

Abstract
The majority of the animal models of acute lung injury (ALI) are focused on the acute phase. This limits the studies of the mechanisms involved in later phases and the effects of long-term treatments. Thus the goal of this study was to develop an experimental ALI model of aspiration pneumonia, in which diffuse alveolar damage continues for 72 h. Rats were intratracheally instilled with one dose of HCl (0.1 mol/l) followed by another instillation of one dose of LPS (0, 10, 20, 30, or 40 μg/g body weight) 2 h later, which models aspiration of gastric contents that progresses to secondary lung injury from bacteria or bacterial products. The rats were euthanized at 24, 48, and 72 h after the last instillation. The results showed that HCl and LPS at all doses caused activation of inflammatory responses, increased protein permeability and apoptosis, and induced mild hypoxemia in rat lungs at 24 h postinstillation. However, this lung damage was present at 72 h only in rats receiving HCl and LPS at the doses of 30 and 40 μg/g body wt. Mortality (∼50%) occurred in the first 48 h and only in the rats treated with HCl and LPS at the highest dose (40 μg/g body wt). In conclusion, intratracheal instillation of HCl followed by LPS at the dose of 30 μg/g body wt results in severe diffuse alveolar damage that continues at least 72 h. This rat model of aspiration pneumonia-induced ALI will be useful for testing long-term effects of new therapeutic strategies in ALI.
Keywords: rat model, gastric aspiration pneumonia, acute lung injury, LPS, VEGF
gastric aspiration is a recognized complication in ICU patients with altered states of consciousness, such as patients with stroke or head trauma, or patients with alterations in sensorium due to alcohol or drugs. Gastric aspiration is a major cause of direct acute respiratory distress syndrome (ARDS) (17, 26, 31), which is associated with high mortality rates (5). Aspiration of sterile oropharyngeal or gastric contents with or without food contents into the lower respiratory tract initially leads to aspiration pneumonitis. In some cases, aspiration pneumonitis can progress to pneumonia when aspiration of bacteria from the oral cavity or nasopharynx to the lower airway occurs on the previously injured lung (17, 26). Aspiration pneumonitis with or without pneumonia frequently leads to diffuse alveolar damage, the histopathological manifestation of ARDS. However, the mechanisms involved in the onset and progression of aspiration-induced ARDS are not fully understood.
Aspiration pneumonitis has been widely modeled in different animals using hydrochloric acid (HCl) (2, 11, 12, 25) or sterile gastric contents instillations with or without particulate food matter from the stomach (4, 13, 14). However, to our knowledge, only a few experimental studies have investigated the pathogenesis of pneumonia in the host with aspiration pneumonitis (18, 23, 27, 30, 32). These previous studies have shown that aspiration of gastric acid strongly enhances pulmonary inflammation induced by lipopolysaccharide (LPS) (32) or bacteria (18, 30). In addition, aspiration of HCl (18, 30) or gastric contents (27) impairs pulmonary bacterial clearance, which can facilitate the development of pneumonia. All these studies, however, have been focused on the mechanisms involved in the pathogenesis of acute lung injury in its early phase, in part, because most of the animal models of ARDS show a spontaneous recovery of lung injury in 24 or 48 h after the initial insult. Therefore, animal models with more lasting lung damage are needed to study the mechanisms involved in the progression of acute lung injury, and also to test the effect of different treatments in later time points after the onset of this disease.
The goal was to develop a rat model of aspiration pneumonitis/endotoxin-induced acute lung injury using intratracheal instillation of HCl combined with different doses of LPS. We expected that this combination of injury would cause rapid development of severe diffuse alveolar damage, lasting at least 72 h. This should be useful for testing new therapeutic strategies in preclinical models of ARDS.
METHODS
Reagents.
Endotoxin (LPS) from Escherichia coli 055:B5 was purchased from Sigma-Aldrich (St. Louis, MO). For immunoblotting, we used anti-cleaved caspase-3 mouse monoclonal antibody from Cell Signaling (Boston, MA) and anti-actin mouse monoclonal antibody from Sigma- Aldrich. The secondary antibody, goat anti-mouse Texas Red, was purchased from Santa Cruz (Dallas, TX).
Animal acute lung injury model.
The rat protocol was approved by the Animal Research Ethics Committee of the Universitat Autònoma de Barcelona and followed the principles of the Generalitat de Catalunya (Art. 32 of decree 214/1997). For the experiments, we used 116 adult male Sprague-Dawley rats (Charles River; Margate, UK) that were housed in standard conditions with food and water ad libitum. Male rats were anesthetized with inhaled isoflurane (2–5%) and treated with one intratracheal instillation of HCl (1.2 μl/g, 0.1 mol/l pH = 1.4) dissolved in 300 μl of sterile saline. Two hours later, these rats received one intratracheal instillation of LPS at different concentrations (10, 20, 30, or 40 μg/g body wt) dissolved in 500 μl of sterile saline, or saline alone. As control, rats received the same volume of saline instead of HCl or LPS via intratracheal instillation. Because the lungs of rats lack pulmonary intravascular macrophages (PIM) (21), high doses of LPS, in the range of micrograms per gram, were used in order to observe manifestation of pulmonary damage.
In summary, animals were randomly distributed to one of the six experimental groups: 1) Control: one instillation of saline followed by one instillation of saline 2 h later; 2) HCl/Saline: one instillation of HCl followed by one instillation of saline 2 h later; 3) HCl/LPS-10: one instillation of HCl followed by one instillation of LPS at the dose of 10 μg/g body wt 2 h later; 4) HCl/LPS-20: one instillation of HCl followed by one instillation of LPS at the dose of 20 μg/g body wt 2 h later; 5) HCl/LPS-30: one instillation of HCl followed by one instillation of LPS at the dose of 30 μg/g body wt 2 h later; and 6) HCl/LPS-40: one instillation of HCl followed by one instillation of LPS at the dose of 40 μg/g body wt 2 h later.
After each instillation, the rats were allowed to recover from anesthesia, returned to their ventilated cages with air, and provided with free access to food and water. After LPS or saline instillation, animal body weight was recorded every 24 h. Animals were also prerandomized to be euthanatized at a specific time point (24, 48, or 72 h), by exsanguination through the abdominal aorta. Also, some rats were euthanized by the same method at 2 h. Animals that died before the specific time point assigned (24, 48, or 72 h) were excluded entirely from the study. The thorax was opened rapidly, and the trachea and lungs were removed in bloc and weighted. Then the left hilum was tied with a suture, and the right lung was removed and flash frozen in liquid nitrogen. The left lung was lavaged with 5 separate 5-ml aliquots of 0.9% NaCl containing 1 mM EDTA. In another series of experiments, the lungs were fixed by intratracheal instillation of 4% paraformaldehyde at a transpulmonary pressure of 15 cmH2O, immersed in the same fixative for 24 ± 12 h, and then embedded in paraffin.
Physiological measurements.
To conduct standardized assessment of pulmonary gas exchange capability and respiratory mechanics, rats receiving HCl and LPS at the dose of 30 μg/g body wt and control rats treated with saline/saline were weighed and underwent surgical preparation for mechanical ventilation at the specified end points of the experiments (24, 48, or 72 h). Mean arterial pressure was first measured using a noninvasive tail-cuff system (Phillips Medizin System; Boeblingen, Germany) in unanesthetized and unrestrained rats (15). Then rats were anesthetized with intramuscular ketamine (75 mg/kg) (ParkeDavis; El Prat de Llobregat, Spain) and xylazine (10 ml/kg) (Rompun Bayer; Barcelona, Spain). At this moment, blood was obtained from the femoral artery for measuring arterial blood gases (Epocal; Ottawa, Canada). Subsequently, rats were secured in supine position, orotracheally intubated with a 22-gauge catheter, tightly tied to avoid air leaks, and connected to a Harvard 7025 rodent ventilator (Ugo Basile; Varesse, Italy). All animals were ventilated for 20 min in volume-controlled ventilation mode with 6 ml/kg Vt, 2 cmH2O positive end-expiratory pressure (PEEP), and 0.21 FiO2. Respiratory mechanics was determined using the end-inspiratory occlusion technique. Static lung compliance was obtained after the assessment of P-V curves. Then rats were euthanized by exsanguination.
Analysis of rat bronchoalveolar lavage fluid.
The BAL fluid samples were processed immediately for total and differential cell counts. Total white cell counts were performed with a hemacytometer, and differential counts were performed on cytospin preparations stained with the Diff-quick method (Pancreac Quimica SAU; Spain). For each rat, a minimum of 200 cells were counted using light microscopy. The remainder of the lavage fluid was centrifuged at 1,000 g for 10 min, and the cell-free supernatant was removed aseptically and stored in individual aliquots at −80°C. The total protein concentration in BAL fluid was determined by the bicinchoninic acid method (Pierce, Thermo Scientific; Rockford, IL), and the concentration of IgM in BAL fluid was measured using an ELISA (Alpha diagnostic International; San Antonio, TX), according to the manufacturer's instructions. The lower limit of detection of the IgM assay was 4.6 ng/ml.
Histological methods in rat lung tissue.
Paraffin-embedded rat lungs were cut into 4-μm thick sections. The lung sections were stained with hematoxylin-eosin (H&E) for light microscopy. The TUNEL (TdT-mediated dUTP-nick end labeling) fluorescent assay for in situ detection of fragmented DNA was performed according to the manufacturer's instructions (Roche Applied Science; Barcelona, Spain) in lung sections from rats receiving HCl and LPS at the dose of 30 μg/g body wt and control rats treated with saline/saline at 24, 48, and 72 h. Briefly, deparaffinized sections were rehydrated, permeabilized, and digested with proteinase K (Dako; Agilent Technologies, Barcelona, Spain) at 37°C for 30 min and then washed in PBS for 15 min. Sections were then incubated with TUNEL reaction mixture that contains TdT and fluorescein-dUTP, at 37°C for 1 h in the dark. Slides were washed again in PBS and mounted with Fluoromount Aqueous Mounting Medium (Sigma; St. Louis, MO). Light and fluorescence microscopy were performed using a Nikon Eclipse Ti microscope, and the images were evaluated using ImageJ software (ImageJ 1.40g; W. Rasband, NIH). Evaluation of lung tissue damage and measurement of TUNEL-positive cells were assessed in a blinded manner on 10 randomly generated visual fields at 200× magnification.
Additional lungs from rats treated with HCl and LPS at the dose of 30 μg/g body wt and from control rats treated with saline/saline at 24, 48, and 72 h were processed for transmission electron microscopy studies. First, the samples were fixed in 2.5% glutaraldehyde in 0.1 mol/l phosphate buffer (pH = 7.4) for 2 h at room temperature and then in 1% osmium tetroxide and 0.8% potassium ferrocyanide for another 2 h. The samples were then washed three times with distilled water and dehydrated with 30%, 50%, 70%, 95%, and 100% acetone. They were embedded in Spurr resin and polymerized at 60°C. The embedded blocks were sectioned using a diamond knife (Diatome) on a Leica Ultracut UCT (Leica Microsystems, Deerfield, IL). Ultrathin sections were placed on copper grids and stained with uranyl acetate and lead citrate before examination under a JEM 1010 (Jeol, Tokyo) equipped with a Gatan/BioScan digital camera (Gatan, Pleasanton, CA).
Lung injury scoring.
Lung injury score (LIS) was quantified by an investigator blinded to the treatment groups using recently published criteria (20). As shown in Table 1, the LIS was obtained by the sum of each of the six independent variables: neutrophils in the alveolar space and/or in the interstitial space, presence of hyaline membranes, proteinaceous debris filling the airspaces, alveolar septal thickening and alveolar congestion. This sum was weighted according to the relevance ascribed to each feature by the American European Consensus Committee (20), and then was normalized to the number of fields evaluated and arbitrarily multiplied by 10 to obtain continuous values between zero and ten (both inclusive). Thus, the resulting lung injury score was derived from the following calculation: LIS: {[(20 × A) + (14 × B) + (7 × C) + (7 × D) + (2 × E) + (2 × F)]/(number of fields × 100)} × 10.
Table 1.
Histologic lung injury results
| Score Per Field |
|||
|---|---|---|---|
| Parameter* | 1 | 2 | 3 |
| A. Neutrophils in the alveolar space | None | 1–5 | >5 |
| B. Neutrophils in the interstitial space | None | 1–5 | >5 |
| C. Hyaline membranes | None | 1 | >1 |
| D. Proteinaceous debris filling the airspaces | None | 1 | >1 |
| E. Alveolar septal thickening | <2× | 2×–4× | >4× |
| F. Alveolar congestion | None | 1–5 | >5 |
Lung injury scoring system [adapted from Matute-Bello et al. (20)]. Reprinted with permission of the American Thoracic Society. Copyright © 2016 American Thoracic Society.
Protein extraction from lung homogenates.
For cytokine and caspase-3 measurements, the right lung was homogenized in lysis buffer containing 1 mM sodium orthovanadate, protease inhibitor cocktail tablets (1 tablet for 250 mg of lung tissue) (Roche; Mannheim, Germany), 0.5% Triton X-100, 150 mM NaCl, 15 mM Tris, 1 mM CaCl2, and 50 mM MgCl2 (pH 7.4) using a hand-held homogenizer. The homogenates were incubated for 30 min at 4°C, centrifuged at 12,000 rpm at 4° C for 20 min, and then filtered with 0.45 μm Nanosep filters (Pall Life Sciences; Madrid, Spain). The total protein concentration in lung homogenates was measured by the bicinchoninic acid method (Pierce; Thermo Scientific; Rockford, IL).
Pulmonary inflammation.
Cytokines IL-1β, TNF-α, IL-10, keratinocyte-derived chemokine (KC), monocyte chemoattractant protein-1 (MCP-1) and vascular endothelial growth factor (VEGF) were measured in lung homogenates using Procarta rat cytokine kit (Affymetrix; Santa Clara, CA) in a multiplex magnetic bead immunoassay (Luminex; Rafer, Spain) according to the manufacturer's instructions.
Western blot of caspase-3 protein.
Equal amounts of proteins from lung homogenates were heat-denatured in Laemmli sample buffer with 2-mercaptoethanol (5%), resolved in 20% SD-PAGE gel and transferred to PVDF membranes (GE Healthcare; Buckinghamshire, UK). Next, the blots were blocked with 5% PBS-nonfat dry milk for 2 h at room temperature and then incubated with mouse monoclonal anti-active caspase-3 (cleaved) primary detection antibody (1:1,000) overnight at 4°C. After thoroughly washing with 1 × PBS 0.05% Tween-20, the membranes were incubated for 2 h at room temperature with a goat anti-mouse Texas Red as a secondary antibody. An anti-actin antibody was used as a loading control. Finally, the fluorescence signal was visualized and analyzed using an LAS4000 system (Fujifilm Life Science; Woodbridge, CT).
Statistics.
The results of the quantitative variables were expressed as means ± SE. Statistical analyses of physiological parameters were analyzed using one-way ANOVA, followed by the post hoc Student-Newman-Keuls test for pairwise comparisons between treatment groups and uninjured control group in each postinstillation time (24, 48, and 72 h). When data failed the normality test in the one-way ANOVA, the Kruskal-Wallis one-way ANOVA on ranks was used. Survival curves were calculated according to the Kaplan-Meier method after the log-rank test. A P value < 0.05 was considered statistically significant. Analyses were done using SigmaPlot 11.0 software (Systat Software) and GraphPad Prism 6.0 software (GraphPad Software).
RESULTS
Effect of intratracheal instillation of HCl on alveolar-capillary barrier integrity at 2 h postinstillation.
Previous studies showed that direct lung injury induced by HCl instillation causes a physiochemical reaction characterized by an increase in protein permeability with no infiltration of neutrophils into the alveolar space at 2 h postinstillation (12, 22). Therefore, we conducted preliminary experiments to confirm the effect of intratracheal instillation of HCl on pulmonary protein permeability at 2 h postinstillation in rat lung in vivo, simulating aspiration pneumonitis. Compared with intratracheal instillation of sterile saline used as control, intratracheal instillation of 0.1 mol/l HCl (pH = 1.4) increased the lung/body weight ratio and the concentrations of IgM and total proteins in BAL fluid at 2 h postinstillation, although not all of the responses reached statistical significance (Fig. 1). In order to model aspiration pneumonitis with lung superinfection by aspiration of gram-negative bacteria, and taking into account that it is still unknown when bacteria aspiration occurs in patients (26), we intratracheally instilled LPS at four different concentrations (10, 20, 30, or 40 μg/g body wt) 2 h after HCl instillation, when pulmonary protein permeability is already increased.
Fig. 1.

Effect of intratracheal instillation of HCl on alveolar-capillary barrier integrity at 2 h postinstillation. Rats were treated with intratracheal instillation of HCl and then studied 2 h later. As control, rats were intratracheally instilled with saline. A: effect of HCl on lung wet weight-to-baseline body weight ratio. B and C: effect of HCl on the concentration of total proteins and IgM in bronchoalveolar lavage (BAL) fluid. Data from 3 independent experiments are reported as mean values ± SE. *P < 0.05, significant differences between rats treated with saline/saline and rats treated with HCl at 2 h postinstillation.
Survival and body weight analysis.
Survival was monitored for 72 h after LPS instillation (Figure 2A). All deaths occurred within 48 h. The survival rates obtained in the HCl and LPS groups at 72 h were 95% for HCl/LPS-10, 80% for HCl/LPS-20, 83% for HCl/LPS-30, and 47% for HCl/LPS-40 groups. The survival rates for control rats treated with saline/saline and HCl/saline were 100%. Compared with control saline/saline and HCl/saline groups, the rats treated with HCl and LPS at the dose of 40 μg/g body wt had a significantly lower survival rate (P < 0.001).
Fig. 2.
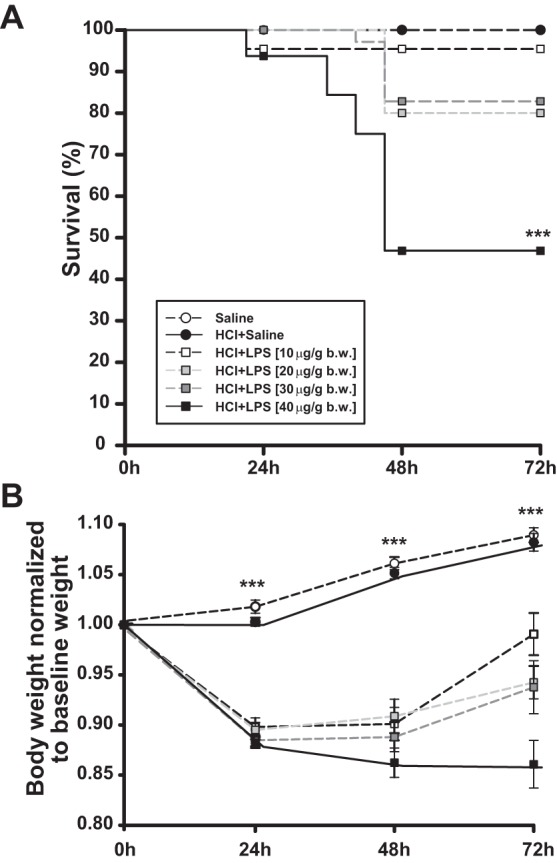
Effect of intratracheal instillation of HCl and LPS at 10, 20, 30, or 40 μg/g body wt on rat mortality and body weight. A: Kaplan-Meier survival curve of rats treated with saline/saline, HCl/saline, or HCl with LPS at 10, 20, 30, or 40 μg/g body wt over 72 h. B: change in rat body weight over time. The rat body weight after euthanasia at 24, 48, or 72 h was normalized by the respective baseline weight. Data from 8 independent experiments are reported as mean values ± SE. ***P < 0.001, significant differences between rats treated with saline/saline and rats treated with HCl and LPS per each time point. No significant differences were found between control rats treated with saline/saline and rats treated with HCl/saline.
Compared with baseline (before instillation), total body weight was significantly reduced in all rats treated with HCl and LPS in the first 24 h and 48 h after the last instillation, with no differences between these two time points. At 24 h postinstillation, the total body weight in the rats declined as shown in Fig. 2B. After 48 h postinstillation, only the rat group treated with HCl and LPS at the highest dose (40 μg/g body wt) continued to lose weight, whereas the total body weights of the rats treated with HCl and LPS at the doses of 10, 20, or 30 μg/g body wt started to gradually increase at this time point compared with baseline. At 72 h, total body weights of the rats treated with HCl and LPS at the dose of 10 μg/g body wt increased near to baseline. Likewise, total body weights of the rats treated with HCl and LPS at the doses of 20 or 30 μg/g body wt increased at 72 h compared with those at 24 and 48 h, although they remained significantly lower than baseline (Fig. 2B). In contrast, the total body weight of the HCl/LPS-40 group remained significantly decreased, compared to baseline. The control rats treated with saline/saline or the rat group treated with HCl/saline progressively gained weight during the first 72 h postinstillation (Fig. 2B).
Alteration of the alveolar capillary barrier permanently.
Compared with the control group, lung/body weight ratio was significantly increased in the rat groups treated with HCl and LPS at all doses and at 24, 48, and 72 h after the last instillation, indicating an increase in lung edema (Fig. 3A). The concentration of total proteins in BAL fluid was significantly increased in rats treated with HCl and LPS at all doses at 24 and 48 h postinstillation, compared with control group (Fig. 3B). This increase lasted 72 h only in rats treated with HCl and LPS at the doses of 30 or 40 μg/g body wt. In line with these results, the concentration of IgM in BAL fluid was significantly increased in rats treated with HCl and LPS at all doses at 24 h postinstillation (Fig. 3C). IgM is a large-molecular-mass protein (900 kDa), thus a good indicator of protein permeability. At 48 and 72 h postinstillation, the IgM levels remained significantly increased in the BAL fluid from rats treated with HCl and LPS at the doses of 20, 30, and 40 μg/g body wt compared with the control group. The concentrations of total protein and IgM in BAL fluid were not different between HCl/saline and saline/saline groups. These data indicate that intratracheal instillation of HCl/LPS-20, -30 and -40 doses caused a significant increase in protein permeability of the alveolar-capillary membrane at 24 h, which remained elevated for 72 h.
Fig. 3.
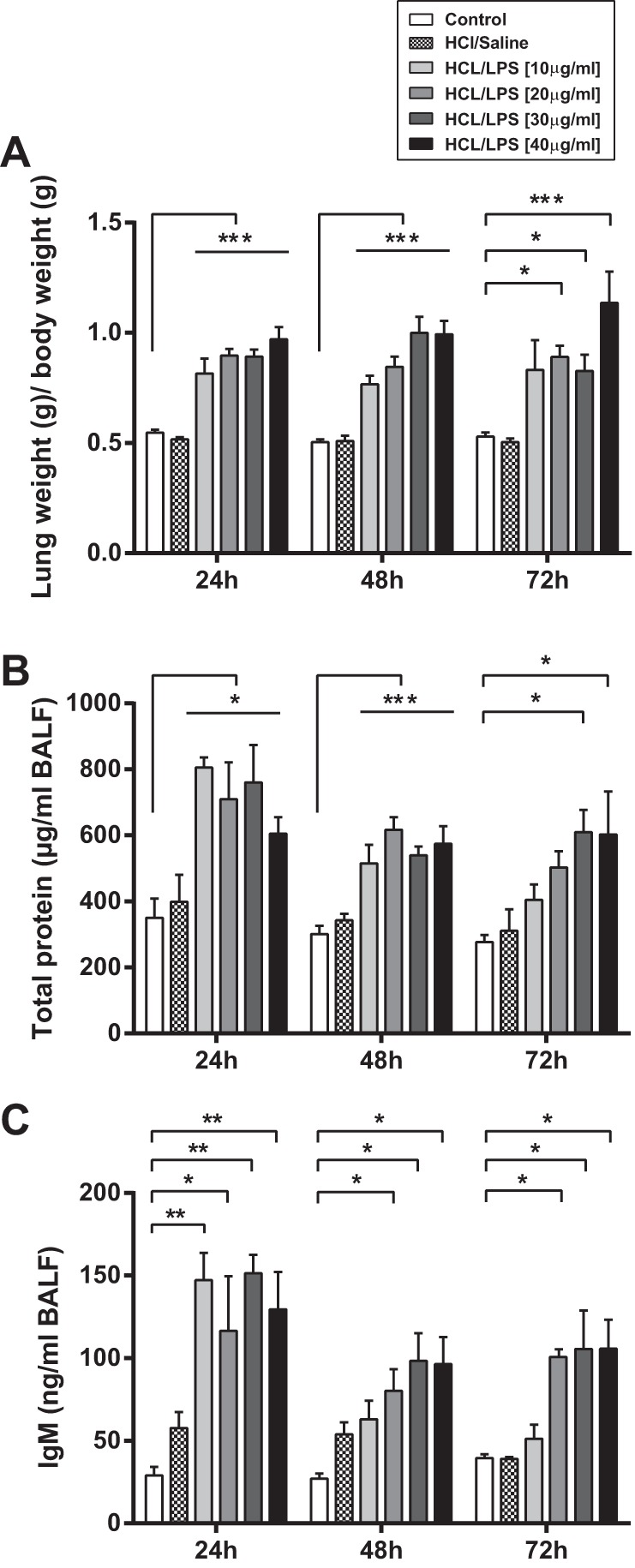
Effect of intratracheal instillation of HCl and LPS at 10, 20, 30, or 40 μg/g body wt on alveolar-capillary barrier integrity. Rats were treated with intratracheal instillation of HCl and LPS at 10, 20, 30, or 40 μg/g body wt and then studied 24, 48, and 72 h later. As control, rats were intratracheally instilled with saline. A: effect of HCl and LPS on lung wet weight-to-baseline body weight ratio. B and C: effect of HCl and LPS at all doses on the concentration of total proteins and IgM in bronchoalveolar lavage (BAL) fluid. Data from 5 independent experiments are reported as mean values ± SE. *P < 0.05, **P <0.01, ***P < 0.001, significant differences between rats treated with saline/saline and rats treated with HCl and LPS per each time point. No significant differences were found between control rats treated with saline/saline and rats treated with HCl/saline.
Histology and transmission electron microscopy.
Intratracheal instillation of HCl and LPS at the concentrations of 20, 30, and 40 μg/g body wt, but not at the concentration of 10 μg/g body wt, caused histologic damage in the rat lungs with lung injury scores significantly higher than those in control rats treated with saline/saline or HCl/saline (Fig. 4B). This damage was observed at 24 h, increased its severity at 48 h, and was still present at 72 h postinstillation. Histologically, the lung damage after HCl/LPS instillation in these rat studies was characterized by alveolar wall thickening, neutrophilic infiltration in the alveolar and the interstitial spaces, proteinaceous debris filling the airspaces, vascular congestion, and some alveolar hemorrhage (see Fig. 4A).
Fig. 4.

Effects of intratracheal instillation of HCl and LPS on rat lungs. Rats were treated with intratracheal instillation of HCl and LPS at 10, 20, 30, or 40 μg/g body wt and then studied 24, 48, and 72 h later. As control, rats were intratracheally instilled with saline. A: representative lung tissue sections stained with hematoxylin and eosin show alveolar wall thickening, vascular congestion, proteinaceous debris, neutrophilic infiltration, and some alveolar hemorrhage only in rats treated with HCl and LPS at 20, 30, and 40 μg/g body wt. This damage increases gradually depending on the LPS dose and time. Original magnification, ×20. B: lung injury score shows a significant injury in rat lungs treated with HCl and LPS at the concentrations of 20, 30, and 40 μg/g body wt, but not at the concentration of 10 μg/g body wt, compared with control rats treated with saline/saline. This damage was observed at 24 h and increased at 48 and 72 h postinstillation. Lung injury was assessed from 3 independent experiments on a scale of 0–2 for each of the following criteria: neutrophils in the alveolar space (A), neutrophils in the interstitial space (B), number of hyaline membranes (C), amount of proteinaceous debris (D), extent of alveolar septal thickening (E), and alveolar congestion (F). The resulting lung injury score was derived from the following calculation: {[(20 × A) + (14 × B) + (7 × C) + (7 × D) + (2 × E) + (2 × F)]/(number of fields × 100)} × 10. *P < 0.05, **P < 0.01, ***P < 0.001 vs. rats treated with saline/saline at each time point. No significant differences were found between control rats treated with saline/saline and rats treated with HCl/saline. C: transmission electron microscopy (TEM) images of lungs of representative animals treated with HCl/LPS-30 showed severe alveolar damage with disruption of the alveolar walls, neutrophilic infiltration, intra-alveolar hemorrhage, and activated macrophages, compared with control rats treated with saline/saline. Yellow asterisks show the neutrophils into the alveolar space. Red arrowheads and blue arrows show the macrophages and their phagosomes, respectively.
In line with these results, transmission electron microscopy (TEM) images (Fig. 4C) showed absence of injury in the lungs of control rats treated with saline/saline. The integrity of the alveolar-capillary barrier was preserved and there was no evidence of hemorrhage or neutrophils recruitment into the alveolar spaces. In contrast, TEM images corresponding to the rat groups treated with HCl/LPS-30, at the three time points studied (24, 48, and 72 h), showed areas of intense alveolar damage, in which there was disruption of the alveolar walls, neutrophils in the alveolar spaces (yellow asterisks), and edema with intra-alveolar hemorrhage. After 24 h, neutrophil recruitment into the alveolar spaces increased and the alveolar hemorrhage was more abundant. Macrophages (red arrowheads) were also observed with higher number of phagosomes (blue arrows), which indicates that these cells were activated. The number of the alveolar type II epithelial cells decreased and their physiological position was lost.
Lung inflammation.
Compared with control group (saline/saline), HCl and LPS at all doses caused a significant increase in the total number of neutrophils and macrophages in BAL fluid at 24, 48, and 72 h (Fig. 5, A and B). Accordingly, the neutrophil chemokine KC and the macrophage chemokine MCP-1 levels in lung homogenates were substantially increased in rat groups treated with HCl and LPS at 24 h compared with control group (Fig. 5, C and D). At 48 and 72 h postinstillation, the levels of KC and MCP-1 in lung homogenates from rats treated with HCl and LPS started to decline, but they remained significantly increased in rats receiving HCl and LPS at the higher doses (30 and 40 μg/g body wt). In line with these results, the lungs of rats treated with HCl and LPS at all doses contained higher concentrations of the pro-inflammatory cytokines IL-1β and TNF-α at the three time points studied compared with control group (Fig. 5, E and F). After 24 h, however, TNF-α concentration started to decrease and remained significantly increased only in the lungs of rats treated with HCl and LPS at 30 or 40 μg/g body wt. No differences were found in the concentration of the anti-inflammatory cytokine IL-10 between control and HCL/LPS groups at any time (Fig. 5G). Concentration of vascular endothelial growth factor (VEGF) remained significantly decreased in the lungs of HCl/LPS treated rats at 24, 48, and 72 h postinstillation compared with control rats (Fig. 5H).
Fig. 5.

Inflammatory responses in the lungs of rats treated with HCl and LPS. Effect of intratracheal instillation of HCl and LPS at 10, 20, 30, or 40 μg/g body wt on polymorphonuclear neutrophils (PMN; A) and macrophages (B) in BAL fluid and in cytokine production (IL-1β, KC, MCP-1, TNF-α, IL-10, and VEGF; C–H, respectively) in lung homogenates 24, 48, and 72 h after HCl instillation. As control, rats were intratracheally instilled with saline. Data from 5 independent experiments are reported as mean values ± SE. *P < 0.05, **P < 0.01, ***P < 0.001, significant differences between rats treated with saline/saline and rats treated with HCl and LPS per each time point. No significant differences were found between control rats treated with saline/saline and rats treated with HCl/saline.
Physiology.
Because serial measurements of physiological parameters have been difficult to obtain, we investigated the effect of intratracheal instillation of HCl/LPS-30 on pulmonary gas exchange capability and respiratory mechanics only at the specified end point times (24, 48, or 72 h). Compared with control group (saline/saline), HCl and LPS at 30 μg/g body wt induced a gradual decrease in the partial pressure of oxygen in arterial blood (PaO2) over time, with a reduction of the PaO2/FiO2 ratio that reached clinical criteria for mild ARDS (PaO2/FiO2 200–300 mmHg) at the three time points studied (Fig. 6A). This decrease in PaO2 was accompanied by a significant reduction of static lung compliance (Fig. 6C) and an increase in plateau airway pressure (Fig. 6D) at all time points, particularly at 48 and 72 h. In contrast, HCl and LPS at 30 μg/g body wt did not change the partial pressure of carbon dioxide in arterial blood (PaCO2) at any of the time points compared with control group (saline/saline) (Fig. 6B).
Fig. 6.

Measurement of arterial blood gases and respiratory mechanics. Assessment of pulmonary gas exchange capability and respiratory mechanics from rats treated with HCl/LPS-30 at 24, 48, and 72 h postinstillation. As control, rats were intratracheally instilled with saline. A and B: effect of HCl/LPS-30 on PaO2/FiO2 ratio and PaCO2, respectively. C and D: effect of HCl/LPS-30 on the static lung compliance and plateau pressure, respectively. Data from three independent experiments are reported as mean values ± SE. *P < 0.05, **P < 0.01, ***P < 0.001, significant differences between rats treated with saline/saline and rats treated with HCl and LPS at 30 μg/g body wt per each time point.
Activation of pro-apoptotic pathways.
Because apoptosis has been shown to be involved in the pathogenesis of ARDS (8, 28, 33), we investigated the effect of intratracheal instillation of HCl/LPS-30 on apoptotic cell death activation. As indicators of apoptosis, we measured the number of TUNEL-positive cells in lung tissue sections and the activity of caspase-3 in lung homogenates (Fig. 7A). Compared with control rats treated with saline/saline, the lungs of the rats treated with HCl/LPS-30 showed higher number of nuclei containing DNA strand breaks (TUNEL positive) at all time points, particularly at 48 and 72 h (Fig. 7B). Also, they showed a significant increase in activated caspase-3 at all time points (Fig. 7C).
Fig. 7.
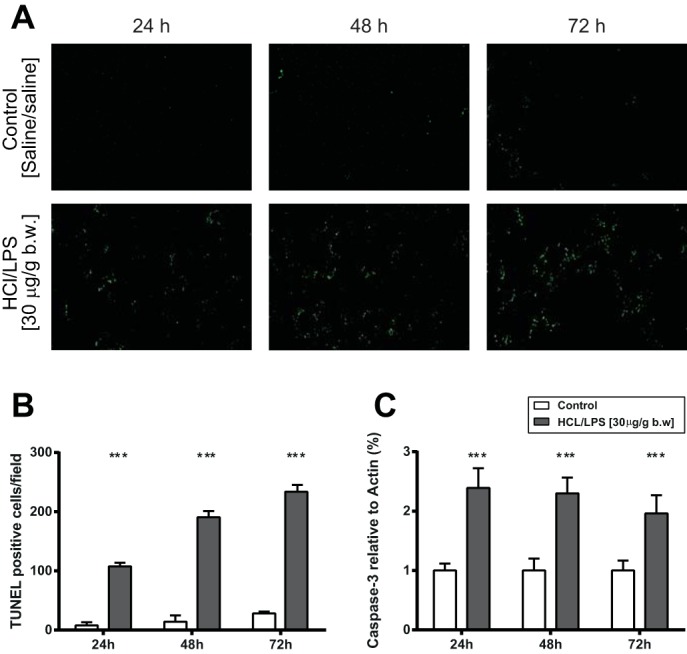
Measurement of apoptosis in lung tissue sections and lung homogenates. Analysis of cell death in lung tissue sections and lung homogenates from rats treated with HCl/LPS-30 at 24, 48, and 72 h postinstillation. A: TUNEL technique shows fluorescence green dots corresponding to nuclei containing DNA strand breaks. Representative results of 3 independent experiments per group. Original magnification, ×200. B: quantification of TUNEL-positive cells in lung sections. Ten fields of lung tissue from each rat were used to quantify the number of the TUNEL-positive cells. Results are reported as means ± SEM. C: caspase-3 activity in lung homogenates was evaluated by Western blot. Actin was used as an invariant control. Data from 3 independent experiments are reported as mean values ± SE. The results are expressed as a percentage of control (rats treated with saline/saline). ***P < 0.001, significant differences between rats treated with saline/saline and rats treated with HCl/LPS-30 per each time point.
DISCUSSION
Animal models of acute lung injury do not reproduce all the features found in humans with clinical ARDS and histological diffuse alveolar damage (21). However, they can provide insights into the underlying mechanisms of acute lung injury and/or its resolution. The main goal of this study was to develop an experimental model of acid- and endotoxin-induced ALI with substantial lung damage that persists at least 72 h. This model should facilitate investigations of the mechanisms involved in the early and later phases of ALI and examination of long-term effect of new therapeutic strategies. Our findings show that intratracheal instillation of HCl and LPS at all doses (10, 20, 30, or 40 μg/g body wt) results in acute lung injury within the first 24 h. This initial lung injury had a variable degree of severity and evolution over time in a dose-dependent manner for LPS. In this way, LPS at 30 μg/g body wt seems to be the most suitable dose to induce severe alveolar damage, lasting for at least 72 h, with an acceptable mortality.
Previous studies showed that intratracheal instillation of HCl alone, using similar doses to the present study, causes lung injury at 2 h postinstillation (12, 22), but, as we also observed in our model, this damage rapidly improves and reaches a complete recovery at 24 h (12). Taken into account that HCl is rapidly neutralized in the lung (3), we instilled LPS as a second hit two hours after instillation of HCl in order to enhance lung injury. In previous studies, different intervals of time between instillations of HCl (or gastric aspirate) and LPS (or live bacteria) have been used, ranging from few minutes (23, 27) to 16–24 h (18, 30, 32). In all cases, the instilled HCl (or gastric aspirate) primes the host to enhance acute inflammatory responses to a subsequent administration of LPS or live bacteria. In patients, aspiration pneumonitis predisposes to lung infection, mainly caused by aspiration of oral and nasopharynx bacteria. However, it is unknown when this bacteria aspiration occurs in patients (26).
In this model, the combination of HCl and LPS caused mortality within 48 h that was particularly high in the group of rats treated with HCl/LPS-40 (∼50%). However, the rats treated with HCl and LPS at 20 or 30 μg/g body wt had a lower and more acceptable mortality rate (∼20%), and lower than that in other models of aspiration pneumonia using live bacteria (18, 23, 27). Also, there was a remarkable loss of total body weight in the first 24 h in all rats treated with HCl/LPS. Then, the total body weight of the rats remained low or started to gradually increase over time depending on the dose of LPS. This loss or gain of body weight in the rats might reflect systemic effects of the lung injury.
Intratracheal instillation of HCl and LPS at all doses also activated inflammatory responses at 24 h, reflected by an increase in the number of PMNs and macrophages in the BAL fluid, and in the expression of KC, MCP-1, IL-1β, TNF-α in lung homogenates. Histologically, there was an early infiltration of inflammatory cells into the alveolar and interstitial spaces, some alveolar hemorrhage and proteinaceous debris filling the airspaces. These changes were accompanied by a significant increase in lung/body weight ratio and in the levels of total protein and IgM in the BAL fluid, suggesting an increase in protein permeability of the alveolar-capillary membrane and formation of protein-rich edema. It has been shown an association between the disruption of the alveolar-capillary barrier and the levels of VEGF in the lung of patients with ARDS (1, 16, 29). VEGF is a growth factor primarily expressed by epithelial cells that promotes alveolar epithelial cell proliferation and plays an important role in lung repair (29). In the lung of patients with ARDS, VEGF is significantly decreased in BAL fluid (16, 29) and lung tissue (1). Similarly, we found decreased VEGF levels at 24 h in lung homogenates of rats exposed to HCl and LPS at all doses, which could hinder lung repair.
From 48 to 72 h postinstillation, only the group of rats treated with HCl/LPS-10 started to improve, as reflected by a decrease in the concentrations of total proteins and IgM, pro-inflammatory cytokine expression and neutrophil recruitment in the lung. On the other hand, the groups of rats treated with HCl and LPS at highest doses (30 and 40 μg/ml body wt) developed severe lung injury that persisted for 72 h.
Altogether, we conclude that LPS at 30 μg/g body wt was the most suitable dose combined with HCl to induce long-lasting diffuse alveolar damage with an acceptable mortality in these rats. Therefore, we extended our study focused on this group. We first assessed arterial blood gases and respiratory mechanics at the specified “end point” times (24, 48, or 72 h). In rats treated with HCL/LPS-30, both arterial oxygenation and respiratory system mechanics were substantially deteriorated compared with control rats treated with saline. Indeed, the degree of hypoxemia observed in the HCl/LPS-30 group achieved clinical criteria for mild ARDS (PaO2/FiO2 200–300 mm Hg) at the three time points studied. As alveolar edema formation in ARDS is associated with epithelial dysfunction (19), the deterioration in respiratory mechanics observed might be a consequence of a combination of alveolar collapse and lung edema. Despite all these alterations caused by HCl/LPS-30, PaCO2 did not change in injured rats compared with rats treated with saline. This could be explained by an increase in their breathing rates, which would improve the CO2 clearance after lung injury.
We also observed a progressive increase in the number of apoptotic cells in the alveolar walls at 24, 48, and 72 h in rats treated with HCl and LPS at 30 μg/g body wt. This might be explained by an increased rate of apoptosis and/or an ineffective clearance of the apoptotic cells by phagocytes such as alveolar macrophages and epithelial cells in the lung (7). There was an increase in intracellular caspase-3 activity, an enzyme that leads to apoptosis, suggesting that the apoptosis rate is augmented after injury. By transmission electron microscopy, we observed that alveolar macrophages had high number of phagosomes at 48 and 72 h postinstillation of HCl/LPS-30, which indicates activation of efferocytosis (phagocytosis of apoptotic/necrotic cells). Secretion of VEGF also occurs in response to apoptotic cell phagocytosis by alveolar epithelial cells and macrophages, perhaps to preserve the alveolar-capillary barrier integrity (6). In this group of rats, the VEGF levels were decreased in all time points. These data suggest that the capacity of apoptotic cell clearance in the lung may also be diminished after injury, although the phagocytic capability of alveolar macrophages may not be totally inhibited. However, further studies are needed to elucidate the role of these processes.
This study has some limitations. The design of this model made it difficult to obtain serial measurements of physiological parameters such as arterial blood gases or lung mechanics. However, we conducted assessment of pulmonary gas exchange capability and respiratory mechanics at the specified “end point” time to evaluate better the degree of lung injury. Then, taken together, we believe that all these data provide strong evidence of sustained lung injury over time which includes the main features recommended by the recent workshop of the American Thoracic Society for ALI models (20). Although lung wet/dry weight ratio is frequently used, we measured lung/body weight ratio, which is also accepted to assess pulmonary edema (24), particularly when comparing different treatment groups (9, 10).
In conclusion, we described a two hit experimental model of ALI caused by instillation of HCl and LPS. Intratracheal instillation of HCl followed 2 h later by an instillation of LPS at the dose of 30 μg/g body wt caused acute lung injury sustained for 72 h that models many of the pathophysiological hallmarks of ARDS. We believe that this model should be useful to study the mechanisms involved in the early and later phases of ALI, as well as to test new therapeutic strategies in this disease.
GRANTS
M. N. Gómez was supported by FIS CA 05/0138. This work was supported by Ministerio de Economía y Competitividad-Instituto de Salud Carlos III (PI12/02548 to A. Artigas and PI12/02451 to R. Herrero), Fundació Parc Taulí (CIR2011/2568 to F. Puig), CIBER de Enfermedades Respiratorias, and National Heart, Lung, and Blood Institute Grant HL-51856 (to M. A. Matthay).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
F.P., R.H., and A.A. conception and design of research; F.P., R.H., R.G.-P., M.N.G., J.T., L.C., O.S., and A.S.-M. performed experiments; F.P., R.H., R.G.-P., M.N.G., J.T., and O.S. analyzed data; F.P., R.H., R.G.-P., and A.A. interpreted results of experiments; F.P., R.H., and R.G.-P. prepared figures; F.P. and R.H. drafted manuscript; F.P., R.H., R.G.-P., L.B., A.S.-M., M.A.M., and A.A. edited and revised manuscript; F.P., R.H., R.G.-P., M.N.G., J.T., L.C., O.S., L.B., A.S.-M., M.A.M., and A.A. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank M. E. Quílez for technical assistance and A. Sánchez-Chardi from Microscopy Facility at the Faculty of Science, Universitat Autònoma de Barcelona, for helpful assistance in transmission electron microscopy images.
REFERENCES
- 1.Abadie Y, Bregeon F, Papazian L, Lange F, Chailley-Heu B, Thomas P, Duvaldestin P, Adnot S, Maitre B, Delclaux C. Decreased VEGF concentration in lung tissue and vascular injury during ARDS. Eur Respir J 25: 139–146, 2005. [DOI] [PubMed] [Google Scholar]
- 2.Amigoni M, Bellani G, Scanziani M, Masson S, Bertoli E, Radaelli E, Patroniti N, Di Lelio A, Pesenti A, Latini R. Lung injury and recovery in a murine model of unilateral acid aspiration: functional, biochemical, and morphologic characterization. Anesthesiology 108: 1037–1046, 2008. [DOI] [PubMed] [Google Scholar]
- 3.Awe WC, Fletcher WS, Jacob SW. The pathophysiology of aspiration penumonitis. Surgery 60: 232–239, 1966. [Google Scholar]
- 4.Bregeon F, Papazian L, Delpierre S, Kajikawa O, Payan MJ, Martin TR, Kipson N, Pugin J. Role of proinflammatory activity contained in gastric juice from intensive care unit patients to induce lung injury in a rabbit aspiration model. Crit Care Med 36: 3205–3212, 2008. [DOI] [PubMed] [Google Scholar]
- 5.Doyle RL, Szaflarski N, Modin GW, Wiener-Kronish JP, Matthay MA. Identification of patients with acute lung injury. Predictors of mortality. Am J Respir Crit Care Med 152: 1818–1824, 1995. [DOI] [PubMed] [Google Scholar]
- 6.Golpon HA, Fadok VA, Taraseviciene-Stewart L, Scerbavicius R, Sauer C, Welte T, Henson PM, Voelkel NF. Life after corpse engulfment: phagocytosis of apoptotic cells leads to VEGF secretion and cell growth. FASEB J 18: 1716–1718, 2004. [DOI] [PubMed] [Google Scholar]
- 7.Henson PM, Cosgrove GP, Vandivier RW. State of the art. Apoptosis and cell homeostasis in chronic obstructive pulmonary disease. Proc Am Thorac Soc 3: 512–516, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Henson PM, Tuder RM. Apoptosis in the lung: induction, clearance and detection. Am J Physiol Lung Cell Mol Physiol 294: L601–L611, 2008. [DOI] [PubMed] [Google Scholar]
- 9.Herrero R, Tanino M, Smith LS, Kajikawa O, Wong VA, Mongovin S, Matute-Bello G, Martin TR. The Fas/FasL pathway impairs the alveolar fluid clearance in mouse lungs. Am J Physiol Lung Cell Mol Physiol 305: L377–L388, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hofstra JJ, Vlaar AP, Cornet AD, Dixon B, Roelofs JJ, Choi G, van der PT, Levi M, Schultz MJ. Nebulized anticoagulants limit pulmonary coagulopathy, but not inflammation, in a model of experimental lung injury. J Aerosol Med Pulm Drug Deliv 23: 105–111, 2010. [DOI] [PubMed] [Google Scholar]
- 11.Jabaudon M, Blondonnet R, Roszyk L, Bouvier D, Audard J, Clairefond G, Fournier M, Marceau G, Dechelotte P, Pereira B, Sapin V, Constantin JM. Soluble receptor for advanced glycation end-products predicts impaired alveolar fluid clearance in acute respiratory distress syndrome. Am J Respir Crit Care Med 192: 191–199, 2015. [DOI] [PubMed] [Google Scholar]
- 12.Kennedy TP, Johnson KJ, Kunkel RG, Ward PA, Knight PR, Finch JS. Acute acid aspiration lung injury in the rat: biphasic pathogenesis. Anesth Analg 69: 87–92, 1989. [PubMed] [Google Scholar]
- 13.Knight PR, Davidson BA, Nader ND, Helinski JD, Marschke CJ, Russo TA, Hutson AD, Notter RH, Holm BA. Progressive, severe lung injury secondary to the interaction of insults in gastric aspiration. Exp Lung Res 30: 535–557, 2004. [DOI] [PubMed] [Google Scholar]
- 14.Knight PR, Rutter T, Tait AR, Coleman E, Johnson K. Pathogenesis of gastric particulate lung injury: a comparison and interaction with acidic pneumonitis. Anesth Analg 77: 754–760, 1993. [DOI] [PubMed] [Google Scholar]
- 15.Krege JH, Hodgin JB, Hagaman JR, Smithies O. A noninvasive computerized tail-cuff system for measuring blood pressure in mice. Hypertension 25: 1111–1115, 1995. [DOI] [PubMed] [Google Scholar]
- 16.Maitre B, Boussat S, Jean D, Gouge M, Brochard L, Housset B, Adnot S, Delclaux C. Vascular endothelial growth factor synthesis in the acute phase of experimental and clinical lung injury. Eur Respir J 18: 100–106, 2001. [DOI] [PubMed] [Google Scholar]
- 17.Marik PE. Aspiration pneumonitis and aspiration pneumonia. N Engl J Med 344: 665–671, 2001. [DOI] [PubMed] [Google Scholar]
- 18.Matt U, Warszawska JM, Bauer M, Dietl W, Mesteri I, Doninger B, Haslinger I, Schabbauer G, Perkmann T, Binder CJ, Reingruber S, Petzelbauer P, Knapp S. Bbeta(15–42) protects against acid-induced acute lung injury and secondary pseudomonas pneumonia in vivo. Am J Respir Crit Care Med 180: 1208–1217, 2009. [DOI] [PubMed] [Google Scholar]
- 19.Matthay MA, Wiener-Kronish JP. Intact epithelial barrier function is critical for the resolution of alveolar edema in humans. Am Rev Respir Dis 142: 1250–1257, 1990. [DOI] [PubMed] [Google Scholar]
- 20.Matute-Bello G, Downey G, Moore BB, Groshong SD, Matthay MA, Slutsky AS, Kuebler WM. An official American Thoracic Society workshop report: features and measurements of experimental acute lung injury in animals. Am J Respir Cell Mol Biol 44: 725–738, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matute-Bello G, Frevert CW, Martin TR. Animal models of acute lung injury. Am J Physiol Lung Cell Mol Physiol 295: L379–L399, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McAuley DF, Frank JA, Fang X, Matthay MA. Clinically relevant concentrations of beta2-adrenergic agonists stimulate maximal cyclic adenosine monophosphate-dependent airspace fluid clearance and decrease pulmonary edema in experimental acid-induced lung injury. Crit Care Med 32: 1470–1476, 2004. [DOI] [PubMed] [Google Scholar]
- 23.Mitsushima H, Oishi K, Nagao T, Ichinose A, Senba M, Iwasaki T, Nagatake T. Acid aspiration induces bacterial pneumonia by enhanced bacterial adherence in mice. Microb Pathog 33: 203–210, 2002. [DOI] [PubMed] [Google Scholar]
- 24.Parker JC, Townsley MI. Evaluation of lung injury in rats and mice. Am J Physiol Lung Cell Mol Physiol 286: L231–L246, 2004. [DOI] [PubMed] [Google Scholar]
- 25.Patel BV, Wilson MR, Takata M. Resolution of acute lung injury and inflammation: a translational mouse model. Eur Respir J 39: 1162–1170, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Raghavendran K, Nemzek J, Napolitano LM, Knight PR. Aspiration-induced lung injury. Crit Care Med 39: 818–826, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rotta AT, Shiley KT, Davidson BA, Helinski JD, Russo TA, Knight PR. Gastric acid and particulate aspiration injury inhibits pulmonary bacterial clearance. Crit Care Med 32: 747–754, 2004. [DOI] [PubMed] [Google Scholar]
- 28.Schmidt EP, Tuder RM. Role of apoptosis in amplifying inflammatory responses in lung diseases. J Cell Death 2010: 41–53, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thickett DR, Armstrong L, Millar AB. A role for vascular endothelial growth factor in acute and resolving lung injury. Am J Respir Crit Care Med 166: 1332–1337, 2002. [DOI] [PubMed] [Google Scholar]
- 30.van Westerloo DJ, Knapp S, van't Veer C, Buurman WA, de Vos AF, Florquin S, van der PT. Aspiration pneumonitis primes the host for an exaggerated inflammatory response during pneumonia. Crit Care Med 33: 1770–1778, 2005. [DOI] [PubMed] [Google Scholar]
- 31.Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med 342: 1334–1349, 2000. [DOI] [PubMed] [Google Scholar]
- 32.Yamada H, Miyazaki H, Kikuchi T, Fujimoto J, Kudoh I. Acid instillation enhances the inflammatory response to subsequent lipopolysaccharide challenge in rats. Am J Respir Crit Care Med 162: 1366–1371, 2000. [DOI] [PubMed] [Google Scholar]
- 33.Yun JH, Henson PM, Tuder RM. Phagocytic clearance of apoptotic cells: role in lung disease. Expert Rev Respir Med 2: 753–765, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]