
It is well known that GR protein undergoes ubiquitination and proteasomal degradation, and this event is stimulated by GR activation.35,37 To determine whether Dex-sensitivity correlated with GR proteasomal degradation, we used the proteasomal inhibitor MG132. We found that MG132 dose dependently increased both basal and Dex-stimulated GRα protein levels in all HUVECs tested and abolished the differences in GRα protein levels between Dex-sensitive and Dex-resistant HUVECs (Figure 2C). MG132 also blocked the effect of Dex on GRα protein degradation in all HUVECs (Figure 2C). Finally, studies on the levels of GRα ubiquitination using an antibody specific for lysine-48 polyubiquitin chains44 revealed that Dex-resistant HUVECs have higher basal and Dex-stimulated GRα protein ubiquitination than Dex-sensitive HUVECs (Figure 2D). Only 4.1% to 9.5% of total GRα was ubiquitinated in untreated cells, and Dex-resistant cells showed 2.4-fold higher basal GRα ubiquitination and increased synthesis of lysine-48 polyubiquitin chains than Dex-sensitive cells (Figure 2D). These data strongly indicate that Dex resistance in HUVECs is associated with increased GRα protein turnover by the ubiquitin-proteasomal system.
Differences in GR Protein Degradation Rates Are Linked to Differential BAG1 Chaperoning
GRα protein is tightly controlled by a complex chaperone system that determines the degradation, maturation, and transcriptional activation of GR complexes.22–26 We performed immunoprecipitation assays to study the protein–protein interactions of GRα with the chaperone system (Figure 3A; Figure IIA in the online-only Data Supplement). GRα basal interaction with HSP90, HSP70, and FKBP51 decreased on Dex stimulation and nuclear translocation, whereas FKBP52–GRα interaction increased in both Dex-sensitive and Dex-resistant HUVECs (Figure IIA in the online-only Data Supplement). We observed a 2- to 3-fold higher basal and Dex-stimulated interaction of GRα with BAG1 isoforms in Dex-resistant cells compared with Dex-sensitive cells (Figure 3A and 3B). GRα protein interacted with the 3 endogenous isoforms of BAG1, in the order of S>M=L (Figure 3A and 3B). We also found increased recruitment of the C terminus of HSC70-interacting protein (CHIP) to the GR complexes after Dex stimulation in all HUVECs, and this interaction was stronger in Dex-resistant HUVECs compared with Dex-sensitive HUVECs (Figure 3A and 3C).
Figure 3.
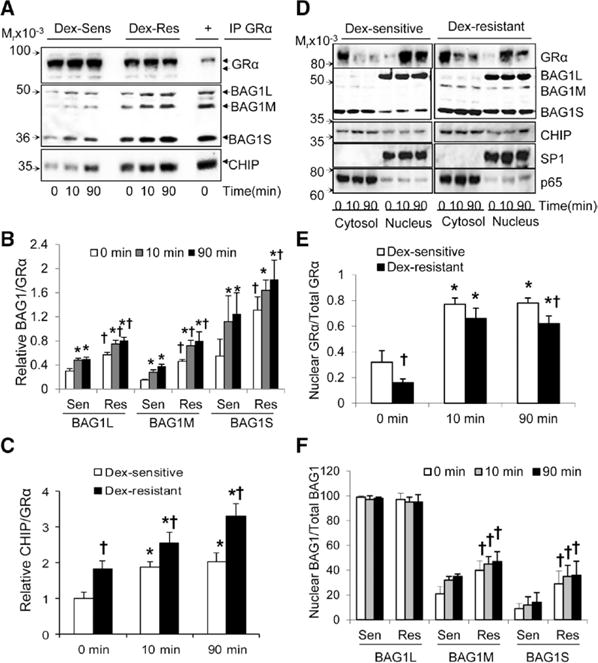
Dexamethasone (Dex) resistance is associated with higher GRα interaction with BCL2-associated athanogene 1 (BAG1) and increased nuclear BAG1 isoform levels. A to C, Analysis of GRα interactions with the chaperone system. Confluent and quiescent human umbilical vein endothelial cells (HUVECs) were treated with Dex (1 μmol/L) for 10 and 90 min, and GRα was immunoprecipitated as described under Methods in the online-only Data supplement. A, Representative immunoblots for GRα, BAG1, and C terminus of HSC70-interacting protein (CHIP). B to C, Bar graphs show the relative ratios of BAG1 isoforms and CHIP bound to GRα. D to F, Analysis of GRα, BAG1, and CHIP cytosolic and nuclear abundance. Cells were treated as described in (A to C), and cytosolic and nuclear proteins were analyzed by immunoblot. D, Representative immunoblots for GRα, BAG1, and CHIP. The specificity protein 1 (SP1) and the nuclear factor-kappa B subunit p65 were used as the positive nuclear and cytosolic markers, respectively. E to F, Bar graphs represent the percentage of nuclear protein over total levels (cytosol+nucleus) for GRα and BAG1. *P<0.05 vs basal levels at time 0 and †P<0.05, Dex-sensitive vs Dex-resistant.
We then investigated the basal and Dex-stimulated expression and subcellular localization of GRα and its chaperones. GRα translocated rapidly into the nucleus on ligand binding in both Dex-sensitive and Dex-resistant cells (Figure 3D and 3E). However, the levels of nuclear GRα protein were higher in Dex-sensitive compared with Dex-resistant HUVECs (Figure 3D and 3E). We found that only BAG1, CHIP, and to a lesser extent FKBP52 and HSP70 were significantly present in the nucleus after Dex stimulation and GRα nuclear translocation, whereas the other chaperones reside mainly in the cytosol (Figure 3D; and Figure IIB in the online-only Data Supplement). Furthermore, Dex-resistant HUVECs show higher levels of all BAG1 isoforms, in particular of nuclear BAG1M and BAG1S (Figure 3D and 3F). There were no significant differences in the cytosolic/nuclear levels of the remaining GR chaperones (Figure 3D; Figure IIB in the online-only Data Supplement). Interestingly, the differences in GR and BAG1 isoform expression were only observed in confluent, but not in proliferating HUVECs (Figure IIC in the online-only Data Supplement). BAG1L and BAG1M expression was similar between Dex-sensitive and Dex-resistant HUVECs in subconfluent cultures (30%–60% confluence), but their expression decreased at confluence in Dex-sensitive HUVECs, whereas the opposite occurred in Dex-resistant HUVECs (Figure IIC in the online-only Data Supplement). Therefore, endogenous expression of BAG1 isoforms shows an inverse correlation with GRα expression, with higher GRα protein levels present in cells with lower BAG1 protein expression.
Overexpression of BAG1L, BAG1M, and BAG1S isoforms significantly decreased basal and Dex-stimulated GRα protein levels in Dex-sensitive cells (Figure 4A and 4B). All BAG1 isoforms significantly decreased Dex-mediated upregulation of PAI1 in Dex-sensitive cells without affecting basal levels of PAI1 protein (Figure 4A and 4C). Furthermore, BAG1 overexpression also blocked Dex-mediated upregulation of FKBP51, another GR-sensitive gene (Figure IIIA and IIIB in the online-only Data Supplement). Finally, overexpression of BAG1 isoforms decreased basal and Dex-stimulated GRE-mediated transactivation as shown by luciferase assays in the order of BAG1L>BAG1M>BAG1S, but did not completely block Dex-mediated transactivation (Figure 4D). Of note, BAG1 isoform overexpression did not significantly increase the expression of other GR chaperones, such as CHIP (Figure IIIA in the online-only Data Supplement).
Figure 4.
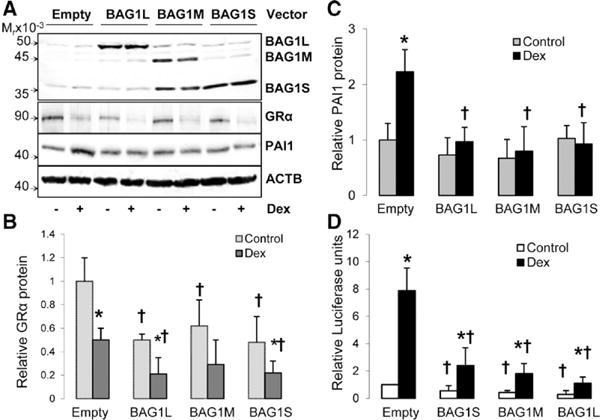
BCL2-associated athanogene 1 (BAG1) overexpression decreases dexamethasone (Dex)-sensitivity in Dex-sensitive human umbilical vein endothelial cells (HUVECs). Dex-sensitive cells were transfected with BAG1L, BAG1M, or BAG1S mammalian expression vectors and then treated with Dex (1 μmol/L) or solvent. The expression of glucocorticoid receptor α (GRα) plasminogen activator inhibitor1 (PAI1), and β-Actin (ACTB) proteins was analyzed by immunoblot. A, Representative immunoblots for BAG1, GRα, PAI1, and ACTB. B, Bar graph for GRα protein expression. C, Bar graph for PAI-1 protein expression. D, Bar graph for glucocorticoid response element–mediated transactivation. Bars represent the mean±SD (n=6). *P<0.05, basal vs Dex-treated and †P<0.05, empty vector vs BAG1 vector.
We then tested the effect of BAG1 silencing on Dex-sensitivity. BAG1 expression silencing using Bag1ORF siRNA decreased the expression of all isoforms with higher silencing of BAG1M and BAG1S (Figure 5A). BAG1 silencing significantly increased basal and Dex-stimulated levels of GRα protein (Figure 5A and 5B), Dex-mediated PAI-1 upregulation (Figure 5A and 5C), and basal and Dex-stimulated GRE transactivation (Figure 5D) in Dex-resistant cells. Bag1ORF siRNA did not silence the expression of other GR chaperones; however, it increased the expression of HSP70 and the Dex-mediated upregulation of FKBP51 (Figure IIIC–IIIE in the online-only Data Supplement).
Figure 5.
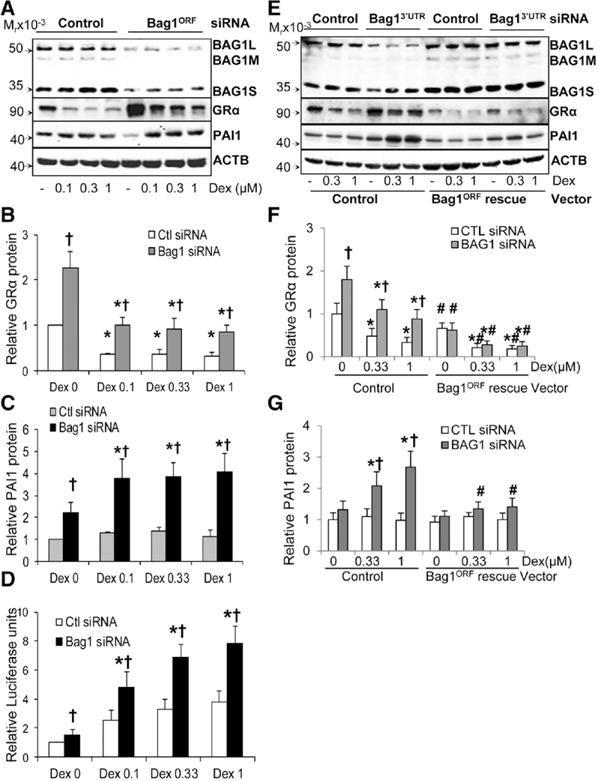
BCL2-associated athanogene 1 (BAG1) silencing increases dexamethasone (Dex)-sensitivity in Dex-resistant human umbilical vein endothelial cells (HUVECs). A to D, Determination of the effect of Bag1ORF silencing on Dex-sensitivity. Dex-resistant HUVECs (n=6) were transfected with Bag1ORF siRNA or control siRNA and then treated with Dex as described under Methods in the online-only Data Supplement. E to G, Rescue siRNA assays. Dex-resistant (n=4) cells were transfected with rescue vectors to generate stable Bag1ORF-expressing cells, and then used to study the effect of Bag13′UTR siRNA on Dex-sensitivity. A, E, Representative immunoblots for BAG1, glucocorticoid receptor α (GRα), plasminogen activator inhibitor-1 (PAI-1), and β-Actin (ACTB). B, F, Bar graph for GRα protein expression. C, G, Bar graph for PAI-1 protein. D, Bar graph for glucocorticoid response element–mediated transactivation. Bars represent the mean±SD. *P<0.05, basal vs Dex-treated; †P<0.05, control siRNA vs Bag1 siRNA; and #P<0.05 control vector vs Bag1ORF rescue vector.
To confirm our overexpression and silencing studies, we performed rescue siRNA assays. Dex-resistant HUVECs were manipulated to stably express a Bag1ORF rescue gene. Further exposure to Bag13′UTR siRNAs led to decreased endogenous BAG1 expression (in particular of BAG1L) in control cells, but not in Bag1ORF rescue expressing cells (Figure 5E). We then confirmed that BAG1 silencing via Bag13′UTR siRNA increased GRα protein expression and Dex-sensitivity (as shown by Dex-mediated upregulation of PAI-1) in control cells, but not in Bag1ORF rescued cells (Figure 5E and 5G).
Finally, we studied BAG1 and GRα protein expression in human coronary artery endothelial cells (HCAEC), HCA smooth muscle cells (HCASMC), human cardiomyocytes (HCM), and human monocytic leukemia cell line THP1. Human coronary vascular cells and cardiomyocytes express significantly higher levels of GRα protein compared with HUVECs (Figure 6A and 6B). However, Dex stimulation only decreased GRα protein expression in HCAECs and HCASMCs, but not in HCM and THP1 cells. Furthermore, HCAECs have similar expression of BAG1 isoforms (S>L>M) to HUVECs, whereas HCASMC and HCM express mostly BAG1S, and THP1 cells only express BAG1L (Figure 6A and 6C). In addition, primary human vascular cells of various origins express all the components of the GR chaperone system, although significant differences were observed in the expression of HSP70, HSP90, and FKBP51 (Figure IVA–IVD in the online-only Data Supplement). For instance, HCAECs, HCASMCs, and HCMs expressed higher levels of HSP70 than HUVECs (Figure IVB in the online-only Data Supplement). HCAECs, HCMs, and THP1 cells expressed lower levels of HSP90 than HUVECs (Figure IVC in the online-only Data Supplement). Finally, HCASMC and HCM expressed about 4-fold higher levels of FKBP51 compared with HUVECs (Figure IVD in the online-only Data Supplement).
Figure 6.
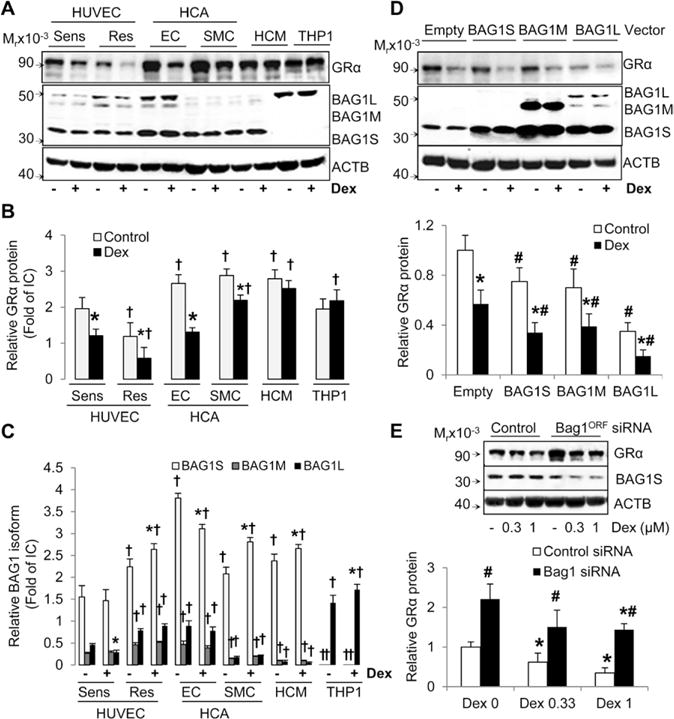
BCL2-associated athanogene 1 (BAG1) and glucocorticoid receptor α (GRα) expression studies in human vascular and immune cells. HCAEC, HCASMC, HCM, and THP1 cells were cultured and treated with dexamethasone (Dex) as described under Methods in the online-only Data Supplement. A to C, Expression of glucocorticoid receptor α (GRα) and BAG1 isoforms in basal and Dex (1 μmol/L)-treated cells. A, Representative immunoblots. B, Bar graph of relative GRα protein levels. C, Bar graph of relative protein levels of BAG1 isoforms. D, BAG1 overexpression decreases GRα protein expression in HCASMCs. Representative immunoblots and bar graph of relative GRα protein levels. E, Bag1ORF silencing increases GRα protein expression in HCASMCs. Representative immunoblots and bar graph of relative GRα protein levels. Primary cells were tested at different passage number for a total number of 3. Bars represent the mean±SD (n=3). *P<0.05, untreated vs Dex-treated; †P<0.05 vs basal levels in Dex-sensitive human umbilical vein endothelial cells; and #P<0.05, control vs Bag1 vector/siRNA.
To further investigate the role of BAG1 in GRα protein expression in another primary vascular cell type, we manipulated the expression of BAG1 in HCASMCs (Figure 6D and 6E). Overexpression of all BAG1 isoforms decreased the levels of basal and Dex-stimulated GRα protein in HCASMCs; however, in contrast with HUVECs, BAG1L induced the strongest effect (Figure 6D). Silencing of BAG1S significantly increased the protein levels of GRα in HCASMCs (Figure 6E). Altogether, our data indicate that BAG1 is an important regulator of GRα protein turnover in HUVECs, and other vascular cells, and that significant differences in BAG1–GR interactions are correlated with in vitro sensitivity to GCs.
Discussion
In the cardiovascular system, endogenous and synthetic glucocorticoids have been associated with adverse cardiovascular events, in particular myocardial infarct and heart failure.3–13 Nevertheless, the molecular mechanisms involved in the development of cardiovascular side effects have only been partially elucidated. The complexity of interindividual variability in GC responses is magnified because of genetic variability in both GC pathway–related genes and target genes. Studies on GC sensitivity variability have identified multiple factors as modifiers of GC response, including (1) regulation of GC metabolizing enzymes, (2) GR polymorphisms, (3) GR isoform expression regulation, (4) inflammatory mediator levels and polymorphisms, (5) coactivator and corepressor availability, and (5) differences in the GR chaperoning system.5–9,31–33 To unravel the mechanisms within the GC signaling pathway that modulate endothelial responses to GCs, we investigated Dex-sensitivity in a panel of 42 HUVECs. Our studies demonstrated significant variability in HUVEC response to Dex, with about one fourth (12/42) demonstrating a generalized lack of sensitivity to Dex, and one third (16/42) demonstrating generalized sensitivity to Dex in a dose-dependent manner. Our results were reproducible over the span of various passages and various in vitro assays, suggesting that individual endothelial sensitivity to GCs is determined by genetic and epigenetic mechanisms. Our results could explain the confounding results from multiple research groups showing beneficial, adverse, or no effect of GCs on cardiovascular disease.5–8
A key factor in determining GC sensitivity is the availability and regulation of the GRα protein.22–28 Dose–response studies demonstrated that all HUVECs were sensitive to Dex-mediated decreases in GRα protein levels; however, the sensitivity to Dex-mediated gene expression regulation correlated with basal and stimulated levels of GRα protein. These differences are attributable to faster GRα protein turnover rates in Dex-resistant than Dex-sensitive HUVECs. Increased GRα turnover in Dex-resistant HUVECs correlated with increased levels of BAG1 L, M, and S in the nucleus, increased GRα interaction with both BAG1 and CHIP, and increased GRα ubiquitination. Silencing of BAG1 expression restored Dex-sensitivity in Dex-resistant HUVECs, whereas BAG1 overexpression decreased Dex-sensitivity in Dex-sensitive cells. Therefore, our data suggest that increased expression of BAG1 isoforms leads to higher interaction with the GRα protein with subsequent recruitment of ubiquitin ligases, such as CHIP and proteasomal degradation of GRα.
BAG1 can repress endothelial GRα function by at least 3 mechanisms: (1) increased GRα degradation by proteasomal complex recruitment, (2) GRα transactivation inhibition via direct interaction with DNA, and (3) decreased GRα maturation by HSP70–BAG1 interactions.24–26,45–48 Our studies suggest that, in HUVECs, BAG1 isoforms stimulate GRα protein ubiquitination and proteasomal degradation of both cytosolic and nuclear GRα. Interestingly, others have reported that BAG1S resides exclusively in the cytosol and does not inhibit GR function.24–26 However, we identified a fraction of BAG1S in the nuclear extract, and the levels of nuclear BAG1S were higher in Dex-resistant cells. We are the first to report that BAG1S inhibits GRα in endothelial cells. Multiple reports have shown that only BAG1L and BAG1M can inhibit GRα-mediated transactivation.24–26 These studies were performed in noncardiovascular cells using overexpression assays where only nuclear BAG1L and BAG1M interact with DNA and directly inhibit GR-mediated gene transcription.24–26 However, our data have clearly shown that a significant amount of endothelial BAG1S is present in the nucleus, and that BAG1S overexpression significantly blunts Dex-sensitivity. Although BAG1S does not contain a nuclear localization signal, we hypothesize that BAG1S could be translocated into the nucleus bound to the GR complex where it targets nuclear GRα for proteasomal degradation.
BAG1 also stimulates cell survival processes in response to stress in a GRα-independent manner.49–51 For example, BAG1S and BAG1L overexpression was neuroprotective in an animal model of stroke,52 and BAG1S was cardioprotective in an in vivo model of ischemia/reperfusion.53 The neuroprotective and cardioprotective effects of BAG1 have been attributed to BAG1-mediated upregulation of HSP70.49–55 Furthermore, BAG1L and BAG1M overexpression have shown strong antiapoptotic and cell mitotic effects in cancerous cells, by activating BCL2 and RAF1.56 It has been reported that BAG1 clients compete with each other to sequester BAG1 proteins.48,49 Because BAG1 protein levels are much lower than its clients, a small change in BAG1 isoform expression, or that of HSP70, RAF1, and BCL2, could alter the interaction of BAG1 with GRα, RAF1, BCL2, or HSP70 and, subsequently, alter vascular homeostasis.
Another novel finding of our study is the differential expression of both GRα and BAG1 isoforms in proliferating versus quiescent cells. Interestingly, we did not observe significant interindividual differences in GRα and BAG1 isoform expression in proliferating HUVECs. These data suggest that significant interindividual differences in vascular BAG1 expression are more likely to be observed in quiescent healthy endothelium than proliferating endothelium at angiogenic and wound healing sites, and that decreased vascular BAG1 protein expression could be an early risk factor of stress-induced or GC-induced cardiovascular disease. We found a report on GC resistance associated with decreased GR expression in SMC derived from human vascular lesions but not from healthy tissue,57 which supports our observations of differential regulation of GR and its chaperones in various vascular cell phenotypes. It is important to note that BAG1 isoforms L, M, and S derive from a single mRNA,48,49 thus, BAG1 mRNA levels may not reflect changes in BAG1 isoform expression. Therefore, studies on the expression of BAG1 and GR protein isoforms in healthy and diseased vascular tissues are required to confirm the protective role of BAG1 on vascular health.
For these reasons, we studied the expression of GRα and BAG1 isoforms in adult human vascular cells HCAEC, HCASMC, and HCM. Of relevance, manipulation of BAG1 isoform expression in HCASMCs also resulted in significant changes in GRα protein levels, suggesting an important role of BAG1 isoforms in HCASMC Dex-sensitivity. HCAECs were most similar to Dex-sensitive HUVECs in the expression pattern of BAG1 isoforms (S>L>M), in GRα protein levels, and in GC sensitivity shown by GRα protein decreases and FKBP51 upregulation. HCASMCs were less sensitive, and HCM and THP1 cells were resistant to Dex-induced GRα protein decreases. Resistance to Dex-induced GRα degradation correlated with increased levels of FKBP51 and is consistent with the inhibitory role of this chaperone on GR nuclear translocation and function.26–28 Altogether, our studies highly suggest that differential expression of BAG1 isoforms could have an important regulatory role on the expression levels and function of vascular GR. Unfortunately, human studies on the role of chaperones in vascular GC sensitivity are missing. However, several studies have revealed an important role of the heat-shock proteins on cardiovascular disease, and the dysregulation of HSP70 expression in atherosclerosis and coronary artery disease has been confirmed.58–61 Therefore, future studies in HCAECs and HCASMCs are necessary to determine whether differential protein expression of BAG1, HSP70, HSP90, or FKBP51 is associated with differences in glucocorticoid-sensitivity in these vascular cells.
Finally, we found of interest the association of Dex-sensitivity in HUVECs with higher maternal body mass index and blood pressure. Dex-sensitivity has been often associated with the metabolic syndrome and hypertension, and our small study confirms the importance of these associations. Several studies have linked the +646C>G and the N365S polymorphisms with increased glucocorticoid-sensitivity and the R23K polymorphism with glucocorticoid-resistance.62,63 These polymorphisms, however, were not significantly associated with Dex-sensitivity in this study (Table VII in the online-only Data Supplement). We are cautious to mention that our study has important limitations, including the small number of subjects, which limits the association studies of rare polymorphisms with GC sensitivity. Another limitation of our study is the exclusion of important endogenous factors (such as blood flow, extracellular matrix, and cell–cell interaction) and their impact on GC sensitivity.
In summary, we have shown that endothelial in vitro Dex-sensitivity varies within individuals, with about one fourth of these individuals showing Dex resistance. Decreased GC sensitivity in endothelial cells is attributable to increased GRα protein degradation, a mechanism that can potentially protect the endothelium from harmful steroid effects. Our results, obtained with fetal cells, suggest that endothelial GC sensitivity could be determined before birth because of intrauterine environment factors, and therefore, could lead to differential individual GC susceptibility to endothelial activation, damage, and repair. We are the first to report that Dex resistance in HUVECs is associated with higher GRα degradation and higher BAG1 isoform expression. In addition, we have shown for the first time that human primary endothelial cells are capable of expressing all 3 BAG1 isoforms, and that all isoforms are capable of inhibiting both basal and Dex-stimulated GRα function. Uncovering novel molecular and genetic markers in association with GC sensitivity could prove useful in identifying individuals that are predisposed to GC-induced cardiovascular disease and warrants further investigation.
Supplementary Material
Significance.
Glucocorticoids are potent anti-inflammatory agents. However, increased sensitivity to endogenous cortisol or synthetic glucocorticoids has been associated with higher risk of cardiovascular diseases, including myocardial infarct and heart failure. The genetic and environmental factors that regulate sensitivity to glucocorticoids in vascular tissues remain undetermined. This study identified differential BCL2-associated athanogene 1 protein expression in association with differential in vitro glucocorticoid sensitivity of human endothelial cells. Increased BCL2-associated athanogene 1 expression prevented glucocorticoid-induced procoagulant and proatherogenic gene expression by increasing the degradation of the glucocorticoid receptor protein. Therefore, vascular BCL2-associated athanogene 1 expression is a protective factor against glucocorticoid-induced adverse effects in the cardiovascular system.
Acknowledgments
We are grateful to Dr John Cidlowski for providing GR vectors, to Dr John Reed for BAG1 vectors, and to Dr Dongbao Chen for initial setup design.
Sources of Funding
This work was funded by the American Heart Association Scientist Development Grant award #0630297 N to Dr Mata-Greenwood, and by internal sources from Loma Linda University.
Footnotes
The online-only Data Supplement is available with this article at http://atvb.ahajournals.org/lookup/suppl/doi:10.1161/ATVBAHA.113.301247/-/DC1
Disclosures
None.
References
- 1.Barnes PJ. Anti-inflammatory actions of glucocorticoids: molecular mechanisms. Clin Sci. 1998;94:557–572. doi: 10.1042/cs0940557. [DOI] [PubMed] [Google Scholar]
- 2.Auphan N, DiDonato JA, Rosette C, Helmberg A, Karin M. Immunosuppression by glucocorticoids: inhibition of NF-kappa B activity through induction of I kappa B synthesis. Science. 1995;270:286–290. doi: 10.1126/science.270.5234.286. [DOI] [PubMed] [Google Scholar]
- 3.Walker BR. Glucocorticoids and cardiovascular disease. Eur J Endocrinol. 2007;157:545–559. doi: 10.1530/EJE-07-0455. [DOI] [PubMed] [Google Scholar]
- 4.Yang S, Zhang L. Glucocorticoids and vascular reactivity. Curr Vasc Pharmacol. 2004;2:1–12. doi: 10.2174/1570161043476483. [DOI] [PubMed] [Google Scholar]
- 5.Sholter DE, Armstrong PW. Adverse effects of corticosteroids on the cardiovascular system. Can J Cardiol. 2000;16:505–511. [PubMed] [Google Scholar]
- 6.Girod JP, Brotman DJ. Does altered glucocorticoid homeostasis increase cardiovascular risk? Cardiovasc Res. 2004;64:217–226. doi: 10.1016/j.cardiores.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 7.Hadoke PW, Iqbal J, Walker BR. Therapeutic manipulation of glucocorticoid metabolism in cardiovascular disease. Br J Pharmacol. 2009;156:689–712. doi: 10.1111/j.1476-5381.2008.00047.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Zaane B, Nur E, Squizzato A, Gerdes VE, Büller HR, Dekkers OM, Brandjes DP. Systematic review on the effect of glucocorticoid use on procoagulant, anti-coagulant and fibrinolytic factors. J Thromb Haemost. 2010;8:2483–2493. doi: 10.1111/j.1538-7836.2010.04034.x. [DOI] [PubMed] [Google Scholar]
- 9.Wallerath T, Witte K, Schäfer SC, Schwarz PM, Prellwitz W, Wohlfart P, Kleinert H, Lehr HA, Lemmer B, Förstermann U. Down-regulation of the expression of endothelial NO synthase is likely to contribute to glucocorticoid-mediated hypertension. Proc Natl Acad Sci USA. 1999;96:13357–13362. doi: 10.1073/pnas.96.23.13357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lou YK, Wen C, Li M, Adams DJ, Wang MX, Yang F, Morris BJ, Whitworth JA. Decreased renal expression of nitric oxide synthase isoforms in adrenocorticotropin-induced and corticosterone-induced hypertension. Hypertension. 2001;37:1164–1170. doi: 10.1161/01.hyp.37.4.1164. [DOI] [PubMed] [Google Scholar]
- 11.Belvisi MG. Regulation of inflammatory cell function by corticosteroids. Proc Am Thorac Soc. 2004;1:207–214. doi: 10.1513/pats.200402-002MS. [DOI] [PubMed] [Google Scholar]
- 12.Muzaffar S, Shukla N, Angelini GD, Jeremy JY. Prednisolone augments superoxide formation in porcine pulmonary artery endothelial cells through differential effects on the expression of nitric oxide synthase and NADPH oxidase. Br J Pharmacol. 2005;145:688–697. doi: 10.1038/sj.bjp.0706235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yamamoto Y, Ishizu A, Ikeda H, Otsuka N, Yoshiki T. Dexamethasone increased plasminogen activator inhibitor-1 expression on human umbilical vein endothelial cells: an additive effect to tumor necrosis factor-alpha. Pathobiology. 2004;71:295–301. doi: 10.1159/000081724. [DOI] [PubMed] [Google Scholar]
- 14.Kimura H, Li X, Torii K, Okada T, Kamiyama K, Mikami D, Takahashi N, Yoshida H. Dexamethasone enhances basal and TNF-alpha-stimulated production of PAI-1 via the glucocorticoid receptor regardless of 11beta-hydroxysteroid dehydrogenase 2 status in human proximal renal tubular cells. Nephrol Dial Transplant. 2009;24:1759–1765. doi: 10.1093/ndt/gfn756. [DOI] [PubMed] [Google Scholar]
- 15.Brown NJ, Kim KS, Chen YQ, Blevins LS, Nadeau JH, Meranze SG, Vaughan DE. Synergistic effect of adrenal steroids and angiotensin II on plasminogen activator inhibitor-1 production. J Clin Endocrinol Metab. 2000;85:336–344. doi: 10.1210/jcem.85.1.6305. [DOI] [PubMed] [Google Scholar]
- 16.Heitzer MD, Wolf IM, Sanchez ER, Witchel SF, DeFranco DB. Glucocorticoid receptor physiology. Rev Endocr Metab Disord. 2007;8:321–330. doi: 10.1007/s11154-007-9059-8. [DOI] [PubMed] [Google Scholar]
- 17.Kino T. Tissue glucocorticoid sensitivity: beyond stochastic regulation on the diverse actions of glucocorticoids. Horm Metab Res. 2007;39:420–424. doi: 10.1055/s-2007-980193. [DOI] [PubMed] [Google Scholar]
- 18.Bamberger CM, Schulte HM, Chrousos GP. Molecular determinants of glucocorticoid receptor function and tissue sensitivity to glucocorticoids. Endocr Rev. 1996;17:245–261. doi: 10.1210/edrv-17-3-245. [DOI] [PubMed] [Google Scholar]
- 19.Whorwood CB, Donovan SJ, Wood PJ, Phillips DI. Regulation of glucocorticoid receptor alpha and beta isoforms and type I 11beta-hydroxysteroid dehydrogenase expression in human skeletal muscle cells: a key role in the pathogenesis of insulin resistance? J Clin Endocrinol Metab. 2001;86:2296–2308. doi: 10.1210/jcem.86.5.7503. [DOI] [PubMed] [Google Scholar]
- 20.Webster JC, Oakley RH, Jewell CM, Cidlowski JA. Proinflammatory cytokines regulate human glucocorticoid receptor gene expression and lead to the accumulation of the dominant negative beta isoform: a mechanism for the generation of glucocorticoid resistance. Proc Natl Acad Sci USA. 2001;98:6865–6870. doi: 10.1073/pnas.121455098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ito K, Chung KF, Adcock IM. Update on glucocorticoid action and resistance. J Allergy Clin Immunol. 2006;117:522–543. doi: 10.1016/j.jaci.2006.01.032. [DOI] [PubMed] [Google Scholar]
- 22.Pratt WB, Morishima Y, Murphy M, Harrelll M. Chaperoning of glucocorticoid receptors. Handb Exp Pharmacol. 2006;172:111–138. doi: 10.1007/3-540-29717-0_5. [DOI] [PubMed] [Google Scholar]
- 23.Grad I, Picard D. The glucocorticoid responses are shaped by molecular chaperones. Mol Cell Endocrinol. 2007;275:2–12. doi: 10.1016/j.mce.2007.05.018. [DOI] [PubMed] [Google Scholar]
- 24.Schmidt U, Wochnik GM, Rosenhagen MC, Young JC, Hartl FU, Holsboer F, Rein T. Essential role of the unusual DNA-binding motif of BAG-1 for inhibition of the glucocorticoid receptor. J Biol Chem. 2003;278:4926–4931. doi: 10.1074/jbc.M212000200. [DOI] [PubMed] [Google Scholar]
- 25.Takayama S, Reed JC. Molecular chaperone targeting and regulation by BAG family proteins. Nat Cell Biol. 2001;3:E237–E241. doi: 10.1038/ncb1001-e237. [DOI] [PubMed] [Google Scholar]
- 26.Cato AC, Mink S. BAG-1 family of cochaperones in the modulation of nuclear receptor action. J Steroid Biochem Mol Biol. 2001;78:379–388. doi: 10.1016/s0960-0760(01)00114-5. [DOI] [PubMed] [Google Scholar]
- 27.Adcock IM, Ito K, Barnes PJ. Glucocorticoids: effects on gene transcription. Proc Am Thorac Soc. 2004;1:247–254. doi: 10.1513/pats.200402-001MS. [DOI] [PubMed] [Google Scholar]
- 28.Kassel O, Herrlich P. Crosstalk between the glucocorticoid receptor and other transcription factors: molecular aspects. Mol Cell Endocrinol. 2007;275:13–29. doi: 10.1016/j.mce.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 29.Bruzdzinski CJ, Johnson MR, Goble CA, Winograd SS, Gelehrter TD. Mechanism of glucocorticoid induction of the rat plasminogen activator inhibitor-1 gene in HTC rat hepatoma cells: identification of cis-acting regulatory elements. Mol Endocrinol. 1993;7:1169–1177. doi: 10.1210/mend.7.9.8247019. [DOI] [PubMed] [Google Scholar]
- 30.Clark AR. Anti-inflammatory functions of glucocorticoid-induced genes. Mol Cell Endocrinol. 2007;275:79–97. doi: 10.1016/j.mce.2007.04.013. [DOI] [PubMed] [Google Scholar]
- 31.Revollo JR, Cidlowski JA. Mechanisms generating diversity in glucocorticoid receptor signaling. Ann N Y Acad Sci. 2009;1179:167–178. doi: 10.1111/j.1749-6632.2009.04986.x. [DOI] [PubMed] [Google Scholar]
- 32.Lu NZ, Cidlowski JA. Translational regulatory mechanisms generate N-terminal glucocorticoid receptor isoforms with unique transcriptional target genes. Mol Cell. 2005;18:331–342. doi: 10.1016/j.molcel.2005.03.025. [DOI] [PubMed] [Google Scholar]
- 33.Biddie SC, Conway-Campbell BL, Lightman SL. Dynamic regulation of glucocorticoid signalling in health and disease. Rheumatology (Oxford) 2012;51:403–412. doi: 10.1093/rheumatology/ker215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Niu N, Manickam V, Kalari KR, Moon I, Pelleymounter LL, Eckloff BW, Wieben ED, Schaid DJ, Wang L. Human glucocorticoid receptor alpha gene (NR3C1) pharmacogenomics: gene resequencing and functional genomics. J Clin Endocrinol Metab. 2009;94:3072–3084. doi: 10.1210/jc.2008-2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pedersen KB, Geng CD, Vedeckis WV. Three mechanisms are involved in glucocorticoid receptor autoregulation in a human T-lymphoblast cell line. Biochemistry. 2004;43:10851–10858. doi: 10.1021/bi049458u. [DOI] [PubMed] [Google Scholar]
- 36.Chrousos GP, Kino T. Glucocorticoid signaling in the cell. Expanding clinical implications to complex human behavioral and somatic disorders. Ann N Y Acad Sci. 2009;1179:153–166. doi: 10.1111/j.1749-6632.2009.04988.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wallace AD, Cidlowski JA. Proteasome-mediated glucocorticoid receptor degradation restricts transcriptional signaling by glucocorticoids. J Biol Chem. 2001;276:42714–42721. doi: 10.1074/jbc.M106033200. [DOI] [PubMed] [Google Scholar]
- 38.van Zonneveld AJ, Curriden SA, Loskutoff DJ. Type 1 plasminogen activator inhibitor gene: functional analysis and glucocorticoid regulation of its promoter. Proc Natl Acad Sci USA. 1988;85:5525–5529. doi: 10.1073/pnas.85.15.5525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Juhan-Vague I, Alessi MC, Mavri A, Morange PE. Plasminogen activator inhibitor-1, inflammation, obesity, insulin resistance and vascular risk. J Thromb Haemost. 2003;1:1575–1579. doi: 10.1046/j.1538-7836.2003.00279.x. [DOI] [PubMed] [Google Scholar]
- 40.Hoekstra T, Geleijnse JM, Schouten EG, Kluft C. Plasminogen activator inhibitor-type 1: its plasma determinants and relation with cardiovascular risk. Thromb Haemost. 2004;91:861–872. doi: 10.1160/TH03-08-0546. [DOI] [PubMed] [Google Scholar]
- 41.Levy AS, Chung JC, Kroetsch JT, Rush JW. Nitric oxide and coronary vascular endothelium adaptations in hypertension. Vasc Health Risk Manag. 2009;5:1075–1087. doi: 10.2147/vhrm.s7464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu Y, Mladinov D, Pietrusz JL, Usa K, Liang M. Glucocorticoid response elements and 11 beta-hydroxysteroid dehydrogenases in the regulation of endothelial nitric oxide synthase expression. Cardiovasc Res. 2009;81:140–147. doi: 10.1093/cvr/cvn231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zerr-Fouineau M, Chataigneau M, Blot C, Schini-Kerth VB. Progestins overcome inhibition of platelet aggregation by endothelial cells by downregulating endothelial NO synthase via glucocorticoid receptors. FASEB J. 2007;21:265–273. doi: 10.1096/fj.06-6840com. [DOI] [PubMed] [Google Scholar]
- 44.Newton K, Matsumoto ML, Wertz IE, et al. Ubiquitin chain editing revealed by polyubiquitin linkage-specific antibodies. Cell. 2008;134:668–678. doi: 10.1016/j.cell.2008.07.039. [DOI] [PubMed] [Google Scholar]
- 45.Liman J, Faida L, Dohm CP, Reed JC, Bähr M, Kermer P. Subcellular distribution affects BAG1 function. Brain Res. 2008;1198:21–26. doi: 10.1016/j.brainres.2008.01.010. [DOI] [PubMed] [Google Scholar]
- 46.Takayama S, Krajewski S, Krajewska M, Kitada S, Zapata JM, Kochel K, Knee D, Scudiero D, Tudor G, Miller GJ, Miyashita T, Yamada M, Reed JC. Expression and location of Hsp70/Hsc-binding anti-apoptotic protein BAG-1 and its variants in normal tissues and tumor cell lines. Cancer Res. 1998;58:3116–3131. [PubMed] [Google Scholar]
- 47.Gehring U. Biological activities of HAP46/BAG-1. The HAP46/BAG-1 protein: regulator of HSP70 chaperones, DNA-binding protein and stimulator of transcription. EMBO Rep. 2004;5:148–153. doi: 10.1038/sj.embor.7400083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kanelakis KC, Morishima Y, Dittmar KD, Galigniana MD, Takayama S, Reed JC, Pratt WB. Differential effects of the hsp70-binding protein BAG-1 on glucocorticoid receptor folding by the hsp90-based chaperone machinery. J Biol Chem. 1999;274:34134–34140. doi: 10.1074/jbc.274.48.34134. [DOI] [PubMed] [Google Scholar]
- 49.Takayama S, Sato T, Krajewski S, Kochel K, Irie S, Millan JA, Reed JC. Cloning and functional analysis of BAG-1: a novel Bcl-2-binding protein with anti-cell death activity. Cell. 1995;80:279–284. doi: 10.1016/0092-8674(95)90410-7. [DOI] [PubMed] [Google Scholar]
- 50.Nollen EA, Morimoto RI. Chaperoning signaling pathways: molecular chaperones as stress-sensing ‘heat shock’ proteins. J Cell Sci. 2002;115:2809–2816. doi: 10.1242/jcs.115.14.2809. [DOI] [PubMed] [Google Scholar]
- 51.Song J, Takeda M, Morimoto RI. Bag1-Hsp70 mediates a physiological stress signalling pathway that regulates Raf-1/ERK and cell growth. Nat Cell Biol. 2001;3:276–282. doi: 10.1038/35060068. [DOI] [PubMed] [Google Scholar]
- 52.Kermer P, Digicaylioglu MH, Kaul M, Zapata JM, Krajewska M, Stenner-Liewen F, Takayama S, Krajewski S, Lipton SA, Reed JC. BAG1 overexpression in brain protects against stroke. Brain Pathol. 2003;13:495–506. doi: 10.1111/j.1750-3639.2003.tb00480.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Townsend PA, Cutress RI, Carroll CJ, Lawrence KM, Scarabelli TM, Packham G, Stephanou A, Latchman DS. BAG-1 proteins protect cardiac myocytes from simulated ischemia/reperfusion-induced apoptosis via an alternate mechanism of cell survival independent of the proteasome. J Biol Chem. 2004;279:20723–20728. doi: 10.1074/jbc.M400399200. [DOI] [PubMed] [Google Scholar]
- 54.Liman J, Ganesan S, Dohm CP, Krajewski S, Reed JC, Bähr M, Wouters FS, Kermer P. Interaction of BAG1 and Hsp70 mediates neuroprotectivity and increases chaperone activity. Mol Cell Biol. 2005;25:3715–3725. doi: 10.1128/MCB.25.9.3715-3725.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Frebel K, Wiese S, Funk N, Pühringer D, Sendtner M. Differential modulation of neurite growth by the S- and the L-forms of bag1, a co-chaperone of Hsp70. Neurodegener Dis. 2007;4:261–269. doi: 10.1159/000101850. [DOI] [PubMed] [Google Scholar]
- 56.Liu HY, Wang ZM, Bai Y, Wang M, Li Y, Wei S, Zhou QH, Chen J. Different BAG-1 isoforms have distinct functions in modulating chemotherapeutic-induced apoptosis in breast cancer cells. Acta Pharmacol Sin. 2009;30:235–241. doi: 10.1038/aps.2008.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bray PJ, Du B, Mejia VM, Hao SC, Deutsch E, Fu C, Wilson RC, Hanauske-Abel H, McCaffrey TA. Glucocorticoid resistance caused by reduced expression of the glucocorticoid receptor in cells from human vascular lesions. Arterioscler Thromb Vasc Biol. 1999;19:1180–1189. doi: 10.1161/01.atv.19.5.1180. [DOI] [PubMed] [Google Scholar]
- 58.Willis MS, Patterson C. Hold me tight: Role of the heat shock protein family of chaperones in cardiac disease. Circulation. 2010;122:1740–1751. doi: 10.1161/CIRCULATIONAHA.110.942250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xu Q, Metzler B, Jahangiri M, Mandal K. Molecular chaperones and heat shock proteins in atherosclerosis. Am J Physiol Heart Circ Physiol. 2012;302:H506–H514. doi: 10.1152/ajpheart.00646.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.He M, Guo H, Yang X, Zhang X, Zhou L, Cheng L, Zeng H, Hu FB, Tanguay RM, Wu T. Functional SNPs in HSPA1A gene predict risk of coronary heart disease. PLoS ONE. 2009;4:e4851. doi: 10.1371/journal.pone.0004851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Almeida MB, do Nascimento JL, Herculano AM, Crespo-López ME. Molecular chaperones: toward new therapeutic tools. Biomed Pharmacother. 2011;65:239–243. doi: 10.1016/j.biopha.2011.04.025. [DOI] [PubMed] [Google Scholar]
- 62.Witchel SF, DeFranco DB. Mechanisms of disease: regulation of glucocorticoid and receptor levels–impact on the metabolic syndrome. Nat Clin Pract Endocrinol Metab. 2006;2:621–631. doi: 10.1038/ncpendmet0323. [DOI] [PubMed] [Google Scholar]
- 63.Manenschijn L, van den Akker EL, Lamberts SW, van Rossum EF. Clinical features associated with glucocorticoid receptor polymorphisms. An overview. Ann N Y Acad Sci. 2009;1179:179–198. doi: 10.1111/j.1749-6632.2009.05013.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.