

Abstract
We (66) have previously described an NSAID-insensitive intramitochondrial biosynthetic pathway involving oxidation of the polyunsaturated mitochondrial phospholipid, cardiolipin (CL), followed by hydrolysis [by calcium-independent mitochondrial calcium-independent phospholipase A2-γ (iPLA2γ)] of oxidized CL (CLox), leading to the formation of lysoCL and oxygenated octadecadienoic metabolites. We now describe a model system utilizing oxidative lipidomics/mass spectrometry and bioassays on cultured bovine pulmonary artery endothelial cells (BPAECs) to assess the impact of CLox that we show, in vivo, can be released to the extracellular space and may be hydrolyzed by lipoprotein-associated PLA2 (Lp-PLA2). Chemically oxidized liposomes containing bovine heart CL produced multiple oxygenated species. Addition of Lp-PLA2 hydrolyzed CLox and produced (oxygenated) monolysoCL and dilysoCL and oxidized octadecadienoic metabolites including 9- and 13-hydroxyoctadecadienoic (HODE) acids. CLox caused BPAEC necrosis that was exacerbated by Lp-PLA2. Lower doses of nonlethal CLox increased permeability of BPAEC monolayers. This effect was exacerbated by Lp-PLA2 and partially mimicked by authentic monolysoCL or 9- or 13-HODE. Control mice plasma contained virtually no detectable CLox; in contrast, 4 h after Pseudomonas aeruginosa (P. aeruginosa) infection, 34 ± 8 mol% (n = 6; P < 0.02) of circulating CL was oxidized. In addition, molar percentage of monolysoCL increased twofold after P. aeruginosa in a subgroup analyzed for these changes. Collectively, these studies suggest an important role for 1) oxidation of CL in proinflammatory environments and 2) possible hydrolysis of CLox in extracellular spaces producing lysoCL and oxidized octadecadienoic acid metabolites that may lead to impairment of pulmonary endothelial barrier function and necrosis.
Keywords: cardiolipin, pulmonary endothelium, octadecadienoic acid, lineolic acid, lipoprotein-associated phospholipase A2
regulation and coordination of many biological functions are known to occur through the following multitude of oxygenated fatty acids as signaling molecules: oxygen-containing eicosanoids including prostaglandins, thromboxanes, leukotrienes, lipoxenes, resolvins, and protectins (2, 3, 6, 7, 59). In the canonical pathway, these oxidized free fatty acids are produced in the cytosol by Ca2+-dependent PLA2 hydrolysis of membrane phospholipids, releasing polyunsaturated fatty acids such as arachidonic or docosahexanoic acids and their subsequent enzymatic oxidation by COX-1 or COX-2, various LOX, p450, or peroxidases. Modulation of this pathway by NSAIDs is a traditional and vital component of pharmacotherapy. Recently, we (63) demonstrated an alternative, NSAID-insensitive, pathway for oxidized free fatty acid biosynthesis that resides in the mitochondria and involves iPLA2γ hydrolysis of oxidized mitochondrial-specific phospholipid CL. The potential importance of this pathway was revealed in this study by profile of oxidized free fatty acids formed in the brain (after closed concussion injury) or small intestine (after whole body irradiation) that was modified by an iPLA2γ inhibitor (e.g., R-BEL). In a model of global cerebral ischemia-reperfusion in young rats, we (30) noted that a mitochondria-targeted nitroxide electron scavenger (70) inhibited CL oxidation (presumably by cytochrome c peroxidase) and improved neurocognitive outcomes. We (30) subsequently showed in a rotenone model of early Parkinson's disease in rats that linoleic acid in sn-1 position was the major oxidation substrate yielding its mono-hydroxy and epoxy derivatives (rather than oxidized arachidonic or docosahexanoic acid). Collectively, these studies 1) suggest an important role for a mitochondria-specific, calcium-independent PLA2-mediated hydrolysis of oxidized CL producing primarily oxygen-modified octadecadienoic acid metabolites that is NSAID insensitive; and 2) add to our observations regarding the role of cytochrome c peroxidase-mediated oxidation of CL and its metabolites in apoptosis (33), mitophagy (15) and other cellular functions (31).
In contrast to the novel intracellular processes noted above, considerably less is known regarding the effect of extracellular CL, CLox, or its hydrolytic products. Many processes are in place to maintain CL in its normal restricted location to the inner membrane of mitochondria (31), and thus elevations in extracellular CL may be perceived as an indication of dyshomeostasis. This may be particularly important in lung, as 1) elevations in CL in airspace secondary to alterations in lipid transporter, ATP8b1, were critical etiologic factors in lung dysfunction (decreased compliance, altered surfactant biosynthesis) in experimental and clinical pneumonia (53); and 2) CL per se was noted to be an active component of experimental group B streptococcus-induced pulmonary hypertension (17). Mitochondria are known to be an important source of DAMP molecules, and infusion of a preparation of mitochondrial DAMPs caused an increase in endothelial cell permeability and severe ALI (73). Beyond the contribution of mitochondrial DNA and formyl peptides, the precise biochemical nature of mitochondrial DAMPs remains uncertain but may include CL. Although prokaryotic CL tends to be fully saturated, the polyunsaturated nature of eukaryotic CL favors oxidation, and we (63) noted that oxidized (but not native or reduced form) CL is a substrate for Lp-PLA2 or PAF-AH that can be released from activated neutrophils or monocytes and act as an extracellular iPLA2. Accordingly, we modified a schema from Ji et al. (30) and present our hypothesis (Fig. 1) that CL that appears in the extracellular space may be oxidized then hydrolyzed by Lp-PLA2, and targets (such as endothelium) may be exposed to a mixture of CL, CLox, oxidized free fatty acids, and lysoCLs.
Fig. 1.

Alternative biosynthesis of lipid mediators. The canonical pathway of eicosanoids and other known lipid mediator biosynthesis by PLA2 hydrolysis followed by enzymatic oxidation of the released free fatty acids (left) is presented in contrast to the alternative pathway (right) of first oxidation of an unsaturated lipid (CL in this example) followed by enzymatic hydrolysis. This alternative pathway represents biosynthesis of lipid mediators through pathways of nontraditional enzymatic targets. PC, phosphatidylcholine; PE, phosphatidylethanolamine; PS, phosphatidylserine; PI, phosphatidylinositol.
The focus on CL as precursor phospholipid seemed reasonable because we previously used oxidative lipidomics to indicate selective oxidation of intrapulmonary CL (along with the other anionic phospholipid, phosphatidylserine) in intact mice exposed to hyperoxia (65), ionizing radiation (64), or single-walled carbon nanotubes (62). In the present study, we used a combination of mass spectrometry (oxidative lipidomics) and bioassays on cultured BPAECs exposed to reaction products of bovine heart CL (primarily tetralinoleoyl) with or without free radical generator, AAPH, and exposure to authentic Lp-PLA2. Although there are a wide range of CL species in mammalian tissue, including lung, the choice of BHCL helped focus our study around linoleoyl enhanced CL, which is the most abundant species in the lung (60) and is likely to result in biosynthesis of octadecadienoic acids, a relatively understudied group of free fatty acids, and their oxidized metabolites that are important in lung injury (22, 38). We used a nondiscriminant and potent water-soluble azo initiator, AAPH, as an oxidizing agent to enhance the likelihood of generating numerous potentially oxidized forms of CL. We noted that oxidation of relatively high levels of BHCL caused necrosis (but not apoptosis) of BPAECs and the percentage of necrotic cells was further increased by adding Lp-PLA2. Oxidation of lower doses of BHCL decreased barrier function of BPAECs (as assessed by transendothelial cell electrical resistance), and this decrement was exacerbated by the addition of Lp-PLA2 and could be mimicked by exposure to several authentic octadecadienoic acid metabolites as well as monolysoCL. Finally, we noted that, in control mice, virtually none of the detectable levels of plasma CL were either oxidized or hydrolyzed; in contrast, 4 h after infection with P. aeruginosa, 34 ± 7.9 mol% (n = 6; P < 0.02) of plasma CL was oxidized. In a subset of two infected mice in which monolysoCL was analyzed, there was an approximate doubling of its molar content. As such, the present study supports further investigation into the role of extracellular CLox and iPLA2-mediated hydrolytic products as DAMPs and effectors of endothelial cell dysfunction in ALI in vivo.
MATERIALS AND METHODS
BHCL and DOPC were purchased from Avanti Polar lipids. 1:1 BHCL to DOPC liposomes were formed by adding molar equivalents of lipids dissolved in chloroform to a glass test tube and then drying under an argon stream to prevent spontaneous oxidation. The lipid wax was then suspended in HBSS and scraped from the sides of the test tube and probe sonicated for six 10-s pulses.
Preparation of AAPH.
The water-soluble azo-initiator, AAPH, was purchased from Sigma-Aldrich and prepared in stock concentrations (5 mM) with HBSS as the solvent and vortexed until clear. Dilutions were made immediately before protocols.
LC-MS.
LC ESI MS/MS was performed on 1:1, BHCL:DOPC liposomes before and after AAPH incubation (5 mM) and after AAPH with addition of authentic Lp-PLA2 (200 ng/ml) (Cayman Chemical) to determine the products of oxidation and hydrolysis. Lipids were extracted by the Folch procedure (21) under nitrogen atmosphere. LC ESI MS/MS analysis was performed on a Dionex HPLC system (utilizing the Chromeleon software), consisting of a Dionex UltiMate 3000 mobile phase pump, equipped with an UltiMate 3000 degassing unit and UltiMate 3000 autosampler (sampler chamber temperature was set at 4°C). The Dionex HPLC system was coupled to an LXQ ion trap mass spectrometer (ThermoFisher) within an Xcalibur operating system. The instrument was operated in negative ion mode (at a voltage differential of −3.5–5.0 kV), and source temperature was maintained at 150°C. Spectra were acquired in negative ion mode using a full-range zoom (370-1,600 m/z, for PL and 200–400 m/z for free fatty acids) or ultra-zoom (selected ion monitoring and selected reaction monitoring) scans. MS/MS analysis of individual phospholipid species was used to determine the fatty acid composition and was carried out with a relative collision energy ranging from 20–40% and with an activation q value at 0.25 for collision-induced dissociation and q value at 0.7 for pulsed-Q dissociation technique. MS/MS analysis was performed using an isolation width of 1 m/z, 5 micro-scans with a maximum injection time of 1,000 ms. Identified lipids were quantitated using appropriate internal standards.
Normal-phase column separation of phospholipids was performed on a Luna 3 μm Silica (2) 100 Å 150 × 1 mm column (Phenomenex). The analysis was performed using gradient solvent A (hexane:propanol:water, 47:57:1, vol/vol) and solvent B (hexane:propanol:water, 47:57:10, vol/vol) each containing 5 mM ammonium acetate and 0.01% formic acid. The column was eluted at a flow rate of 0.05 ml/min as follows: 0–3 min, linear gradient, 10–37% solvent B; 3.0–12.5 min, isocratic at 37% solvent B; 12.5–20.0 min, linear gradient, 37–100% solvent B; 20–45 min, isocratic at 100% solvent B; 45–60 min, isocratic at 10% solvent B.
Reverse-phase column separation of free fatty acids was performed on a Luna 3 μm C18 (2) 100 Å, 150 × 1 mm column. The analysis was conducted using gradient solvent A (tetrahydrofuran:methanol:water:acetic acid, 25.0:30.0:44.9:0.1, vol/vol) and solvent B (methanol:water, 9:1, vol/vol) each containing 5 mM ammonium acetate. The column was eluted at a flow rate of 0.05 ml/min as follows: 0–3 min, isocratic at 50% solvent B; 3–23 min, linear gradient, 50–98% solvent B; 23–40 min, isocratic at 98% solvent B; 40–42 min, linear gradient, 98-50% solvent B; 42–60 min, isocratic at 50% solvent B.
EPR.
EPR detection of ascorbate radicals was performed on a JEOL-RE1X spectrometer at 25°C. Spectra of ascorbate radicals were recorded upon addition of ascorbic acid (25 μM) to the reaction mixture containing HBSS (Life Technologies Biotechnology) (pH 7.4) and 100 μM DTPA to chelate adventitious metals. The measurements were performed in a gas-permeable Teflon tubing (0.8-mm internal diameter, 0.013-mm thickness) obtained from Alpha Wire. The Teflon tube (∼8 cm in length) was filled with 65 μl of the reaction mixture, folded into halves, and placed into an open EPR quartz tube (inner diameter of 3.0 mm) in such a way that the sample was entirely within the microwave radiation area. In a typical experiment, the spectra of ascorbate radicals were recorded under the following conditions: center field 3,354 G, power 20 mW, field modulation 0.79 G, sweep times 10 and 20 s, sweep width 2.5 G, receiver gain 4,000, time constant 0.1 s. Spectra were collected using EPRware software (Scientific Software Services).
Cultured BPAECs.
BPAECs were purchased from VEC Technologies and grown in MCDB-131 COMPLETE (VEC Technologies) at 37°C in an atmosphere with 5% O2-5% CO2 in a Coy Hypoxic Chamber to more closely reproduce Po2 of mixed venous blood that cells were exposed to in situ. All BPAECs used in experiments were between passages 4–8.
AlamarBlue Assay.
Cell viability was determined by a quantifying reduction of the fluorogenic indicator alamarBlue (Alamar Biosciences). BPAECs (1 × 105) were allowed to attach to 96-well tissue culture dishes (3–4 wells/condition). All reactions were carried out in 100-μl volumes total. BHCL was prepared as above and then exposed to 5 mM AAPH and 200 ng/ml Lp-PLA2 in a reaction vial for 2 h depending on experimental conditions. This lipid suspension was then added to the wells and incubated at 37°C for 2 h. After 2 h of exposure, the reaction suspension was removed, and HBSS with alamarBlue was added to the treated wells and allowed to incubate for 3 h. Fluorescence was determined with a Perkin Elmer Fusion Alpha plate reader. Oxidized alamarBlue has previously been shown to be absorbed by living cells and reduced by intracellular dehydrogenases. Changes in the fluorescence emission at 590 nm are used as a reflection of viability (20, 48).
Flow cytometry.
Cell viability was determined by phosphatidylserine externalization and/or propidium iodide internalization. Phosphatidylserine externalization was determined with an annexin V-FITC apoptosis detection kit (Biovision). Treated cells were rinsed in HBSS, trypsinized, and centrifuged at 900 revolutions/min for 5 min. For annexin V assay, the cell pellet was resuspended in 400 μl of binding buffer and supplemented with 2 μl of FITC-annexin V and 2 μl of propidium iodide. A FACS Canto (BD Biosciences) was used to perform flow cytometric analysis. Ten thousand events were recorded and analyzed for each sample.
Endothelial cell barrier function.
ECIS was performed on model Zθ from Applied BioPhysics and was used to monitor monolayer permeability on 8W10E+ arrays. BPAECs were trypsinized, and ∼3 × 105 cells per well were plated in the 8W10E+ arrays and placed in a 5% CO2-5% O2 hypoxic chamber. The ECIS array station instrument was placed in a different 37°C incubator with 5% CO2-21% O2. ECIS software scanned multiple frequencies repeatedly at 4, 32, and 64 mHz to collect resistance, impedance, and capacitance, respectively. To assure confluency of monolayers, capacitance data from all wells were below 10 nF at 64,000 Hz. Subsequently, media were removed from wells and replaced with 200 μl of HBSS (Ca2+/Mg2+). Cells were allowed to reequilibrate at 37°C, and then various experimental condition mixtures were created, as described above, and were applied by adding 200 μL of experimental condition mixture in HBSS (Ca2+/Mg2+) to 200 μL of HBSS (Ca2+/Mg2+) already at stable resistance equilibrium in the wells. ECIS software was used for data collection and analysis in real-time throughout the experiment.
Animal studies.
C57BL6 mice were anesthetized, and the larynx was visualized before endotracheal intubation with a 24-gauge plastic catheter (12); P. aeruginosa (strain PA103, 106 CFU/mouse) was administered intratracheally. Four hours later, animals were killed, and plasma was obtained for determination of CL and molar percentage of CLox (n = 6 infected mice) and monolysoCL (n = 2). All procedures were approved by the University of Pittsburgh Institutional Animal Care and Use Committee.
Statistics.
Prism software version 6.0f (GraphPad) was used to perform all final statistical analysis with two-way ANOVA followed by Bonferroni post hoc tests. Assumptions are that all treatment groups and test samples are independent.
RESULTS
In vitro model.
To establish a cell-free system to determine whether CLox was a substrate for Lp-PLA2 (Fig. 2), it was critical to assure that polyunsaturated CL was stable in HBSS and that free radical generation by azo initiator, AAPH, did not directly affect Lp-PLA2. In Fig. 3, we note that addition of 25 μM sodium ascorbate to HBSS resulted in production of ascorbyl radical (by EPR) that was quenched by the addition of the metal chelator, DTPA, consistent with the presence of adventitious metal in our preparation of HBSS (Fig. 3, A and B). Accordingly, DTPA (100 μM) was routinely added to HBSS for all experiments to avoid potential oxidation of CL at baseline. We then added ascorbate to HBSS containing AAPH (5 mM) and noted an intense production of ascorbyl radical (Fig. 3C) that was partially quenched by DTPA (Fig. 3D) and completely quenched with the addition of BHT (100 μM) (Fig. 3E). We concluded from these preliminary experiments that CL could be maintained in reduced form at baseline by adding DTPA to HBSS and that AAPH was still capable of generating free radicals (Fig. 3D) to potentially oxidize CL. Addition of BHT before Lp-PLA2 protected the enzyme from being oxidized by any remnant AAPH in medium (Fig. 3E).
Fig. 2.

In vitro oxidation and hydrolysis of tetralinoleoyl CL. A cell-free in vitro system was developed with CL oxidation by the azo initiator, AAPH. Subsequently the CLox was hydrolyzed by the Ca++-independent Lp-PLA2, producing monolyso- and dilyso- (not shown) CL species, as well as oxygenated free fatty acids.
Fig. 3.

EPR reveals the presence of free ascorbyl radicals generated by HBSS in a cell-free system. EPR spectra shown of a cell-free in vitro system containing HBSS were recorded 10 and 20 s after addition of 25 μM ascorbate (A), then metal chelator, DTPA (100 μM; B). C shows the addition of the free radical generator AAPH (5 mM) to 25 μM ascorbate, then addition of 100 μM DTPA (D), followed by the addition of 100 μM BHT (E).
Under these conditions, we analyzed BHCL using LC-MS and noted (Fig. 4, bottom) that BHCL was primarily in its reduced form with a major cluster at m/z of 1,447 (and minor cluster at m/z 1,469). AAPH-induced oxidation of BHCL was evident by the appearance of signals from the products with additional multiple oxygens at intervals of +16 m/z from the major cluster at 1,447 m/z (Fig. 4, top). Addition of either AAPH or Lp-PLA2, alone, did not affect overall content of BHCL in DOPC/BHCL liposomes; however, sequential exposure of BHCL to AAPH (e.g., oxidation) and then Lp-PLA2 (hydrolysis) resulted in a significant loss of substrate (Fig. 5, bottom, left). These data suggest that CLox but not CL is a substrate for Lp-PLA2, and this was further confirmed by LC-MS showing potential products of this reaction (see Fig. 2) readily detectable (Fig. 5, top) after sequential exposure of BHCL to AAPH and Lp-PLA2 (but not to either alone). Indeed, the accumulation of monolysoCL and dilysoCL and the other reaction products (Fig. 5, bottom, right) after treatment of DOPC/BHCL liposomes with AAPH/Lp-PLA2 accounted for a significant portion of the lost substrate (Fig. 5 bottom, left). Further analysis of products of oxidized free fatty acids formed in this reaction showed multiple oxo, hydroxy, epoxy, and hydroperoxy metabolites of linoleic acid (Fig. 6). A partial list of detectable octadecadienoic metabolites and their identities after fragmentation analyses are summarized in Table 1.
Fig. 4.

BHCL in the presence of AAPH produced multiple oxidation products as shown by LC-MS/MS. LC-MS/MS was performed on the reduced form on BHCL (A) and BHCL + AAPH (B). A indicates 2 CL clusters at m/z 1,469 (minor), 1,447 (major). B indicates multiple CL clusters, the largest at m/z 1,447, 1,469, 1,479, 1,495 and 1,511, 1,527, 1,543, and 1,559. There was a decrease in the major m/z 1,447 CL species and increases in species at the other m/z displayed in gray representing various oxidized forms of CL with or without adducts. Each of these oxidized forms of CL having an additional oxygen molecule added was determined by the m/z increase of 16.
Fig. 5.

Lp-PLA2 hydrolyzes BHCL only after oxidation. LC-MS profiles of monolysoCL and dilysoCL species in experimental conditions of BHCL with or without AAPH and subsequent hydrolysis by Lp-PLA2 are shown (top). LC-MS comparison of molar percentage of intact tetralinoleoyl CL (bottom, left) and monolysoCL (MCL) species, dilysoCL (DCL) species, and free fatty acids (FFA) (bottom, right) are shown for experimental conditions of BHCL with or without AAPH and subsequent hydrolysis by Lp-PLA2 (bottom, left).
Fig. 6.

Identification of free fatty acid products. LC-MS was performed on experimental conditions of BHCL with or without AAPH and subsequent hydrolysis by Lp-PLA2. Molar percentages of the various free fatty acids products are shown (top). LC-MS base profiles of only a few of the formed oxygenated free fatty acids, m/z 327, 311, 295, and 293 are shown in the conditions of BHCL + AAPH and BHCL + AAPH with addition of Lp-PLA2 after treatment with DTPA and BHT to quench Fe and AAPH free radicals (bottom).
Table 1.
Free fatty acid oxygenated products formed in CL exposed to AAPH and Lp-PLA2
| m/z | Name | Abbreviation |
|---|---|---|
| 293 | 9-oxo, 10,12-octadecadienoic acid | 9-KODE |
| 293 | 13-oxo, 9,11-octadecadienoic acid | 13-KODE |
| 295 | 9-hydroxy, 10,12-octadecadienoic acid | 9-HODE |
| 295 | 13-hydroxy-9,11-octadecadienoic acid | 13-HODE |
| 295 | 9,10-epoxy, 11-octadecenoic acid | 9,10-EpOME |
| 295 | 12,13-epoxy, 9-octadecenoic acid | 12,13-EpOME |
| 309 | 9-oxo,14-hydroxy-octadecenoic acid | 9,14-KHODE |
| 309 | 8-hydroxy, 13-oxo-octadecenoic acid | 8,13-HKODE |
| 309 | 9-10-epoxy, 13-oxo-octadecenoic acid | 9,10-EKODE |
| 309 | 12-13-epoxy, 9-oxo-octadecenoic acid | 12,13-EKODE |
| 309 | 9-10-epoxy, 13-hydroxy-octadecenoic acid | |
| 309 | 12-13-epoxy, 9-hydroxy-octadecenoic acid | |
| 311 | 9-hydroperoxy-octadecadienoic acid | |
| 311 | 13-hydroperoxy-octadecadienoic acid | |
| 311 | 8,13-dihydroxy-9,11-octadecadienoic acid | 8,13-diHODE |
| 311 | 9,14-dihydroxy-10,12-octadecadienoic acid | 9,14-diHODE |
| 327 | 9-hydroperoxy, 12,13-epoxy-octadecadienoic acid | 9,12,13-HpEpOME |
Toxicity of CLox and its Lp-PLA2-mediated metabolites on cultured BPAECs.
In addition to avoiding untoward inactivation of Lp-PLA2 by persistent AAPH-generated free radical production (see addition of BHT above), we needed to identify a concentration of AAPH that was capable of oxidizing BHCL but was not directly toxic to BPAECs, as the next series of experiments involved transferring reaction products directly on to BPAECs. In Fig. 7, we note the relationship between AAPH concentration and viability of BPAECs after 2-h exposure, as assessed by alamarBlue, and thus we chose to oxidize BHCL with a concentration (5 μM) of AAPH that was not toxic to BPAECs but was capable of oxidizing BHCL (Fig. 4). After first noting that BHCL was not toxic (1–10 nmol/100 μl), we observed a concentration-dependent toxicity of AAPH-treated BHCL (e.g., CLox) that was significantly greater at 10 nmol dose of AAPH-treated BHCL in the presence of Lp-PLA2 (Fig. 8). Liposomes without CL (i.e., DOPC alone) with or without AAPH did not affect cell viability (data not shown). Further examination of the mode of cell death by analytical flow cytometry revealed that AAPH-treated BHCL increased propidium iodide staining (e.g., necrosis) from <1% at control to 36% on average, and this was further increased to 72% after addition of Lp-PLA2-treated oxidized BHCL (Fig. 9). As positive controls (data not shown), BPAECs exposed to hypotonic shock and H2O2 increased events in quadrant 1 (necrosis), whereas BPAECs treated with staurosporine increased events primarily in quadrant 4 (annexin V binding and apoptosis). A summary of several subcultures exposed in this manner indicates that necrosis of BPAECs with Lp-PLA2-treated liposomes containing CLox was greater than either Lp-PLA2-treated BHCL or CLox itself (Fig. 9).
Fig. 7.

AAPH toxicity to BPAECs. BPAEC survival after exposure to 0–10 mM AAPH. Viability of 10,000 BPAECs was determined by alamarBlue assay.
Fig. 8.

Survival of BPAECs after exposure to BHCL, AAPH, and Lp-PLA2. BPAECs (10,000 cells) were exposed to BHCL with or without Lp-PLA2 plus 5 mM of AAPH and subsequent addition of Lp-PLA2 in HBSS for 4 h, and cell viability was determined by alamarBlue assay.
Fig. 9.

Lp-PLA2 exacerbates necrotic cell death in the presence of oxidized CL. Representative flow cytometry plots of annexin V and propidium iodide staining for cell death are shown for control and 2-h exposure of 10 nmol/100 μl doses of BHCL, Lp-PLA2, CLox, and CLox + Lp-PLA2 in BPAECs (top). Bar graph representation of percentage of cells in Q1 (viable) and Q3 (necrotic) for all experiments (bottom). FACS, fluorescence-activated cell sorting.
Reaction products of CLox and Lp-PLA2 decrease endothelial cell barrier function.
BPAECs were grown on an ECIS system, and endothelial cell permeability was assessed by transendothelial cell resistance in real time. Nonlethal doses of CLox caused a concentration-dependent decrease in resistance at 1 and 10 nM. Addition of Lp-PLA2 to 1 nmol/100 μl CLox increased its barrier impairment to that of 10 nmol/100 μL CLox (Fig. 10). DOPC liposomes without CL with or without AAPH had no effect (data not shown). A positive control of thrombin is shown to provide sense of physiological changes with a predictable barrier-decreasing agent.
Fig. 10.

Lp-PLA2 exacerbated the AAPH-treated BHCL decrease in transendothelial cell electrical resistance (increase monolayer permeability). BPAECs (30,000 cells) were plated and grown to a confluent monolayer and exposed to BHCL in HBSS for 1 h, and permeability was determined by ECIS array experiments run with the 8W10E+ polyethylene terephthalate sterile disposable electrode array, with or without a nontoxic dose of AAPH with or without 2,000 ng/ml Lp-PLA2. Thrombin was used as a positive control for an increase in monolayer permeability (green line). Data from each trial were averaged at specific time points and then plotted vs. time with ± error bars reflecting standard errors from the trials. *P < 0.05.
In Fig. 11, we compare the real-time effects of addition of equimolar amounts of authentic reaction products (13-HODE, 9-HODE, or monolysoCL) to the effect of reaction mixture of 1 nmol/100 μl BHCLox and Lp-PLA2. Parts of the overall decrease in resistance attributable to the latter could be mimicked by addition of individual potential reaction products including octadecadienoic acids and monolysoCL, suggesting potential contributions of multiple species, alone and/or in combination, on endothelial cell permeability.
Fig. 11.

Lp-PLA2 hydrolysis products cause decrease in transendothelial cell electrical resistance (increase monolayer permeability). ECIS experiments for a few of the commercially available Lp-PLA2 hydrolysis products we identified (purple, orange, and blue lines) were performed. *P < 0.05.
Animal studies.
Normalized retention times on liquid chromatography (left-hand spectra) and ESI MS/MS (right-hand spectra) for CL, CLox, and monolysoCL are depicted in Fig. 12, A–C, respectively. Within each panel, we contrast results from one control mouse with a P. aeruginosa (PA103)-infected mouse. Detectable levels of CL with a retention time of 41 m and m/z of 1,447.9 (major cluster of circulating murine CL) are apparent in plasma of both control and infected mice (Fig. 12A). After infection, detectable levels of oxidized CL with an earlier retention time (26 m) and m/z of 1,463.8 are apparent only in the infected mouse (Fig. 12B). In control mice, circulating levels of monolysoCL were minimally detectable (retention time 18 m; m/z 1,185.7), whereas a more robust signal is apparent for this species after infection (Fig. 12C). In the two controls and two infected mice in which a complete spectrum including monolysoCL was examined, there was an increase from 20 mol% to 40 mol% of circulating CL appearing as monolysoCL. In Fig. 13, we show complete data from all animals regarding molar percentage of CLox and note that there was virtually no detectable CLox in the plasma of four control mice, whereas CLox molar content increased in all six infected mice to 34 ± 7.9 mol% (n = 6; P < 0.02).
Fig. 12.

CLox and monolysoCL are present in Pseudomonas aeruginosa infection. LC-MS profiles of plasma from control and mice 4 h after Pseudomonas aeruginosa infection are shown for CL (A), CLox (B), and monolysoCL (C) with chemical structures of the hypothesized Lp-PLA2 hydrolysis pathway on the left.
Fig. 13.
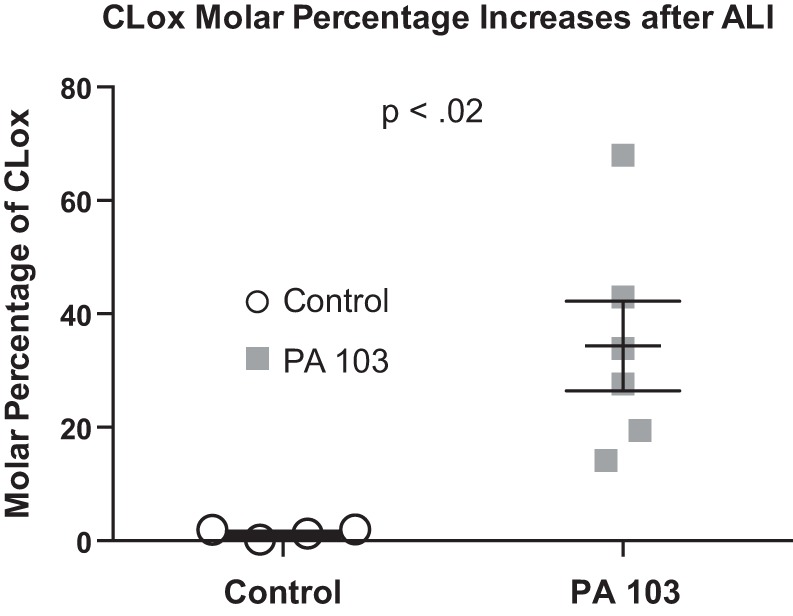
CLox molar percentage is increased in Pseudomonas aeruginosa infection. Molar percentage of CLox/(CLox + CL) was calculated from plasma of control and mice 4 h after Pseudomonas aeruginosa infection.
DISCUSSION
We developed an in vitro model system to confirm (63) that CLox (but not CL) is a substrate for Lp-PLA2 (in addition to iPLA2γ) and then assessed the biological activity of the products made on cultured BPAECs (see Fig. 2). When adventitious metals were chelated with DTPA (Fig. 3) and an antioxidant (BHT) was used to quench free radicals that may have inadvertently inactivated the enzyme (Lp-PLA2) of interest (Fig. 3), liposomes containing BHCL and DOPC would remain stable in HBSS, and the potent and promiscuous water-soluble azo initiator, AAPH, could be used to oxidize CL without affecting Lp-PLA2. Under these conditions, we noted that AAPH added a number of oxygen-containing groups to CL (Fig. 4), and the oxidizable polyunsaturated BHCL (but not fully saturated bacterial CL, data not shown) decreased (Fig. 5, bottom, left). Elements of the reaction predicted in Fig. 2 were noted by appearance of monolysoCL and dilysoCL (Fig. 5), and oxidized free fatty acids (Fig. 6) after sequential exposure to AAPH and Lp-PLA2 and a partial list of oxygenated octadecadienoic fatty acids is shown in Table 1. The requisite need for highly sensitive mass spectrometry underscores the complexity of the products made and suggests that the list of oxygenated monolysoCL, dilysoCL (Fig. 5), and octadecadienoic acids (Table 1) is a small component of this mixture based on limits of detection and purposeful focus of analytical approaches. The subsequent bioassays of reaction material on BPAECs revealed that these lipid products (lysophospholipids and oxidized free fatty acids) were toxic, resulting in a dose-dependent spectrum of changes from increased permeability (Figs. 10 and 11) to cell death by necrosis (Figs. 8 and 9). Although CLox was toxic to BPAECs, Lp-PLA2 increases in percentage of cell death (by necrosis) and permeability suggest that the hydrolytic products formed by this phospholipase, singularly or in combination, also need to be considered. Evidence suggestive that this pathway exists in vivo was noted in spectra of mice after P. aeruginosa infection showing increases in both CLox and monolysoCL (Fig. 12). CLox was only detectable in infected mice and reached almost 40% of molar content of CL (Fig. 13).
Polyunsaturated fatty acids, polyunsaturated phospholipids, and lung injury.
The focus of most efforts on lipids and lung injury involves the canonical pathway depicted in Fig. 1. In this regard, calcium-dependent activation of cytosolic PLA2 liberates arachidonic acid from its esterified position in membrane phospholipids, and arachidonic acid becomes oxidized by COX-1, COX-2, or various LOXs or P450s. The array of NSAID (and LOX- or P450-sensitive inhibitors)-sensitive oxidized free fatty acids (prostaglandins, leukotrienes, thromboxane, resolvins, alarmins, etc.) contribute in various fashion to the pathogenesis (8, 47), repair, and remodeling in ALI. Although linoleic acid (c18:2 η-6) is the most abundant of polyunsaturated fatty acids (66), it is less studied in the above context than the arachidonic or docosahexanoic acid pathways, yet linoleic acid is known to undergo lipid peroxidation enzymatically (e.g., 15-lipoxygenase) or nonenzymatically (72). Polyunsaturated phospholipids are historically viewed in the context of lipid peroxidation in conditions of oxidative stress in ALI and act as useful biomarkers with secondary products such as malonyldialdehyde (27) or along with arachidonic acid, as a source of isoprostanes (28).
CL is a disphosphatidylglycerol with four acyl chains (most commonly enriched in linoleic acid) and is normally restricted to the inner mitochondrial membrane. The specificity of CL in lipid composition of mitochondria, its unique structure (57), and its localization to the inner mitochondrial membrane (54) where it interacts with a large number of proteins account for its functional contributions as activators (13) of enzymes (oxidative phosphorylation and proton and ATP production) and regulators (24, 34) of mitochondrial transporters (phosphate and carboxylate carriers). These functions subserve metabolic interaction of mitochondria with cytosol and define CL in the context of endosymbiont in which the prokaryotic origin of mitochondria facilitates such a relationship between this organelle and the eukaryotic cytoplasm. We noted (31) that collapse of its asymmetric distribution and its ability to undergo oxidation reactions accounts for its role in quality control of damaged mitochondria (mitophagy) (35), apoptosis in prooxidant environments (33), and inflammation (30, 63). The present study extends our recent study (63) regarding oxidation of CL and hydrolysis by calcium-independent intramitochondrial iPLA2γ and inflammation. We noted in lung tissue that the reaction of CL oxidation by cytochrome c is highly selective for CL over considerably more abundant phospholipids (phosphatidylcholine or phosphatidylethanolamine) and predominantly oxidized C18:2 molecular species even though a longer chain (C20:4, C22:5, or C22:6) would predictably be more reactive (64, 65). Although CL is not a good substrate for either calcium-independent phospholipases (26), oxidized CL can be hydrolyzed by iPLA2γ or Lp-PLA2 (63). This latter work emphasized the intracellular events that would arise with oxidation of CL and hydrolysis of oxidized linoleic acid and suggested that such pathways may be important in the extracellular environment, too. Previous reports showed that inhibition of iPLA2γ by R-BEL reduced ALI following trauma and hemorrhagic shock (43) in intact rats and that thrombin and tryptase stimulated proinflammatory phenotype in cultured human lung microvascular endothelial cells (52), suggesting that such a pathway may indeed be operative. As such, we now describe an extracellular pathway involving release of CLox (or CL that subsequently gets oxidized) and then hydrolysis by Lp-PLA2 forming (oxygenated) octadecadienoic acid and lysoCL (Fig. 1) and provide a simple model system to assess the central dogma (Fig. 2) of this pathway.
Lp-PLA2.
The plasma form of Lp-PLA2 is the single source of hydrolysis of PAF, is derived from hematopoietic cells, and is largely but not exclusively synthesized and secreted by macrophages (61). Its expression is regulated at a transcriptional level by a number of pro- and anti-inflammatory molecules and ultimately distributed in association with lipoproteins. It is a novel inflammatory marker of cardiovascular disease (39), and short-term studies with darapladib in humans revealed that this agent indeed was a well-tolerated inhibitor of Lp-PLA2 (32). Nonetheless, in longer-term studies in patients with stable coronary heart disease, darapladib did not significantly reduce the risk of the primary composite end point of cardiovascular death, myocardial infarction, or stroke (69), suggesting that such strategy may not be useful for these end points and in this context, relegating Lp-PLA2 to useful biomarker status.
Before this latter experience, a number of preclinical and human trials regarding the role of PAF and its receptor (PAF-R) as well as Lp-PLA2 have been reported. As PAF is a potent neutrophil activator and proinflammatory lipid, pharmacological (11, 14, 42) and genetic (45) inhibition of PAF-R has been shown to attenuate lung injury in experimental animals. To date, however, clinical trials with PAF-R antagonists have not affected mortality of patients with acute respiratory distress syndrome (18, 50, 67). PAF levels were found to be elevated in bronchoalveolar lavage of human subjects with ALI (40, 46) and in the serum of septic patients (25). As its original name sake (PAF-acetyl hydrolase), Lp-PLA2 is conceived to be a major regulator of circulating levels of PAF (51), and noted decreases in Lp-PLA2 in sepsis (10, 16) prompted some clinical trials to infuse recombinant Lp-PLA2 and limit the course of ALI (58). Lp-PLA2 activity has been reported to be elevated in plasma and decreased in bronchoalveolar lavage in hyperoxic lung injury (29), and the opposite was the case in oleic acid injury (56). Although Lp-PLA2 is the single enzyme involved in hydrolysis of circulating PAF, Liu et al. (36) noted that transport of PAF, rather than enzymatic inactivation, dominates its early disposition in intact mice. Accordingly, it is possible that Lp-PLA2-mediated hydrolysis of oxidized phospholipids such as CLox may be a more critical function of this enzyme, and other mediators (such as octadecadienoic acids as shown in Table 1) may account for its role as inflammatory (or anti-inflammatory) in ALI.
Extracellular CLox, lysoCL, and octadecadienoic acid.
The infusion of CL into intact animals has produced several provocative findings in lung, suggesting that a pathological elevation in extracellular CL may contribute to pathophysiological aspects of ALI. Intravenous injection of CL isolated from group B streptococcus by Curtis et al. (17) was shown to mimic transient pulmonary hypertension produced by heat-killed microorganisms in newborn lambs. Ray et al. (53) reported that CL impaired surface tension lowering of surfactant and, when given intratracheally, adversely affected lung mechanics by impairing surfactant activity and ultimately producing edema and disruption of alveolar lining cells in intact mice. In addition, in this study, exogenous CL was toxic to cultured mouse lung epithelium. Neither of these early studies assessed the oxidation state of infused CL although the material prepared by Curtis et al. (17) was presumably fully saturated (e.g., bacterial CL) and somewhat immune to oxidation. As in our study, addition of oxidized (but not native) CL has proinflammatory effects on cultured endothelial cells that are blocked by annexin A5 (5, 68). The selective toxicity of CLox vs. CL is not limited to endothelial cells, as these investigators (5) observed this selective toxicity in cultured macrophages and we noted (data not shown) toxicity of CLox (but not CL) on human A549 cells.
Exacerbation of the toxicity of CLox by authentic Lp-PLA2 (Figs. 8–11) suggests that hydrolytic products (Fig. 2) including lysoCL and (oxygenated) octadecadienoic acids can contribute to the overall toxicity of extracellular CL. Relatively little is known regarding the activity of exogenous lysoCL (mono or dilyso) although elevations in plasma monolysoCL to total CL levels are vital diagnostic markers of Barth's syndrome. Barth's syndrome is an x-linked genetic disorder secondary to mutations in tafazzin, a CL-remodeling enzyme, with a potentially profound phenotype (cardiomyopathy, neutropenia, skeletal myopathy, prepubertal growth delay, and dysmorphic features). In a limited aspect of our study, exogenous monolysoCL increased permeability of BPAECs (Fig. 11). Further examination of this effect requires synthesis of a range of monolysoCL (and possibly dilysoCL) with various oxidation states of different acyl chains. In contrast, there is modest literature on the effect of extracellular octadecadienoic acids, including a profiling of their effects on airway epithelial cells (37). We noted that 9- or 13-HODE increased permeability of BPAECs (Fig. 11). This effect may be coupled to their ability to activate transient receptor potential vanilloid-type I channels (1) or may be nonreceptor mediated (disorganization of the plasma membrane). The mechanism and the specific metabolites of Lp-PLA2-mediated enhanced cell death of BPAECs (Figs. 8 and 9, Table 1) remain unclear, but the end effect is reminiscent of liponecrosis described by Titorenko and colleagues (55).
Mito-DAMP molecules.
Since the original hypothesis by Matzinger (41) regarding the paramount importance of the immune system in combating danger, it is now well accepted that PAMPs and endogenous molecules released upon tissue injury (DAMPs) signal threats of infectious and/or aseptic injurious origin, independent of discriminatory efforts of nonself- or self-identity (49). Intra- and extracellular DAMPs affect function of antigen-presenting cells and multiple other cell types. Oftentimes DAMPS are recognized by PRRs (including TLR, nucleotide oligomerization domain-like receptors, etc.) that also signal PAMPs. DAMPs may be derived from plasma membrane, secreted, or be end-stage degradation products of mitochondria (mito-DAMP), where they recapitulate incomplete symbiosis, taking on prokaryotic features (mitochondrial DNA, N-formyl peptides) (9, 19). Mito-DAMPs released after different modes of death can communicate with cellular components of innate and adaptive immune systems and have profound inflammatory and immunomodulatory effects (73). Circulating mitochondrial DAMPs cause inflammatory responses to injury, linking trauma and inflammation, oxidative stress and ischemic injury (4, 23). Overlapping activities of PAMPs, DAMPs, and PRRs are apparent in ALI, including critical roles for pulmonary endothelium and neutrophils in ALI after hemorrhagic shock (71) and Toll receptors in aseptic hyperoxic lung injury (44). It is possible that mitochondrial lipids (i.e., CL and CLox) are themselves mito-DAMPs and in the process of cell death are extruded to the extracellular space. As noted above, we (5) recently found that membranes with mitochondrial or bacterial CLs on their surface were engulfed through phagocytosis, which depended on the scavenger receptor CD36. Distinct from this process, the copresentation of CL with TLR4 agonist lipopolysaccharide dampened TLR4-stimulated production of cytokines. These data suggest that externalized, extracellular CLs play a dual role in host-host and host-pathogen interactions by promoting phagocytosis and attenuating inflammatory immune responses and underscore their potential role as mito-DAMPs. The proinflammatory effects of extracellular CLox (68) and the toxicity on BPAECs noted in our present study suggest that circulating plasma CLox (and its hydrolytic products), originally derived from its mitochondrial location, may interact with its likely target, e.g., endothelium, and contribute to the interface of cell injury and innate and adaptive immunity. In this regard, we noted that CLox comprised a significantly large fraction of circulating CL after infection with P. aeruginosa in intact mice (Fig. 13), and monolysoCL increased as a fraction of total CL in plasma of the two mice examined after P. aeruginosa (Fig. 12). From these simple steady-state measurements, we could not deduce whether CL was oxidized (and hydrolyzed) within cells and then released or whether CL was released and postsynthetically modified in the vascular space. As such, formation of increased amounts of monolysoCL may have been catalyzed by either of two iPLA2 isoforms, iPLA2γ or Lp-PLA2. These studies will require genetic and pharmacological manipulations as well as more detailed analysis within the plasma space of oxygenated forms of linoleic acid.
In conclusion, a simple in vitro model showed that CLox (but not native or fully saturated CL) and its Lp-PLA2-mediated hydrolytic products (mono- and dilysoCL; oxygenated octadecadienoic acid) were toxic to BPAECs. At low doses, there was an increase in permeability of representative products and its mixture; at higher doses, death occurred by necrosis (not apoptosis). It is possible that release of CL (native and oxidized) from its normal restricted location in the inner mitochondrial membrane (either by secretion or cell death, itself) and release of iPLA2γ-mediated hydrolytic products to the extracellular milieu and/or hydrolysis of CLox by extracellular Lp-PLA2 are important signaling pathways in inflammation, akin to mito-DAMP. Along with our original description (63) of a mitochondrial NSAID insensitivity biosynthetic pathway for oxidized free fatty acid production (Fig. 1), this pathway (Fig. 2) greatly enhances the possible modes of communication of CLox beyond its clear role in cellular apoptosis (33), mitophagy (15), and other aspects of inflammation (30, 63) and immunity (5).
GRANTS
This work was supported in part by grants from the University of Pittsburgh Vascular Medicine Institute, Children's Hospital of Pittsburgh Scholars Award, NIH PO1 114,453, UL1 TR000005, and K12HD052892.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
JRB was involved in conception and design of research, collected, analyzed, interpreted data, and drafted the manuscript. KAW was involved in the conception and design of research, collected, analyzed, interpreted data, and edited the manuscript. VT was involved in the design of research, collected, analyzed, interpreted data, and edited the manuscript. YT was involved in the design of research, collected, analyzed, interpreted data, and edited the manuscript. AAA collected, analyzed, and interpreted data and edited the manuscript. SOA was involved in conception and design of research. RKM was involved in the design of research and collected, analyzed, and interpreted data. BBC was involved in the design of research. JZ was involved in the design of research and collected, analyzed, and interpreted data. YZ was involved in the design of research collected, analyzed, interpreted data, and edited the manuscript. VEK was involved in conception and design of research, interpretation of data, editing, and revising of the final manuscript. BRP was involved in conception and design of research, interpretation of data, and editing, revising, and approval of final manuscript.
ACKNOWLEDGMENTS
The authors thank Danielle Crosby and Diane Lenhart for technical assistance and Samantha Taylor and Ying Shan for assistance with statistical analysis.
Glossary
- AAPH
2,2′Azobis (2-methylpropionamidine) dihydrochloride
- ALI
Acute lung injury
- BHCL
Bovine heart cardiolipin
- BHT
Butylated hydroxytoluene
- BPAECs
Bovine pulmonary artery endothelial cells
- CL
Cardiolipin
- CLox
Oxidized cardiolipin
- COX
Cyclooxygenase
- DAMPS
Damage-associated molecular patterns
- DilysoCL
Dilyso cardiolipin
- DTPA
Diethylenetriaminepentaacetic acid
- DOPC
Dioleoylphosphatidyl choline
- ECIS
Electric cell impedance sensing
- EPR
Electron paramagnetic resonance spectroscopy
- ESI MS/MS
Electrospray ionization-tandem mass spectrometry
- HODE
Hydroxyoctadecadienoic
- iPLA2γ
Calcium-independent phospholipase A2-γ
- LC-MS
Liquid chromatography-mass spectrometry
- LOX
Lipoxygenase
- Lp-PLA2
Lipoprotein phospholipase A2
- MonolysoCL
Monolyso cardiolipin
- P. aeruginosa
Psuedomonas aeruginosa
- PA103
Pseudomonas aeruginosa strain PA103
- PAF
Platelet-activating factor
- PAF-AH
Platelet-activating factor acetylhydrolase
- PAMPs
Pathogen-associated molecular patterns
- PLA2
Phospholipase A2
- PRRs
Pattern recognition receptors
- R-BEL
R-bromoenol lactone
- TLR
Toll-like receptor
REFERENCES
- 1.Alsalem M, Wong A, Millns P, Arya PH, Chan MS, Bennett A, Barrett DA, Chapman V, Kendall DA. The contribution of the endogenous TRPV1 ligands 9-HODE and 13-HODE to nociceptive processing and their role in peripheral inflammatory pain mechanisms. Br J Pharmacol 168: 1961–1974, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aoki H, Hisada T, Ishizuka T, Utsugi M, Kawata T, Shimizu Y, Okajima F, Dobashi K, Mori M. Resolvin E1 dampens airway inflammation and hyperresponsiveness in a murine model of asthma. Biochem Biophys Res Commun 367: 509–515, 2008. [DOI] [PubMed] [Google Scholar]
- 3.Ariel A, Serhan CN. Resolvins and protectins in the termination program of acute inflammation. Trends Immunol 28: 176–183, 2007. [DOI] [PubMed] [Google Scholar]
- 4.Arslan F, de Kleijn DP, Pasterkamp G. Innate immune signaling in cardiac ischemia. Nat Rev Cardiol 8: 292–300, 2011. [DOI] [PubMed] [Google Scholar]
- 5.Balasubramanian K, Maeda A, Lee JS, Mohammadyani D, Dar HH, Jiang JF, St Croix CM, Watkins S, Tyurin VA, Tyurina YY, Kloditz K, Polimova A, Kapralova VI, Xiong Z, Ray P, Klein-Seetharaman J, Mallampalli RK, Bayir H, Fadeel B, Kagan VE. Dichotomous roles for externalized cardiolipin in extracellular signaling: Promotion of phagocytosis and attenuation of innate immunity. Sci Signal 8: ra95, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bannenberg G, Arita M, Serhan CN. Endogenous receptor agonists: resolving inflammation. ScientificWorldJournal 7: 1440–1462, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bannenberg G, Serhan CN. Specialized pro-resolving lipid mediators in the inflammatory response: An update. Biochim Biophys Acta 1801: 1260–1273, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bonnans C, Levy BD. Lipid mediators as agonists for the resolution of acute lung inflammation and injury. Am J Respir Cell Mol Biol 36: 201–205, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Calfee CS, Matthay MA. Clinical immunology: Culprits with evolutionary ties. Nature 464: 41–42, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cao Y, Stafforini DM, Zimmerman GA, McIntyre TM, Prescott SM. Expression of plasma platelet-activating factor acetylhydrolase is transcriptionally regulated by mediators of inflammation. J Biol Chem 273: 4012–4020, 1998. [DOI] [PubMed] [Google Scholar]
- 11.Chang SW, Feddersen CO, Henson PM, Voelkel NF. Platelet-activating factor mediates hemodynamic changes and lung injury in endotoxin-treated rats. J Clin Invest 79: 1498–1509, 1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen BB, Coon TA, Glasser JR, McVerry BJ, Zhao J, Zhao Y, Zou C, Ellis B, Sciurba FC, Zhang Y, Mallampalli RK. A combinatorial F box protein directed pathway controls TRAF adaptor stability to regulate inflammation. Nat Immunol 14: 470–479, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chicco AJ, Sparagna GC. Role of cardiolipin alterations in mitochondrial dysfunction and disease. Am J Physiol Cell Physiol 292: C33–C44, 2007. [DOI] [PubMed] [Google Scholar]
- 14.Christman BW, Lefferts PL, Blair IA, Snapper JR. Effect of platelet-activating factor receptor antagonism on endotoxin-induced lung dysfunction in awake sheep. Am Rev Respir Dis 142: 1272–1278, 1990. [DOI] [PubMed] [Google Scholar]
- 15.Chu CT, Ji J, Dagda RK. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat Cell Biol 15: 1197–1205, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Claus RA, Russwurm S, Dohrn B, Bauer M, Losche W. Plasma platelet-activating factor acetylhydrolase activity in critically ill patients. Crit Care Med 33: 1416–1419, 2005. [DOI] [PubMed] [Google Scholar]
- 17.Curtis J, Kim G, Wehr NB, Levine RL. Group B streptococcal phospholipid causes pulmonary hypertension. Proc Natl Acad Sci USA 100: 5087–5090, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dhainaut JF, Tenaillon A, Hemmer M, Damas P, Le Tulzo Y, Radermacher P, Schaller MD, Sollet JP, Wolff M, Holzapfel L, Zeni F, Vedrinne JM, de Vathaire F, Gourlay ML, Guinot P, Mira JP. Confirmatory platelet-activating factor receptor antagonist trial in patients with severe gram-negative bacterial sepsis: a phase III, randomized, double-blind, placebo-controlled, multicenter trial. BN 52021 Sepsis Investigator Group. Crit Care Med 26: 1963–1971, 1998. [DOI] [PubMed] [Google Scholar]
- 19.Dieude M, Striegl H, Tyznik AJ, Wang J, Behar SM, Piccirillo CA, Levine JS, Zajonc DM, Rauch J. Cardiolipin binds to CD1d and stimulates CD1d-restricted gammadelta T cells in the normal murine repertoire. J Immunol 186: 4771–4781, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Erukhimov JA, Tang ZL, Johnson BA, Donahoe MP, Razzack JA, Gibson KF, Lee WM, Wasserloos KJ, Watkins SA, Pitt BR. Actin-containing sera from patients with adult respiratory distress syndrome are toxic to sheep pulmonary endothelial cells. Am J Respir Crit Care Med 162: 288–294, 2000. [DOI] [PubMed] [Google Scholar]
- 21.Folch J, Lees M, Sloane Stanley GH. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem 226: 497–509, 1957. [PubMed] [Google Scholar]
- 22.George CL, Fantuzzi G, Bursten S, Leer L, Abraham E. Effects of lisofylline on hyperoxia-induced lung injury. Am J Physiol Lung Cell Mol Physiol 276: L776–L785, 1999. [DOI] [PubMed] [Google Scholar]
- 23.Gill R, Tsung A, Billiar T. Linking oxidative stress to inflammation: Toll-like receptors. Free Radic Biol Med 48: 1121–1132, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gohil VM, Greenberg ML. Mitochondrial membrane biogenesis: phospholipids and proteins go hand in hand. J Cell Biol 184: 469–472, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Graham RM, Stephens CJ, Silvester W, Leong LL, Sturm MJ, Taylor RR. Plasma degradation of platelet-activating factor in severely ill patients with clinical sepsis. Crit Care Med 22: 204–212, 1994. [DOI] [PubMed] [Google Scholar]
- 26.Hsu YH, Dumlao DS, Cao J, Dennis EA. Assessing phospholipase A2 activity toward cardiolipin by mass spectrometry. PLoS One 8: e59267, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jamieson D, Chance B, Cadenas E, Boveris A. The relation of free radical production to hyperoxia. Annu Rev Physiol 48: 703–719, 1986. [DOI] [PubMed] [Google Scholar]
- 28.Janssen LJ. Isoprostanes and lung vascular pathology. Am J Respir Cell Mol Biol 39: 383–389, 2008. [DOI] [PubMed] [Google Scholar]
- 29.Jehle R, Schlame M, Buttner C, Frey B, Sinha P, Rustow B. Platelet-activating factor (PAF)-acetylhydrolase and PAF-like compounds in the lung: effects of hyperoxia. Biochim Biophys Acta 1532: 60–66, 2001. [DOI] [PubMed] [Google Scholar]
- 30.Ji J, Baart S, Vikulina AS, Clark RS, Anthonymuthu TS, Tyurin VA, Du L, St Croix CM, Tyurina YY, Lewis J, Skoda EM, Kline AE, Kochanek PM, Wipf P, Kagan VE, Bayir H. Deciphering of mitochondrial cardiolipin oxidative signaling in cerebral ischemia-reperfusion. J Cereb Blood Flow Metab 35: 319–328, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jiang JF, Tyurina YY, Kapralov AA, Tyurin VA, Yanamala N, Shrivastava IH, Mohammadyani D, Qiang Wang KZ, Zhu J, Klein-Seetharaman J, Balasubramanian K, Amoscato AA, Borisenko G, Huang Z, Gusdon AM, Cheikhi A, Steer EK, Wang R, Baty C, Watkins S, Bahar I, Bayir H, Kagan VE, Kagan VE, Tyurina YY, Tyurin VA, Mohammadyani D, Angeli JP, Baranov SV, Klein-Seetharaman J, Friedlander RM, Mallampalli RK, Conrad M, Bayir H. Cardiolipin signaling mechanisms: collapse of asymmetry and oxidation. Nat Cell Biol 22: 1667–1680, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Johnson JL, Shi Y, Snipes R, Janmohamed S, Rolfe TE, Davis B, Postle A, Macphee CH. Effect of darapladib treatment on endarterectomy carotid plaque lipoprotein-associated phospholipase A2 activity: a randomized, controlled trial. PLoS One 9: e89034, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kagan VE, Tyurin VA, Jiang J, Tyurina YY, Ritov VB, Amoscato AA, Osipov AN, Belikova NA, Kapralov AA, Kini V, Vlasova II, Zhao Q, Zou M, Di P, Svistunenko DA, Kurnikov IV, Borisenko GG. Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat Chem Biol 1: 223–232, 2005. [DOI] [PubMed] [Google Scholar]
- 34.Klingenberg M. Cardiolipin and mitochondrial carriers. Biochim Biophys Acta 1788: 2048–2058, 2009. [DOI] [PubMed] [Google Scholar]
- 35.Levy BD, Serhan CN. Resolution of acute inflammation in the lung. Annu Rev Physiol 76: 467–492, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu J, Chen R, Marathe GK, Febbraio M, Zou W, McIntyre TM. Circulating platelet-activating factor is primarily cleared by transport, not intravascular hydrolysis by lipoprotein-associated phospholipase A2/ PAF acetylhydrolase. Circ Res 108: 469–477, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mabalirajan U, Ahmad T, Rehman R, Leishangthem GD, Dinda AK, Agrawal A, Ghosh B, Sharma SK. Baicalein reduces airway injury in allergen and IL-13 induced airway inflammation. PLoS One 8: e62916, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mabalirajan U, Rehman R, Ahmad T, Kumar S, Singh S, Leishangthem GD, Aich J, Kumar M, Khanna K, Singh VP, Dinda AK, Biswal S, Agrawal A, Ghosh B. Linoleic acid metabolite drives severe asthma by causing airway epithelial injury. Sci Rep 3: 1349, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Macphee CH, Nelson JJ, Zalewski A. Lipoprotein-associated phospholipase A2 as a target of therapy. Curr Opin Lipid 16: 442–446, 2005. [DOI] [PubMed] [Google Scholar]
- 40.Matsumoto K, Taki F, Kondoh Y, Taniguchi H, Takagi K. Platelet-activating factor in bronchoalveolar lavage fluid of patients with adult respiratory distress syndrome. Clin Exp Pharmacol Physiol 19: 509–515, 1992. [DOI] [PubMed] [Google Scholar]
- 41.Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol 12: 991–1045, 1994. [DOI] [PubMed] [Google Scholar]
- 42.Miotla JM, Jeffery PK, Hellewell PG. Platelet-activating factor plays a pivotal role in the induction of experimental lung injury. Am J Respir Cell Mol Biol 18: 197–204, 1998. [DOI] [PubMed] [Google Scholar]
- 43.Morishita K, Aiboshi J, Kobayashi T, Yokoyama Y, Mikami S, Kumagai J, Onisawa K, Otomo Y. Group VIB Ca(2+)-independent phospholipase A(2gamma) is associated with acute lung injury following trauma and hemorrhagic shock. J Trauma Acute Care Surg 75: 767–774, 2013. [DOI] [PubMed] [Google Scholar]
- 44.Murray LA, Knight DA, McAlonan L, Argentieri R, Joshi A, Shaheen F, Cunningham M, Alexopolou L, Flavell RA, Sarisky RT, Hogaboam CM. Deleterious role of TLR3 during hyperoxia-induced acute lung injury. Am J Respir Crit Care Med 178: 1227–1237, 2008. [DOI] [PubMed] [Google Scholar]
- 45.Nagase T, Ishii S, Kume K, Uozumi N, Izumi T, Ouchi Y, Shimizu T. Platelet-activating factor mediates acid-induced lung injury in genetically engineered mice. J Clin Invest 104: 1071–1076, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nakos G, Kitsiouli EI, Tsangaris I, Lekka ME. Bronchoalveolar lavage fluid characteristics of early intermediate and late phases of ARDS. Alterations in leukocytes, proteins, PAF and surfactant components. Intens Care Med 24: 296–303, 1998. [DOI] [PubMed] [Google Scholar]
- 47.Ott J, Hiesgen C, Mayer K. Lipids in critical care medicine. Prostaglandins Leukot Essent Fatty Acids 85: 267–273, 2011. [DOI] [PubMed] [Google Scholar]
- 48.Page B, Page M, Noel C. A new fluorometric assay for cytotoxicity measurements in-vitro. Int J Oncol 3: 473–476, 1993. [PubMed] [Google Scholar]
- 49.Piccinini AM, Midwood KS. DAMPening inflammation by modulating TLR signalling. Med Inf (Lond) 2010: 672395, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Poeze M, Froon AH, Ramsay G, Buurman WA, Greve JW. Decreased organ failure in patients with severe SIRS and septic shock treated with the platelet-activating factor antagonist TCV-309: a prospective, multicenter, double-blind, randomized phase II trial. TCV-309 Septic Shock Study Group. Shock 14: 421–428, 2000. [DOI] [PubMed] [Google Scholar]
- 51.Prescott SM, Zimmerman GA, Stafforini DM, McIntyre TM. Platelet-activating factor and related lipid mediators. Annu Rev Biochem 69: 419–445, 2000. [DOI] [PubMed] [Google Scholar]
- 52.Rastogi P, McHowat J. Inhibition of calcium-independent phospholipase A2 prevents inflammatory mediator production in pulmonary microvascular endothelium. Respir Physiol Neurobiol 165: 167–174, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ray NB, Durairaj L, Chen BB, McVerry BJ, Ryan AJ, Donahoe M, Waltenbaugh AK, O'Donnell CP, Henderson FC, Etscheidt CA, McCoy DM, Agassandian M, Hayes-Rowan EC, Coon TA, Butler PL, Gakhar L, Mathur SN, Sieren JC, Tyurina YY, Kagan VE, McLennan G, Mallampalli RK. Dynamic regulation of cardiolipin by the lipid pump Atp8b1 determines the severity of lung injury in experimental pneumonia. Nat Med 16: 1120–1127, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Renner LD, Weibel DB. Cardiolipin microdomains localize to negatively curved regions of Escherichia coli membranes. Proc Natl Acad Sci USA 108: 6264–6269, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Richard VR, Beach A, Piano A, Leonov A, Feldman R, Burstein MT, Kyryakov P, Gomez-Perez A, Arlia-Ciommo A, Baptista S, Campbell C, Goncharov D, Pannu S, Patrinos D, Sadri B, Svistkova V, Victor A, Titorenko VI. Mechanism of liponecrosis, a distinct mode of programmed cell death. Cell Cycle 13: 3707–3726, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Salluh JI, Pino AV, Silva AR, Gomes RN, Souza HS, e Silva JR, Jandre FC, Giannella-Neto A, Zimmerman GA, Stafforini DM, Prescott SM, Castro-Faria-Neto HC, Bozza PT, Bozza FA. Lung production of platelet-activating factor acetylhydrolase in oleic acid-induced acute lung injury. Prostaglandins Leukot Essent Fatty Acids 77: 1–8, 2007. [DOI] [PubMed] [Google Scholar]
- 57.Schlame M. Cardiolipin synthesis for the assembly of bacterial and mitochondrial membranes. J Lipid Res 49: 1607–1620, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schuster DP, Metzler M, Opal S, Lowry S, Balk R, Abraham E, Levy H, Slotman G, Coyne E, Souza S, Pribble J. Recombinant platelet-activating factor acetylhydrolase to prevent acute respiratory distress syndrome and mortality in severe sepsis: Phase IIb, multicenter, randomized, placebo-controlled, clinical trial. Crit Care Med 31: 1612–1619, 2003. [DOI] [PubMed] [Google Scholar]
- 59.Schwab JM, Chiang N, Arita M, Serhan CN. Resolvin E1 and protectin D1 activate inflammation-resolution programmes. Nature 447: 869–874, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sparvero LJ, Amoscato AA, Dixon CE, Long JB, Kochanek PM, Pitt BR, Bayir H, Kagan VE. Mapping of phospholipids by MALDI imaging (MALDI-MSI): realities and expectations. Chem Phys Lipids 165: 545–562, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stafforini DM. Biology of platelet-activating factor acetylhydrolase (PAF-AH, lipoprotein associated phospholipase A2). Cardiovasc Drugs Ther 23: 73–83, 2009. [DOI] [PubMed] [Google Scholar]
- 62.Tyurina YY, Kisin ER, Murray A, Tyurin VA, Kapralova VI, Sparvero LJ, Amoscato AA, Samhan-Arias AK, Swedin L, Lahesmaa R, Fadeel B, Shvedova AA, Kagan VE. Global phospholipidomics analysis reveals selective pulmonary peroxidation profiles upon inhalation of single-walled carbon nanotubes. ACS Nano 5: 7342–7353, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tyurina YY, Poloyac SM, Tyurin VA, Kapralov AA, Jiang J, Anthonymuthu TS, Kapralova VI, Vikulina AS, Jung MY, Epperly MW, Mohammadyani D, Klein-Seetharaman J, Jackson TC, Kochanek PM, Pitt BR, Greenberger JS, Vladimirov YA, Bayir H, Kagan VE. A mitochondrial pathway for biosynthesis of lipid mediators. Nat Chem 6: 542–552, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tyurina YY, Tyurin VA, Kapralova VI, Wasserloos K, Mosher M, Epperly MW, Greenberger JS, Pitt BR, Kagan VE. Oxidative lipidomics of gamma-radiation-induced lung injury: mass spectrometric characterization of cardiolipin and phosphatidylserine peroxidation. Radiat Res 175: 610–621, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tyurina YY, Tyurin VA, Kaynar AM, Kapralova VI, Wasserloos K, Li J, Mosher M, Wright L, Wipf P, Watkins S, Pitt BR, Kagan VE. Oxidative lipidomics of hyperoxic acute lung injury: mass spectrometric characterization of cardiolipin and phosphatidylserine peroxidation. Am J Physiol Lung Cell Mol Physiol 299: L73–L85, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vangaveti VN, Jansen H, Kennedy RL, Malabu UH. Hydroxyoctadecadienoic acids: Oxidised derivatives of linoleic acid and their role in inflammation associated with metabolic syndrome and cancer. Eur J Pharmacol. In press. [DOI] [PubMed] [Google Scholar]
- 67.Vincent JL, Spapen H, Bakker J, Webster NR, Curtis L. Phase II multicenter clinical study of the platelet-activating factor receptor antagonist BB-882 in the treatment of sepsis. Crit Care Med 28: 638–642, 2000. [DOI] [PubMed] [Google Scholar]
- 68.Wan M, Hua X, Su J, Thiagarajan D, Frostegard AG, Haeggstrom JZ, Frostegard J. Oxidized but not native cardiolipin has pro-inflammatory effects, which are inhibited by Annexin A5. Atherosclerosis 235: 592–598, 2014. [DOI] [PubMed] [Google Scholar]
- 69.White HD, Held C, Stewart R, Tarka E, Brown R, Davies RY, Budaj A, Harrington RA, Steg PG, Ardissino D, Armstrong PW, Avezum A, Aylward PE, Bryce A, Chen H, Chen MF, Corbalan R, Dalby AJ, Danchin N, De Winter RJ, Denchev S, Diaz R, Elisaf M, Flather MD, Goudev AR, Granger CB, Grinfeld L, Hochman JS, Husted S, Kim HS, Koenig W, Linhart A, Lonn E, Lopez-Sendon J, Manolis AJ, Mohler ER 3rd, Nicolau JC, Pais P, Parkhomenko A, Pedersen TR, Pella D, Ramos-Corrales MA, Ruda M, Sereg M, Siddique S, Sinnaeve P, Smith P, Sritara P, Swart HP, Sy RG, Teramoto T, Tse HF, Watson D, Weaver WD, Weiss R, Viigimaa M, Vinereanu D, Zhu J, Cannon CP, Wallentin L. Darapladib for preventing ischemic events in stable coronary heart disease. N Engl J Med 370: 1702–1711, 2014. [DOI] [PubMed] [Google Scholar]
- 70.Wipf P, Xiao J, Jiang J, Belikova NA, Tyurin VA, Fink MP, Kagan VE. Mitochondrial targeting of selective electron scavengers: synthesis and biological analysis of hemigramicidin-TEMPO conjugates. J Am Chem Soc 127: 12460–12461, 2005. [DOI] [PubMed] [Google Scholar]
- 71.Xiang M, Fan J. Pattern recognition receptor-dependent mechanisms of acute lung injury. Mol Med 16: 69–82, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yoshida Y, Umeno A, Akazawa Y, Shichiri M, Murotomi K, Horie M. Chemistry of lipid peroxidation products and their use as biomarkers in early detection of diseases. J Oleo Sci 64: 347–356, 2015. [DOI] [PubMed] [Google Scholar]
- 73.Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, Brohi K, Itagaki K, Hauser CJ. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464: 104–107, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]