

Abstract
VX-770 (Ivacaftor) has been approved for clinical usage in cystic fibrosis patients with several CFTR mutations. Yet the binding site(s) on CFTR for this compound and other small molecule potentiators are unknown. We hypothesize that insight into this question could be gained by comparing the effect of potentiators on CFTR channels from different origins, e.g., human, mouse, and Xenopus (frog). In the present study, we combined this comparative molecular pharmacology approach with that of computer-aided drug discovery to identify and characterize new potentiators of CFTR and to explore possible mechanism of action. Our results demonstrate that 1) VX-770, NPPB, GlyH-101, P1, P2, and P3 all exhibited ortholog-specific behavior in that they potentiated hCFTR, mCFTR, and xCFTR with different efficacies; 2) P1, P2, and P3 potentiated hCFTR in excised macropatches in a manner dependent on the degree of PKA-mediated stimulation; 3) P1 and P2 did not have additive effects, suggesting that these compounds might share binding sites. Also 4) using a pharmacophore modeling approach, we identified three new potentiators (IOWH-032, OSSK-2, and OSSK-3) that have structures similar to GlyH-101 and that also exhibit ortholog-specific potentiation of CFTR. These could potentially serve as lead compounds for development of new drugs for the treatment of cystic fibrosis. The ortholog-specific behavior of these compounds suggest that a comparative pharmacology approach, using cross-ortholog chimeras, may be useful for identification of binding sites on human CFTR.
Keywords: human CFTR, murine CFTR, Xenopus CFTR, potentiator, blocker, pharmacophore model
cystic fibrosis (CF), an inherited disease found mainly in the Caucasian population, is caused by mutations in CFTR, the cystic fibrosis transmembrane conductance regulator. CFTR is a member of the ABC Transporter Superfamily but is unique in its function as a chloride ion channel. Experimental data and available homology models suggest that CFTR contains five functional domains: two membrane-spanning domains (MSDs) with six transmembrane helices (TM) in each MSD, two nucleotide-binding domains (NBD1, NBD2), and a unique regulatory (R) domain containing multiple protein kinase A (PKA) and C (PKC) consensus sites (2, 12, 31).
In addition to the human gene, CFTR clones have been produced from other species, including Xenopus laevis (frog) (35), pig, mouse (14), and sheep (6). We and others have demonstrated that these orthologs have different biophysical characteristics. For example, wild-type (WT) murine CFTR (mCFTR) differs from WT human CFTR (hCFTR) in its chloride conductance, ATP-dependent gating, and PKA sensitivity. As another example, the unitary conductance of Xenopus CFTR (xCFTR) is larger than that of hCFTR although they exhibit similar single-channel behavior with a prominent full open state and rare subconductance states. On the other hand, all three orthologs are regulated by ATP and PKA (11, 30).
Irrespective of their origin, the activities of CFTR channels can be modulated by many small drug-like compounds. These could be broadly divided into CFTR potentiators, CFTR correctors, and CFTR inhibitors. In particular, many potentiators have been discovered in the last decade, including P1 (VRT-532), P2 (PG-01), and P3 (SF-03), which are made available by the CF Foundation. P1 has been proposed to directly potentiate hCFTR and a few mutants via binding from the intracellular side (7, 38). In addition, some CFTR blockers also have been found to bear potentiator function. These include GlyH-101, NPPB, and glibenclamide. NPPB, a well-known small-molecule hCFTR blocker, was discovered to potentiate hCFTR when the channel is incompletely phosphorylated, i.e., when activated by a low concentration of PKA (44). Extremely high concentrations of NPPB also were reported to potentiate hCFTR from the intracellular side (9). We previously demonstrated that hCFTR blockers GlyH-101 and glibenclamide not only block but also potentiate mCFTR (11).
Efforts toward the development of CFTR potentiators have culminated in the groundbreaking work of Van Goor and colleagues (37) at Vertex Pharmaceuticals that led to the identification of the FDA-approved potentiator VX-770 (Ivacaftor). This compound has provided proof of principle that the function of CFTR proteins can be specifically and selectively modified by small-molecule drugs to improve lung function to a pharmaceutically relevant degree. However, the binding sites of this compound as well as of the above potentiators of CFTR remain unknown.
This lack of information hampers the rational design of yet more efficacious CFTR potentiators or compounds that could potentiate additional CFTR mutants. This is required since Ivacaftor only benefits a limited number of CF patients (∼5%) at least when administered by itself (32). The vast majority of CF patients, bearing the F508del mutation, are likely to require a combination therapy as exemplified by the recently approved Orkambi (VX-770/VX-809) combination therapeutic (19, 41). There is a clear need to develop both new correctors to repair defective trafficking and more efficacious potentiators to activate the channels that have been rescued to the apical plasma membrane (38).
With this in mind, the goals of this study were to identify new potentiators and to understand the ortholog-specific activities of the more established compounds. Here, we report on the use of a pharmacophore model, derived from known potentiators, for the virtual screening of a large database of commercially available compounds that led to the identification of three new lead compounds with potentiator activity in vitro. Additionally, we show that these compounds as well as the previously identified potentiators have ortholog-specific activity at human, murine, and Xenopus CFTR proteins. Finally, we describe initial aspects of the mechanisms of action of these compounds. These results set the stage for further application of these approaches to identify new potentiator compounds and the sites to which they bind.
MATERIALS AND METHODS
Preparation of oocytes and cRNA.
Human CFTR cRNAs used in electrophysiology experiments were prepared from constructs encoding WT-hCFTR in the pGEMHE vector (hCFTR/pGEMHE kindly provided by Dr. D. Gadsby, Rockefeller University). Murine CFTR (mCFTR/pcDNA 3.1) was kindly provided by Dr. A. P. Naren (University of Cincinnati) and subcloned into the pGEMHE vector (mCFTR/pGEMHE). Xenopus CFTR (xCFTR/pTM) was kindly provided by Dr. M. Welsh (University of Iowa) and subcloned into the pcDNA3.1 vector (xCFTR/pcDNA 3.1) The mutants of hCFTR used in this study were prepared by using site-directed mutagenesis with the Quikchange protocol (Stratagene, La Jolla, CA), and all mutant constructs were verified by sequencing across the entire open reading frame before use. Xenopus laevis oocytes were injected with 0.4–10 ng of CFTR cRNAs and were incubated at 17°C in modified Liebovitz's L-15 medium with the addition of HEPES (pH 7.5), penicillin, and streptomycin. Recordings were made typically 24–96 h after the injection of cRNAs. Methods of animal handling and oocyte collection were in accordance with NIH guidelines and the protocol was approved by the Institutional Animal Care and Use Committee of Emory University (10, 13).
Electrophysiology.
For inside-out macropatch recording, pipettes were pulled from borosilicate glass (Sutter Instrument, Novato, CA) and pipette resistances were 1–2 MΩ when filled with chloride-containing pipette solution (in mM) 150 NMDG-Cl, 5 MgCl2, 10 TES (pH 7.5). Channels were activated by excision into cytoplasmic solution containing (in mM) 150 NMDG-Cl, 1.1 MgCl2, 2 Tris-EGTA, 10 TES, 1 MgATP (adenosine 5′-triphosphate magnesium), and different concentrations of PKA (pH 7.5). Hence, our experimental conditions were designed to mimic the native physiological conditions in terms of ATP concentration, typically estimated to be 1–10 mM (3, 4). CFTR channels were studied in excised, inside-out patches at room temperature (22–23°C) (22, 24, 42). Oocytes were prepared for study by shrinking in hypertonic solution followed by manual removal of the vitelline membrane. Macropatch recordings were performed with an Axopatch 200B amplifier operated by pClamp 8.2 software; data were filtered at 100 Hz with a four-pole Bessel filter and acquired at 2 kHz. The voltage protocol used in this project was applied every 5 s: hold at Vm = 0 mV, then step to +100 mV for 50 ms followed by a ramp down to −100 mV over 300 ms prior to return to 0 mV.
Standard two-electrode voltage clamp (TEVC) techniques were used to study the effects of compounds on hCFTR, mCFTR, and xCFTR with application of reagents to the extracellular bath. Each oocyte was injected with CFTR cRNA along with cRNA encoding the β2-adrenergic receptor (β2AR). Electrode resistances measured 0.5–1.4 MΩ when filled with 3 M KCl and measured in standard ND96 bath solution that contained (in mM) 96 NaCl, 2 KCl, 1 MgCl2, and 5 HEPES (pH 7.5). CFTR channels were activated by exposure to 10 μM isoproterenol (ISO) in ND96 and alternatively assayed in the presence or absence of different concentrations of compounds in the bath solution, typically in the continuing presence of ISO. Currents were acquired with an Axoclamp 900A amplifier and Clampex 10.2 software, and current data were digitized at 2 kHz.
Pharmacophore modeling.
A pharmacophore model was constructed using LigandScout 4.0 (45), from the structures of three known active compounds: IOWH-032, GlyH-101, and glibenclamide. Each of these compounds was represented by a set of 500 conformations generated with the LigandScout implementation of the OMEGA program applying the “best setting” option. The pharmacophore model was generated using the “merged features” option. Thanks to the high similarity between the three compounds we were able to identify five features common to all (by setting the parameter “number of omitted features” to zero). Thus the final pharmacophore model consisted of aromatic ring (AR), hydrophobic ring (H), negative ionizable moiety (NI), H-bond acceptor (HBA), and H-bond donor (HBD). In addition, shape constraints were introduced using the excluded volume option.
Virtual screening and similarity analysis.
The pharmacophore model was used to virtually screen ∼7.37 million commercially available drug-like compounds available from the ZINC database (17, 18). Each compound was represented by 25 conformers generated with the OMEGA program and assembled into a multiconformer database. During screening, we required that each compound would match four pharmacophoric features (aromatic ring, negative ionizable moiety, H-bond acceptor, and H-bond donor), leaving the hydrophobic ring as optional. Under these conditions, 7,835 hits were identified (0.1% hit rate), from which only 387 compounds had higher pharmacophore score values than those for IOWH-032, GlyH-101, and glibenclamide. These 387 hits were further processed by the Canvas program (Canvas, version 2.6, 2015, Schrodinger, New York, NY), to identify compounds most similar to the known actives (using dendritic fingerprints and the Tanimoto similarity index). A short list of similar compounds was compiled from which two compounds were purchased and submitted to biological evaluation.
Source of reagents.
Unless otherwise noted, all reagents were obtained from Sigma Chemical (St. Louis, MO). L-15 medium was obtained from GIBCO-BRL (Gaithersburg, MD). PKA was obtained from Promega (Madison, WI). Under “standard conditions” in patch experiments, PKA was used at 1 μl/ml (127.6 U/ml) final concentration; solutions with PKA at lower concentrations (25.5 or 6.4 U/ml) were prepared by dilution in intracellular solution. GlyH-101 was obtained from Calbiochem (Billerica, MA) and prepared as a stock solution at 50 mM in DMSO. VX-770 was obtained from Selleckchem (Houston, TX) and was prepared as a stock solution at 10 mM in DMSO. NPPB was initially prepared as a 0.5 M stock in DMSO as previously reported (47). P1, P2, and P3 were provided by Cystic Fibrosis Foundation Therapeutics. IOWH-032 was obtained from Medchem Express (MCE, Princeton, NJ). OSSK-630513 and OSSK-674842 were obtained from Princeton Biomolecular Research (Monmouth Junction, NJ). VX-770, P1, P2, P3, IOWH-032, OSSK-630513 (OSSK-3), and OSSK-674842 (OSSK-2) were initially prepared as 10 mM stocks in DMSO. CFTRinh172 was initially prepared as 50 mM stock in DMSO. Cact-A1 was kindly provided from Dr. W. Namkung (Yonsei University, South Korea) and prepared as 10 mM stocks in DMSO. All chemicals were diluted to a final concentration in recording solution immediately before use.
Statistical analysis.
Unless otherwise noted, values given are means ± SE. Statistical analyses were performed by using the t-test for unpaired or paired measurements in SigmaPlot 12.3 (San Jose, CA). For multiple comparisons, we performed one-way ANOVA plus Bonferroni's multiple comparison tests using Prism (La Jolla, CA). P < 0.05 was considered significantly different (*P < 0.05; **P < 0.01; ***P < 0.001).
RESULTS
Human, murine, and Xenopus CFTR are differentially modulated by GlyH-101, NPPB, and VX-770.
It is not known whether potentiators bind at the membrane domain or at the NBDs of CFTR, but the efficacy of some potentiators appears to depend on CFTR's activation state (43, 44). This phenomenon could reflect either a dependence on the degree of phosphorylation of the R-domain, a dependence on the occupancy of the ATP binding sites at the interface between the two NBDs, or an allosteric effect whereby drug binds to other regions of CFTR (including the MSDs) and stabilizes the open conformation. In this regard, it is important to note that the degree of conservation between the three CFTR orthologs studied here is lowest in the MSDs. Yet there are still many specific residues in the MSDs that are conserved across hCFTR, mCFTR, and xCFTR, and, consequently, expanding the analysis reported in this work to more remote orthologs may enable deeper insight into residues that may contribute to the binding of potentiators.
Murine CFTR and xCFTR share ∼78 and 77% sequence identity with hCFTR, respectively (30, 35). As shown in the partial alignment provided in Fig. 1, despite this high identity, there are several residues conserved between hCFTR and mCFTR that are not conserved in xCFTR (e.g., position 378 in the human sequence). At other sites, human and Xenopus orthologs match, while murine CFTR is divergent. These differences may make it possible to use a comparison of ortholog-specific responses to compounds to identify their binding sites. In accord with this observation, we have shown recently that hCFTR and mCFTR respond differently to VX-770 (11). Similarly, while GlyH-101 and NPPB are well-known human CFTR blockers, it also has been shown recently that NPPB potentiates hCFTR while GlyH-101 potentiates mCFTR.
Fig. 1.

Partial sequence alignment of hCFTR, mCFTR, and xCFTR. The indicated sequence is a contiguous segment starting at the intracellular end of TM5 and extending into NBD1. Domain boundaries are indicated by arrows. Representative sequence differences among the three species are indicated in bold: 1, Residues conserved in hCFTR and mCFTR but not xCFTR; 2, residues conserved in hCFTR and xCFTR but not in mCFTR; 3, residues conserved in mCFTR and xCFTR but not in hCFTR; 4, residues are divergent across the three species.
In this work we have used the TEVC technique to determine the effects of GlyH-101, NPPB, and VX-770 on hCFTR, mCFTR, and xCFTR expressed in Xenopus oocytes and used the excised, inside-out macropatch technique to explore mechanism. These comparative molecular pharmacology studies made use of expression in oocytes so that the experimental background was constant and the only difference between experiments was the origin of CFTR, thus enabling direct comparisons.
Exposure of intact oocytes expressing stably activated xCFTR to 25 μM GlyH-101 for 50 s led to strong block of CFTR current that was reversed immediately upon washout (Fig. 2A); the block of xCFTR by GlyH-101 was similar to that of hCFTR (Fig. 2B). In contrast, GlyH-101 under the same conditions both blocked and potentiated mCFTR as revealed by a large increase in current above baseline upon removal of GlyH-101 from the bath and thereby relief from pore block (summarized in Fig. 2C) (11). Potentiation of CFTR by bath-applied compounds in these experiments reflects the result of permeation of these hydrophobic compounds to the cytoplasmic face of the channel. This conclusion is supported by previous data showing that the degree of potentiation by GlyH-101 is enhanced by longer exposure to compounds in the bath and by the rapid response of CFTR in excised macropatches when compounds were directly applied to the cytoplasmic face of the patch (11). Potentiation by GlyH-101 was not observed in hCFTR or xCFTR; thus potentiation by GlyH-101 exhibited ortholog-specific behavior.
Fig. 2.

hCFTR, mCFTR, and xCFTR reacted to NPPB, VX-770, GlyH-101 differently. Representative whole oocyte two-electrode voltage clamp (TEVC) current traces were recorded at Vm = −60 mV in ND96 solution. In these traces, macroscopic CFTR-mediated current is shown as downward deflections. CFTR channels were activated to steady state (dashed line) by stimulation of the coexpressed β2AR by isoproterenol (ISO). After activation to steady state, oocytes were exposed to 25 μM GlyH-101 (A), 200 μM NPPB (D and E), or 5 μM VX-770 (F and G) for ∼50 s in the continuing presence of ISO. Dashed lines indicate the maximum plateau current in each trace in the presence of ISO alone. Fractional change of current (Fc, fractional block or fractional potentiation) at steady state in response to exposure to compounds was calculated according to the equation: Fc = 1 − Ib/Ic, for block, or Fc = Ib/Ic − 1, for potentiation, where Ic and Ib represent the steady-state control currents and currents in the presence of chemicals, respectively. Summary data for all variants tested under the same conditions are shown in (B, C, H, I, and J). **P < 0.01 compared with hCFTR. ***P < 0.001 compared with hCFTR. nd, Not detected; n = 5–8. The data for block of hCFTR by 25 μM GlyH-101 have been cited from a previous publication (11).
As demonstrated previously (47), exposure to extracellular 200 μM NPPB for ∼50 s only blocked hCFTR (Fig. 2, D and H). Block by NPPB developed more slowly than block by GlyH-101 because NPPB must diffuse across the plasma membrane to reach its binding site from the cytoplasmic end of the channel. In contrast, mCFTR responded to 200 μM NPPB in a complex manner (Fig. 2E). Current first exhibited a slow decrease, similar to kinetics of the slow inhibition of hCFTR in Fig. 2D, followed by a slow relaxation toward the initial steady-state level of activated current. Upon washout of NPPB from the bath, current rebounded to a level even higher than control before application of compound. In prior experiments with GlyH-101, this rebound behavior was determined to represent potentiation of CFTR that is evident after relief from pore block (11). Similarly, the poor steady-state block of mCFTR compared with hCFTR (Fig. 2E) reflects concurrent block and potentiation. Xenopus CFTR also was highly sensitive to both block and potentiation by NPPB (Fig. 2, H and I). From these results, we conclude that mCFTR is less sensitive to NPPB-mediated block than either hCFTR or xCFTR (Fig. 2H), and that xCFTR is potentiated by NPPB much more strongly than is either hCFTR or mCFTR (Fig. 2I). Thus both NPPB-mediated block and NPPB-mediated potentiation exhibited ortholog-specific behavior.
We also studied the effects of 5 μM VX-770 on hCFTR, mCFTR, and xCFTR using the same experimental design. Similar to NPPB, VX-770 vigorously potentiated xCFTR with much smaller effects on hCFTR and mCFTR (Fig. 2, F, G, and J). These results suggest that the sensitivity to block and to potentiation do not follow the same pattern (e.g., compare Fig. 2, B and C vs. H and I), and therefore block and potentiation may reflect binding to different sites. Consistent with this notion, our previous study showed that block and potentiation of mCFTR by GlyH-101 were differentially sensitive to point mutations in the pore domain (11). In summary, the three compounds studied thus far (GlyH-101, NPPB, and VX-770) exhibit different patterns of efficacy in potentiating hCFTR, mCFTR, and xCFTR (Table 1).
Table 1.
Summary data for effects of VX-770, NPPB, GlyH-101, IOWH-032, OSSK-674842, OSSK-630513, P1, P2, and P3 on wild-type and mutant CFTR orthologs: experiments in intact cells
| VX-770 |
NPPB |
GlyH |
IOWH |
OSSK-2 |
OSSK-3 |
P1 |
P2 |
P3 |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| In | Po | In | Po | In | Po | In | Po | In | Po | In | Po | In | Po | In | Po | In | Po | |
| hCFTR | N | ∼ | Y | N | Y | N | Y | Y | Y | N | Y | Y | N | Y | N | Y | N | Y |
| mCFTR | N | Y | Y | Y | Y | Y | Y | N | Y | N | Y | N | ∼ | N | ∼ | N | ∼ | N |
| xCFTR | N | Y | Y | Y | Y | N | N | N | Y | Y | Y | Y | N | Y | N | Y | Y | N |
Data represent effect of extracellularly applied compounds in TEVC experiments performed in oocytes expressing these CFTR orthologs, with channels activated via β2AR-mediated stimulation of PKA. GlyH-101, GlyH; IOWH-032, IOWH; OSSK-630513, OSSK-3; OSSK-674842, OSSK-2; In, inhibition; Po, potentiation; Y, yes; N, no; ∼, effect is variable and small.
P1, P2, and P3 displayed varied effects on hCFTR, mCFTR, and xCFTR.
P1, P2, and P3 have been developed as potentiators of hCFTR and are made available to researchers by Cystic Fibrosis Foundation Therapeutics. However, it is not known whether these compounds also potentiate mCFTR and xCFTR. We tested the effect of ∼50-s exposures of 5 μM P1, 5 μM P2, and 5 μM P3 on hCFTR, mCFTR, and xCFTR expressed in oocytes. Representative TEVC data and summary results are shown in Fig. 3. P1, P2, and P3 potentiated hCFTR similarly to previous reports (Fig. 3, A–C) (28, 38). Surprisingly, none of these compounds potentiated mCFTR under the same experimental conditions (Fig. 3, D and E). Like hCFTR, xCFTR was potentiated by P1 and P2, but unlike hCFTR, P3 only inhibited xCFTR (Fig. 3, F–H). As summarized in Fig. 3, I–K, these results indicate that P1, P2, and P3 also exhibited ortholog-specific effects on CFTR.
Fig. 3.

Effects of 5 μM potentiators P1, P2, and P3 on hCFTR, mCFTR, and xCFTR. Representative current traces recorded by TEVC from oocytes expressing hCFTR (A–C), mCFTR (D and E), and xCFTR (F–H) at Vm = −60 mV. The oocytes were exposed to 5 μM P1, P2, or P3 in the continuing presence of 10 μM ISO for ∼50 s. Dashed lines show the maximum plateau current before addition of P1, P2, or P3. Fractional potentiation of hCFTR, mCFTR, and xCFTR were calculated with the equation described in Fig. 2 and summary data are shown in (I–K). **P < 0.01 and ***P < 0.001 compared with hCFTR; n = 5–12 each.
IOWH-032 affects hCFTR, mCFTR, and xCFTR differently.
IOWH-032 is a synthetic extracellular CFTR inhibitor, originally developed to treat diarrhea, that entered Phase II clinical trials in 2013 but did not progress further in clinical development. IOWH-032 is structurally similar to GlyH-101 but the reported IC50 for pore block (∼8 μM) is higher than that of GlyH-101 (∼1 μM) (26, 34). Because GlyH-101 exhibited ortholog-specific effects, we hypothesized that IOWH-032 might also affect CFTR in an ortholog-specific manner. During a ∼50-s extracellular exposure to IOWH-032, hCFTR was blocked as well as potentiated (Fig. 4A). Extracellular IOWH-032 blocked hCFTR rapidly, in a concentration-dependent manner with apparent Kd = 6.1 nM (Fig. 4B). Through time, IOWH-032 also potentiated hCFTR in a concentration-dependent manner with an apparent Kd = 0.64 nM (Fig. 4C). We note that the apparent Kd for potentiation is likely a great overestimation, since extracellular compound was only applied to cells briefly, making it unlikely that the intracellular concentration had equilibrated. We next tested the effects of IOWH-032 on mCFTR and xCFTR. Unlike GlyH-101, IOWH-032 did not potentiate and only blocked mCFTR with an apparent Kd = 42.9 μM (Fig. 4, D and E). IOWH-032 had no effect on xCFTR, even at concentrations as high as 1 and 5 μM (Fig. 4F). The data suggest that IOWH-032 both blocks and potentiates hCFTR, probably by interaction with different binding sites: one accessible extracellularly and one accessible intracellularly. The newly identified potentiation of hCFTR by IOWH-032 is unique because it was not observed on either mCFTR or xCFTR; hence, IOWH-032 also exhibited ortholog-specific effects for both block and potentiation.
Fig. 4.

hCFTR, mCFTR, and xCFTR reacted to IOWH-032 differently. A: IOWH-032 inhibited (*) as well as potentiated (#) hCFTR expressed in oocytes and recorded with TEVC at Vm = −60 mV. The oocyte was exposed to 5 nM IOWH-032 in the continuing presence of 10 μM ISO for ∼50 s. Dashed lines show the maximum plateau current before IOWH-032 addition. The concentration-dependent block and potentiation of hCFTR by IOWH-032 are shown in B and C, respectively; curves indicate the fit with a ligand-binding, one-site saturation equation, giving apparent Kd = 6.1 nM for block and apparent Kd = 0.64 nM for potentiation by extracellular IOWH-032, with ∼50-s exposure. D: representative current trace recorded by TEVC from an oocyte expressing mCFTR at Vm = −60 mV. The oocyte was exposed to 50 μM IOWH-032 in the continuing presence of 10 μM ISO for ∼50 s. Under this protocol, IOWH-032 only inhibited mCFTR. The concentration-dependent block of mCFTR by IOWH-032 is shown in E; curve indicates the fit with a ligand-binding, one-site saturation equation, giving apparent Kd = 42.9 μM for block by extracellular IOWH-032. F: IOWH-032 had no effect on xCFTR. Representative current trace of xCFTR recorded by TEVC at Vm = −60 mV. The oocyte was exposed to 1 μM IOWH-032 in the continuing presence of 10 μM ISO for ∼50 s; n = 5–8 for each.
Two new potentiator compounds identified by pharmacophore modeling.
Computer-aided drug design combined with experimental confirmation is widely recognized as a successful approach to drug development (21). We performed pharmacophore analysis based on the chemical structures of GlyH-101, IOWH-032, and glibenclamide. As shown in Fig. 5, we identified two new compounds, OSSK-630513 (OSSK-3) and OSSK-674842 (OSSK-2), from the ZINC database that share common pharmacophoric features with the known active compounds. We then tested the effects of OSSK-2 and OSSK-3 on hCFTR. Representative current traces are shown in Fig. 6A for OSSK-3 and Fig. 6D for OSSK-2. OSSK-3 exhibited both block as well as potentiation of hCFTR during a ∼50-s extracellular exposure. At Vm = −60 mV, the apparent Kd for block of hCFTR was 33.0 μM and the apparent Kd for potentiation of hCFTR was 31.7 nM (Fig. 6, B and C). Under the same protocol, OSSK-2 exhibited very weak block of hCFTR without obvious potentiation. The apparent Kd for block of hCFTR at Vm = −60 mV was 71.3 μM (Fig. 6E).
Fig. 5.

Chemical structures of GlyH-101, IOWH-032, glibenclamide, and the two new compounds OSSK-674842 (OSSK-2) and OSSK-630513 (OSSK-3) identified with a computational pharmacophore model derived from the structures of IOWH-032, GlyH-101, and glibenclamide.
Fig. 6.

Effects of new compounds OSSK-630513 (OSSK-3) and OSSK-674842 (OSSK-2) on hCFTR. Representative current traces of hCFTR affected by OSSK-3 (A) and OSSK-2 (D) recorded under the same conditions as in Fig. 1. Dashed lines show the maximum plateau current before addition of compounds. The concentration-dependent block (B) and potentiation (C) of hCFTR by OSSK-3 is shown with curves indicating the fit with a ligand-binding, one-site saturation equation, giving apparent Kd = 33.0 μM for block and apparent Kd = 31.7 nM for potentiation by extracellular OSSK-3. The concentration-dependent block on hCFTR by OSSK-2 is shown in E with apparent Kd = 71.3 μM; curve indicates the fit with a ligand-binding, one-site saturation equation; n = 4–8 for each point.
Since GlyH-101 affects hCFTR, mCFTR, and xCFTR differently and OSSK-2 and OSSK-3 share very similar chemical structures with GlyH-101 (Fig. 5), we hypothesized that OSSK-2 and OSSK-3 might exhibit the same behavior. At Vm = −60 mV, a ∼50-s extracellular exposure to 5 μM OSSK-2 led to very weak block of mCFTR without discernible potentiation (Fig. 7A). In contrast, a ∼50-s extracellular exposure to 5 μM OSSK-2 caused very weak block of xCFTR at first but this was followed by strong potentiation (Fig. 7B; summary data are shown in Fig. 7, C and D). We found that 5 μM OSSK-2 blocked hCFTR, mCFTR, and xCFTR with similarly weak efficacy but potentiated xCFTR significantly more strongly than hCFTR or mCFTR. We also compared the effects of 5 μM OSSK-3 on mCFTR and xCFTR with its effects on hCFTR; representative current traces are shown in Fig. 7, E and F. At Vm = −60 mV, a ∼50-s extracellular exposure to 5 μM OSSK-3 led to very weak block of mCFTR without visible potentiation (Fig. 7E). In contrast, xCFTR responded to the same treatment with initial very weak block that was followed by strong potentiation (Fig. 7F; summary data are shown in Fig. 7, G and H). OSSK-3 at 5 μM exhibited stronger blocking effect on mCFTR than on either hCFTR or xCFTR but potentiated xCFTR markedly better than hCFTR and mCFTR. We conclude that OSSK-3 and OSSK-2 both block and potentiate CFTR and show ortholog-specific behavior.
Fig. 7.

mCFTR and xCFTR reacted to OSSK-2 and OSSK-3 differently from hCFTR. Representative current traces of mCFTR (A and E) and xCFTR (B and F) affected by OSSK-3 or OSSK-2 recorded under the same conditions as in Fig. 6. Dashed lines show the maximum plateau current before the addition of compounds. Fractional potentiation of hCFTR, mCFTR, and xCFTR were calculated with the equation described in Fig. 2. Summary data for effects of 5 μM OSSK-2 on hCFTR, mCFTR, and xCFTR are shown in C and D. Summary data for effects of 5 μM OSSK-3 on hCFTR, mCFTR, and xCFTR are shown in G and H. nd, Not detected. **P < 0.01 and ***P < 0.001 compared with hCFTR and mCFTR; n = 5–7 for each.
Taken together, the results presented thus far indicate that all nine of the compounds tested exhibit ortholog-specific behavior in their ability to potentiate WT CFTR currents in whole cells (Table 1). These compounds and experimental models may therefore serve as excellent tools for identifying potentiator binding sites in CFTR.
The newly identified compounds potentiate F508del-hCFTR expressed in Xenopus oocytes.
Nearly 90% of CF patients in the United States bear at least one F508del-hCFTR allele; the current clinically approved drug Ivacaftor (VX-770) has only very limited effect on F508del-hCFTR in patients when used by itself and long-term use of Ivacaftor may diminish F508del-hCFTR functional expression (40). Therefore, it is extremely important to develop new potentiators that target F508del-hCFTR after it is correctly trafficked to the plasma membrane. Using the TEVC technique, we asked whether the newly identified compounds, IOWH-032, OSSK-3, and OSSK-2, potentiate F508del-hCFTR expressed in oocytes. Oocytes do not exhibit the impaired trafficking of F508del-hCFTR to the plasma membrane (15), enabling potentiation of this mutant to be studied directly. Representative current traces and summary data are shown in Fig. 8. A ∼50-s extracellular exposure to 20 nM IOWH-032 strongly potentiated F508del-hCFTR channels without obvious blocking effect (Fig. 8A). Identical treatment with 5 μM OSSK-3 caused very weak initial block that was followed by significantly stronger potentiation compared with the potentiation of WT-hCFTR (Fig. 8B). Similar results were found with 5 μM OSSK-2 (Fig. 8C). The response to 5 μM VX-770 under identical conditions is shown in Fig. 8D, for comparison. The data suggest that, like VX-770, which only mildly potentiates WT-hCFTR but strongly potentiates multiple CFTR mutants other than F508del-hCFTR, OSSK-2 and OSSK-3 mildly potentiate WT-hCFTR but very strongly potentiate F508del-hCFTR, whereas IOWH-032 potentiates F508del-hCFTR only slightly better than WT-hCFTR. These compounds may therefore be useful for future development of new CFTR potentiators.
Fig. 8.

IOWH-032, OSSK-3, OSSK-2, and VX-770 potentiated F508del-hCFTR. Representative current traces of F508del-hCFTR potentiated by 20 nM IOWH (A), 5 μM OSSK-3 (B), 5 μM OSSK-2 (C), or 5 μM VX-770 (D) recorded under the same experimental conditions as in Fig. 2. Dashed lines show the maximum plateau current before addition of compounds. Fractional potentiation of WT- and F508del-hCFTR was calculated with the equation described in Fig. 2. Summary data are shown at right. *P < 0.05 and ***P < 0.001 compared with WT-hCFTR; n = 4–12 for each.
P1, P2, and P3 do not have additive effects on hCFTR.
As noted above, P1, P2, and P3 are pure potentiators of hCFTR in intact cells, while the rest of the compounds we tested thus far also show inhibitory effects on this protein at high concentrations. We performed competition experiments using P1, P2, and P3 to test whether these potentiators occupy the same binding site or sites in hCFTR. Representative current traces and summary data are shown in Fig. 9; hCFTR channels were activated by 10 μM ISO, then 5 μM P1 was added extracellularly in the presence of ISO. After currents reached the new plateau, cells were exposed to 5 μM P1 plus 5 μM P2 together with ISO (Fig. 9A). Similar experiments are shown in Fig. 9, B and C for other combinations. Under these experimental conditions, there were no additive effects observed among P1, P2, and P3. These results in intact cells suggest that the three compounds might share the same binding site or sites at hCFTR.
Fig. 9.

Comparison of effects of potentiators P1, P2, and P3 on WT-hCFTR. Representative current traces (A–C) recorded from oocytes expressing WT-hCFTR under the same experimental conditions as in Fig. 2. P1 + P2: 5 μM P1 + 5 μM P2. P2 + P3: 5 μM P2 + 5 μM P3. Summary data are shown at right; n = 4 for each.
In these whole-cell conditions, activation of CFTR leads to submaximal open probability. Since the addition of the first potentiator led to less than a doubling of current, it is unlikely that open probability was fully maximized such that the second potentiator could not further activate channels. However, to investigate the sensitivity to these known potentiators under conditions in which CFTR activity was controlled more carefully, we turned to excised, inside-out macropatches. Results are summarized in Table 2. P1 and P2 were previously reported to potentiate WT hCFTR and multiple mutants including D1152H- and G551D-hCFTR, from the intracellular side of the membrane with different efficacies (7, 8, 20, 28, 38). We undertook experiments to test the possible additive effects of P1, P2, and P3 using macropatches pulled from oocytes expressing hCFTR; this approach provides better control of the intracellular concentration of test compounds, which are directly applied to the cytoplasmic face of the channel. In light of recent data suggesting that the efficacy of CFTR potentiation is dependent on degree of PKA-mediated activation (29, 44), use of macropatches also allowed us to more closely control phosphorylation state for these mechanistic experiments. Wild-type hCFTR channels were fully activated with 1 mM MgATP plus 127.6 U/ml PKA (standard PKA conditions) in symmetrical 150 mM Cl− solution; currents were elicited with a ramp protocol between Vm = −100 and +100 mV. P1 at 5 μM failed to potentiate hCFTR currents under standard conditions (Fig. 10A) as well as under low-PKA conditions (25.5 U/ml PKA; data not shown). P2 also did not potentiate hCFTR under standard PKA conditions (Fig. 10B) but strongly potentiated hCFTR under extremely low PKA conditions (6.4 U/ml PKA; Fig. 10C). Surprisingly, 5 μM P3 not only was unable to potentiate CFTR current but actually inhibited CFTR by ∼25% (0.26 ± 0.04 fold change, n = 5) in both standard PKA conditions (not shown) and low-PKA conditions (Fig. 10D). Fractional inhibition was calculated with the equation shown in the Fig. 2 legend, where Ic represents the maximum current in the ATP and PKA alone condition and Ib represents the remaining current in the presence of P3 at Vm = −100 mV.
Table 2.
Summary data for effects of P1, P2, and P3 on wild-type and mutant CFTR orthologs: experiments in excised, inside-out macropatches with indicated PKA concentration
| P1, 5 μM |
P2, 5 μM |
P3, 5 μM |
P3, 10 μM |
||||||
|---|---|---|---|---|---|---|---|---|---|
| In | Po | In | Po | In | Po | In | Po | ||
| WT-hCFTR | 127.6 U/ml PKA | N | N | N | N | Y | N | ||
| WT-hCFTR | 25.5 U/ml PKA | ∼ | N | ∼ | N | Y | N | ||
| WT-hCFTR | 6.4 U/ml PKA | N | Y | N | Y | Y | N | ||
| D1152A-hCFTR | 127.6 U/ml PKA | N | N | N | N | Y | N | ||
| D1152A-hCFTR | 25.5 U/ml PKA | N | N | N | N | Y | N | ||
| D1152A-hCFTR | 6.4 U/ml PKA | N | Y | N | Y | Y | N | N | Y |
Data represent effect of intracellularly applied compounds on wild-type (WT) or mutant CFTR currents in macropatches pulled from oocytes expressing these channels, following activation by 1 mM ATP and the concentration of PKA shown.
Fig. 10.
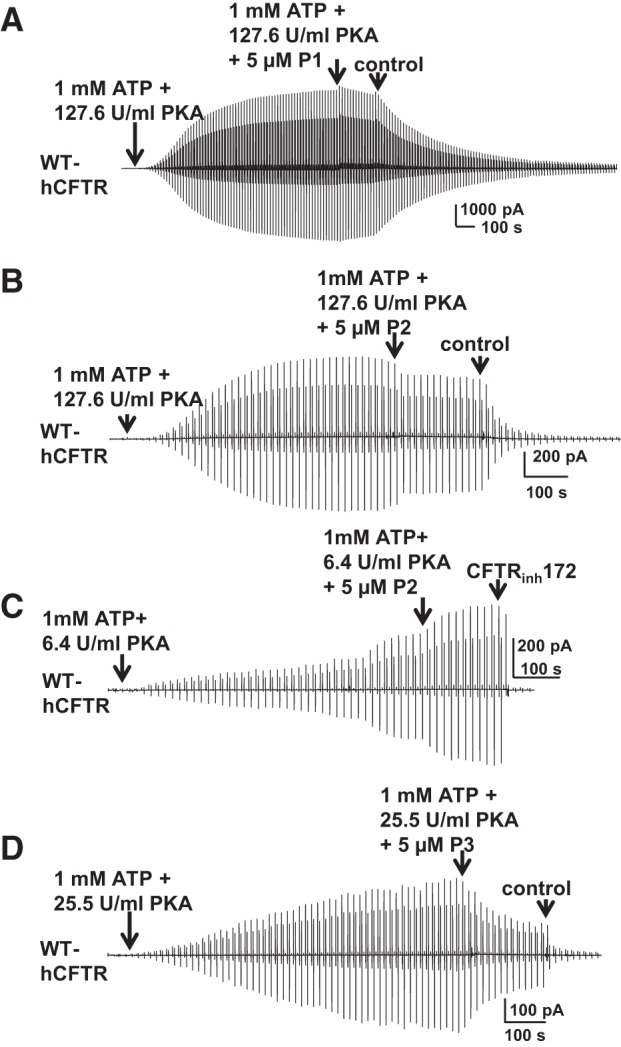
Effects of known potentiators on CFTR in excised, inside-out macropatches. P1, P2, and P3 did not potentiate WT-hCFTR under standard (127.6 U/ml; A and B) or low (25.5 U/ml; D) PKA conditions, while P2 potentiated WT-hCFTR under very low PKA (6.4 U/ml) conditions (C). Representative macropatch currents of WT-hCFTR recorded in inside-out mode with symmetrical 150 mM Cl− solution under the experimental conditions listed in the panels. A voltage ramp protocol described in materials and methods was applied every 5 s; n = 4–5 each.
Since prior publications suggest that P1 is more effective at potentiating hCFTR mutants that exhibit low open probability, compared with potentiation of WT hCFTR, we tested the effects of P1, P2, and P3 on D1152A-hCFTR using inside-out macropatches. P1 and P2 failed to potentiate D1152A-hCFTR currents under standard PKA conditions when applied individually (Fig. 11, A and B). At high concentrations, cytoplasmic P3 inhibited D1152A-hCFTR (data not shown) in a manner similar to inhibition of WT hCFTR (Fig. 10D). Therefore, P3 was tested at a concentration low enough to avoid inhibition; 10 nM P3 was not able to potentiate D1152A-CFTR under standard PKA conditions (Fig. 11C) but did exhibit mild potentiation under extremely low PKA conditions (Fig. 11, D and E). Hence, P3 may act like genistein, which potentiates CFTR at low concentration but inhibits CFTR at high concentration (1, 25). We further confirmed that P3 potentiates D1152A-hCFTR when applied extracellularly in two-electrode experiments (Fig. 11, F and G); the ∼50-s exposure to 5 μM P3 in whole oocyte experiments likely allowed only a very limited amount of P3 to permeate into the oocytes, thus resulting in potentiation without evidence of block.
Fig. 11.

Potentiation of D1152A-hCFTR is dependent on phosphorylation state. P1, P2, and P3 failed to potentiate D1152A-hCFTR under standard conditions (127.6 U/ml PKA; A–C). 10 nM P3 potentiated D1152A-hCFTR under extremely low (6.4 U/ml) PKA conditions in inside-out macropatches (D). Channels were phosphorylated with different concentrations of PKA in the presence of 1 mM MgATP. Representative macropatch currents are shown for D1152A-hCFTR recorded in inside-out mode with symmetrical 150 mM Cl− solution; a voltage ramp protocol described in materials and methods was applied every 5 s. CFTRinh172 concentration is 10 μM. Summary data for 10 nM P3 under extremely low PKA conditions are shown in E. I, maximum current in the presence of ATP + PKA + different compounds; I(ATP+PKA), maximum current in presence of ATP + PKA alone. ***P < 0.001 compared with ATP and PKA alone; n = 4 for each. F: P3 potentiated D1152A-hCFTR recorded in intact cells using TEVC as in Fig. 2. G: summary data for 5 μM P3 potentiation of D1152A-hCFTR in intact cells, calculated with the same equation as described in Fig. 2. *P < 0.05 compared with WT; n = 4–6 for each.
Taken together, these results in both WT and mutant hCFTR support the conclusions that 1) potentiation of CFTR by known compounds P1–P3 is dependent on CFTR phosphorylation state, as observed for the effects of NPPB on hCFTR (44), suggesting that care must be taken in studying the effects of potentiator compounds due to the apparent dependence of mechanism on allosteric regulation of the channel by phosphorylation, and 2) intracellular PKA activity in whole cell conditions may be relatively low following activation by physiological processes (36), because activation of CFTR in oocytes using the β2AR leads to low phosphorylation state such that channels remain sensitive to extracellularly applied potentiators.
Given the enhanced sensitivity of poorly phosphorylated D1152A-hCFTR channels to potentiation by known compounds, we used macropatch recordings to return to the question of whether P1, P2, and P3 may bind to overlapping sites. We first confirmed that P1, P2, and P3 did not potentiate D1152A-hCFTR under standard or low-PKA conditions, when applied individually; under these conditions there was no evidence of interaction between P1 and P2 or P2 and P3 (Fig. 12, A–C). Under extremely low PKA conditions (6.4 U/ml PKA), D1152A-CFTR was potentiated weakly by 5 μM P1 and strongly by 5 μM P2, but inhibited by 5 μM P3. Application of a second potentiator in these experiments indicated that P1 blocked the effects of subsequent addition of P2 (Fig. 11D), P2 did not block the effects of subsequent addition of P3 (Fig. 11E), and P3 did not block the effects of subsequent addition of P1 (Fig. 11F). These results suggest that P1 and P2 may share overlapping binding sites, but the binding site for P3 does not overlap with that of either P1 or P2. Furthermore, these experiments in excised, inside-out macropatches suggest that P1, P2, and P3 modulate CFTR by a mechanism that involves direct interaction with the channel.
Fig. 12.

Evidence for overlap of potentiator binding sites. P1 and P2 failed to potentiate D1152A-hCFTR under standard PKA (127.6 U/ml) conditions (A and B) but potentiated D1152A-hCFTR under extremely low (6.4 U/ml) PKA conditions (D and E) in macropatches recorded with the same protocol as in Fig. 11. D1152A-hCFTR was inhibited by 5 μM P3 in both low-PKA (25.5 U/ml) conditions (C) and extremely low PKA (6.4 U/ml) conditions (F); n = 4–6 for each.
P1, P2, P3, IOWH-032, OSSK-2, and OSSK-3 do not directly activate hCFTR.
Cact-A1 was recently identified as a CFTR activator and produced CFTR current in CFTR-expressing cells in the absence of PKA stimulation (27). We found that indeed Cact-A1 activated hCFTR current in whole oocytes in the absence of ISO (Fig. 13D), which supports the designation of Cact-A1 as an hCFTR activator. We asked whether P1, P2, P3, IOWH-032, OSSK-2, and OSSK-3 also behave as activators like Cact-A1; representative TEVC current traces are shown in Fig. 13. Channels were allowed to close completely at baseline before exposure to test compounds. A ∼100-s extracellular exposure to 5 μM P1 alone failed to activate hCFTR; no current was apparent until activation of CFTR via the β2AR with 10 μM ISO (Fig. 13A). Similar results were found for all other compounds tested (Fig. 13, B–F) other than Cact-A1, which induced significant CFTR activation from baseline, prior to stimulation with ISO. These results suggest that the compounds tested here, unlike Cact-A1, are not hCFTR activators.
Fig. 13.

Unlike Cact-A1, P1, P2, P3, IOWH, OSSK-2, and OSSK-3 did not directly activate WT-hCFTR in the absence of isoproterenol. Representative whole oocyte TEVC current traces were recorded at Vm = −60 mV in ND96 solution, in oocytes expressing CFTR with the β2AR as in Fig. 2. Exposure for ∼100 s to 5 μM P1 (A), 5 μM P2 (B), 5 μM P3 (C), 20 nM IOWH (E), 5 μM OSSK-2 (F), and 5 μM OSSK-3 (G) alone failed to stimulate hCFTR-WT channels whereas exposure to 10 μM Cact-A1 alone significantly activated WT-hCFTR (D). As a positive control for expression, channels were activated upon exposure to 10 μM isoproterenol (ISO) alone. H: summary data for the compounds tested above. I represents current in the presence of compounds alone but absence of ISO; IISO represents the maximum current in the presence of ISO in the same cell. ***P < 0.001 compared with all other compounds; n = 4–7 for each. nd, Not detected.
DISCUSSION
In these studies, we investigated the effects of multiple compounds on hCFTR, mCFTR, and xCFTR (Table 1). We found that, in intact cells, 1) VX-770, P1, P2, and P3 are pure potentiators of hCFTR with varying efficacies and GlyH-101 is a pure blocker of hCFTR and xCFTR but can also potentiate mCFTR; 2) P1, P2, and P3 are not potentiators of mCFTR; 3) P1 and P2 are potentiators of xCFTR, while P3 is a blocker of xCFTR; 4) IOWH-032 and OSSK-3 are blockers as well as potentiators of hCFTR while OSSK-2 only blocks hCFTR; 5) NPPB, OSSK-2, and OSSK-3 are blockers as well as potentiators of xCFTR; and 6) IOWH-032, OSSK-2, and OSSK-3 are blockers of mCFTR but not potentiators. Additionally, in excised patches, 7) cytosolic P1 and P2 potentiate hCFTR in a manner dependent on PKA-mediated phosphorylation and appear to share overlapping binding sites; 8) cytosolic P3 blocks hCFTR at high concentration but potentiates at low concentration; and 9) P1, P2, P3, IOWH-032, OSSK-2, and OSSK-3 are not direct activators of hCFTR. Comparing the activities of these nine potentiator compounds in three CFTR orthologs indicates that each of them exhibits ortholog-specific behavior. It is important to note, as shown in Table 1, that the pattern of responsiveness across the three orthologs is not always the same. For some compounds, only hCFTR was responsive while for others, only mCFTR or only xCFTR was responsive. Hence, the lack of pattern to responsiveness suggests that ortholog-specific effects do not reflect ortholog-specific differences in channel gating.
It was previously reported that various mutations at widely dispersed locations in CFTR confer sensitivity to potentiation by VX-770, suggesting that these mutations, themselves, do not form drug binding sites (23, 39). Instead, these mutations may alter the energetics of channel gating in a way that can be overcome by potentiators acting as allosteric modulators. The data that we present here suggest that P1 and P2 might share a binding site in hCFTR (Fig. 12); this phenomenon was also reported previously for other compounds, including curcumin and genistein, that might bind to related binding sites in hCFTR (5, 8). P3 appears to bind to one site for inhibition and a different site for potentiation (Figs. 11 and 12). The binding sites responsible for the effects of potentiators may represent locations where small molecules work to increase channel activity, thus serving as a target for the development of new therapeutics that may have even greater effect on even more disease-relevant mutants. Once those sites are identified, investigators will be able to use computational drug design to develop compounds that fit the binding pockets to potentiate CFTR channel activity.
Owing to the complexity of multiple potential binding sites in CFTR for potentiators, the traditional method of identifying possible binding sites by making mutations at potential sites and comparing the effect of drugs on mutants and WT proteins is likely to have limited success. The data that we present here clearly show that hCFTR, mCFTR, and xCFTR, despite sharing high amino acid sequence identity, reacted to multiple compounds differently. The ortholog-specific activity of CFTR potentiators will enable the use of chimeric approaches to identify compound-binding sites and study mechanisms of potentiators on CFTR. This strategy not only has been used in the study of CFTR (33) but also has been used widely in multiple other channels, including the pentameric ligand-gated ion channels (16) and calcium-activated Cl− channels (46).
Details of the molecular mechanism(s) by which CFTR potentiators increase channel activity are not yet clear. However, recent studies, along with data presented here, suggest that these compounds may function by an allosteric mechanism that is tied to activation of the channel by PKA-mediated phosphorylation. Wang, Kirk, and colleagues (44) previously reported that NPPB, a well-known small molecule blocker of CFTR, also potentiated poorly phosphorylated hCFTR channels. In our hands, P1, P2, and P3 robustly potentiated hCFTR in intact cells (Fig. 3), while P1- and P2-mediated potentiation in excised patches was highly dependent on phosphorylation state (Figs. 10–12). Cellular activity of PKA is rather low and depends on cell type and conditions (36). These data, together with the previous reports, may explain the differences in results between experiments in intact cells and in excised patches, where phosphorylation levels may not have been the same. However, the availability of nonhuman CFTR orthologs that do not respond to novel or known potentiators of human CFTR, such as mCFTR or xCFTR in the case of IOWH-032, may enable the generation of testable hypotheses to determine the mechanism of action of these compounds on human CFTR.
Increasing evidence, including the data presented here, shows that drugs previously described as blockers of CFTR such as NPPB, GlyH-101, and glibenclamide may also potentiate the channel (Table 1). Blocker activity of these compounds typically reflects occlusion of the pore, as shown by several investigators. In the case of GlyH-101, this arises from occlusion from the extracellular mouth of the pore (26, 34); for NPPB (47) and glibenclamide (13), this arises from occlusion from the cytoplasmic mouth of the pore. However, potentiation requires interaction with cytoplasmic domains in all cases, since this activity develops rapidly upon direct application to the cytoplasmic face of the channel in excised macropatch experiments but arises slowly upon application to the extracellular surface of whole cells. In the case of GlyH-101, our prior study (11) showed that pore block was sensitive to mutations in the pore domain while potentiation was insensitive to the same mutations, suggesting that these two activities arise from binding at different sites. The complexity of these drug effects on CFTR necessitates the development of new drugs that target CFTR with more straightforward function, such as pure potentiators.
As the number of compounds capable of potentiating CFTR activity grows, we gain the opportunity to learn from these compounds to expand the search for new therapeutic drugs. The identification of multiple molecules capable of potentiating CFTR will provide important information regarding the chemical moieties shared by these compounds that may be used to develop other novel potentiators, either through computational design of new compounds or by virtual screening of compound libraries. Efforts are underway now to separate the double effects of IOWH-032, OSSK-2, and OSSK-3 by combining pharmacophore analysis and experimental screening to identify pure potentiators or blockers of hCFTR.
GRANTS
This work was supported by the National Institutes of Health (Grants DK-056481, DK-075016); the Cystic Fibrosis Foundation (CFF MCCART14GO); and an Emory/Children's EECRC 2015 seed grant.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
G.C., N.K., H.S., and N.A.M. conception and design of research; G.C., N.K., and B.R.I. performed experiments; G.C., N.K., and B.S. analyzed data; G.C. and N.A.M. interpreted results of experiments; G.C. prepared figures; G.C. and N.A.M. drafted manuscript; G.C., N.K., B.S., D.T.I., H.S., and N.A.M. edited and revised manuscript; G.C., H.S., and N.A.M. approved final version of manuscript.
REFERENCES
- 1.Ai T, Bompadre SG, Wang X, Hu S, Li M, Hwang TC. Capsaicin potentiates wild-type and mutant cystic fibrosis transmembrane conductance regulator chloride-channel currents. Mol Pharmacol 65: 1415–1426, 2004. [DOI] [PubMed] [Google Scholar]
- 2.Alexander C, Ivetac A, Liu X, Norimatsu Y, Serrano JR, Landstrom A, Sansom M, Dawson DC. Cystic fibrosis transmembrane conductance regulator: using differential reactivity toward channel-permeant and channel-impermeant thiol-reactive probes to test a molecular model for the pore. Biochemistry 48: 10078–10088, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ataullakhanov FI, Vitvitsky VM. What determines the intracellular ATP concentration. Biosci Rep 22: 501–511, 2002. [DOI] [PubMed] [Google Scholar]
- 4.Beis I, Newsholme EA. The contents of adenine nucleotides, phosphagens and some glycolytic intermediates in resting muscles from vertebrates and invertebrates. Biochem J 152: 23–32, 1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berger AL, Randak CO, Ostedgaard LS, Karp PH, Vermeer DW, Welsh MJ. Curcumin stimulates cystic fibrosis transmembrane conductance regulator Cl− channel activity. J Biol Chem 280: 5221–5226, 2005. [DOI] [PubMed] [Google Scholar]
- 6.Bose SJ, Scott-Ward TS, Cai Z, Sheppard DN. Exploiting species differences to understand the CFTR Cl− channel. Biochem Soc Trans 43: 975–982, 2015. [DOI] [PubMed] [Google Scholar]
- 7.Caldwell RA, Grove DE, Houck SA, Cyr DM. Increased folding and channel activity of a rare cystic fibrosis mutant with CFTR modulators. Am J Physiol Lung Cell Mol Physiol 301: L346–L352, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Caputo A, Hinzpeter A, Caci E, Pedemonte N, Arous N, Di Duca M, Zegarra-Moran O, Fanen P, Galietta LJ. Mutation-specific potency and efficacy of cystic fibrosis transmembrane conductance regulator chloride channel potentiators. J Pharmacol Exp Ther 330: 783–791, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Csanady L, Torocsik B. Structure-activity analysis of a CFTR channel potentiator: distinct molecular parts underlie dual gating effects. J Gen Physiol 144: 321–336, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cui G, Freeman CS, Knotts T, Prince CZ, Kuang C, McCarty NA. Two salt bridges differentially contribute to the maintenance of cystic fibrosis transmembrane conductance regulator (CFTR) channel function. J Biol Chem 288: 20758–20767, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cui G, McCarty NA. Murine and human CFTR exhibit different sensitivities to CFTR potentiators. Am J Physiol Lung Cell Mol Physiol 309: L687–L699, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cui G, Rahman KS, Infield DT, Kuang C, Prince CZ, McCarty NA. Three charged amino acids in extracellular loop 1 are involved in maintaining the outer pore architecture of CFTR. J Gen Physiol 144: 159–179, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cui G, Song B, Turki HW, McCarty NA. Differential contribution of TM6 and TM12 to the pore of CFTR identified by three sulfonylurea-based blockers. Pflügers Arch 463: 405–418, 2012. [DOI] [PubMed] [Google Scholar]
- 14.Dong Q, Ostedgaard LS, Rogers C, Vermeer DW, Zhang Y, Welsh MJ. Human-mouse cystic fibrosis transmembrane conductance regulator (CFTR) chimeras identify regions that partially rescue CFTR-DeltaF508 processing and alter its gating defect. Proc Natl Acad Sci USA 109: 917–922, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Drumm ML, Wilkinson DJ, Smit LS, Worrell RT, Strong TV, Frizzell RA, Dawson DC, Collins FS. Chloride conductance expressed by delta F508 and other mutant CFTRs in Xenopus oocytes. Science 254: 1797–1799, 1991. [DOI] [PubMed] [Google Scholar]
- 16.Duret G, Van Renterghem C, Weng Y, Prevost M, Moraga-Cid G, Huon C, Sonner JM, Corringer PJ. Functional prokaryotic-eukaryotic chimera from the pentameric ligand-gated ion channel family. Proc Natl Acad Sci USA 108: 12143–12148, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Irwin JJ, Shoichet BK. ZINC—a free database of commercially available compounds for virtual screening. J Chem Inf Model 45: 177–182, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Irwin JJ, Sterling T, Mysinger MM, Bolstad ES, Coleman RG. ZINC: a free tool to discover chemistry for biology. J Chem Inf Model 52: 1757–1768, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jones AM, Barry PJ. Lumacaftor/ivacaftor for patients homozygous for Phe508del-CFTR: should we curb our enthusiasm? Thorax 70: 615–616, 2015. [DOI] [PubMed] [Google Scholar]
- 20.Liu X, Dawson DC. Cystic fibrosis transmembrane conductance regulator (CFTR) potentiators protect G551D but not DeltaF508 CFTR from thermal instability. Biochemistry 53: 5613–5618, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mandal S, Moudgil M, Mandal SK. Rational drug design. Eur J Pharmacol 625: 90–100, 2009. [DOI] [PubMed] [Google Scholar]
- 22.McCarty NA, Zhang ZR. Identification of a region of strong discrimination in the pore of CFTR. Am J Physiol Lung Cell Mol Physiol 281: L852–L867, 2001. [DOI] [PubMed] [Google Scholar]
- 23.Melani R, Tomati V, Galietta LJ, Zegarra-Moran O. Modulation of cystic fibrosis transmembrane conductance regulator (CFTR) activity and genistein binding by cytosolic pH. J Biol Chem 285: 41591–41596, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mense M, Vergani P, White DM, Altberg G, Nairn AC, Gadsby DC. In vivo phosphorylation of CFTR promotes formation of a nucleotide-binding domain heterodimer. EMBO J 25: 4728–4739, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moran O, Zegarra-Moran O. A quantitative description of the activation and inhibition of CFTR by potentiators: genistein. FEBS Lett 579: 3979–3983, 2005. [DOI] [PubMed] [Google Scholar]
- 26.Muanprasat C, Sonawane ND, Salinas D, Taddei A, Galietta LJ, Verkman AS. Discovery of glycine hydrazide pore-occluding CFTR inhibitors: mechanism, structure-activity analysis, and in vivo efficacy. J Gen Physiol 124: 125–137, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Namkung W, Park J, Seo Y, Verkman AS. Novel amino-carbonitrile-pyrazole identified in a small molecule screen activates wild-type and DeltaF508 cystic fibrosis transmembrane conductance regulator in the absence of a cAMP agonist. Mol Pharmacol 84: 384–392, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pedemonte N, Sonawane ND, Taddei A, Hu J, Zegarra-Moran O, Suen YF, Robins LI, Dicus CW, Willenbring D, Nantz MH, Kurth MJ, Galietta LJ, Verkman AS. Phenylglycine and sulfonamide correctors of defective delta F508 and G551D cystic fibrosis transmembrane conductance regulator chloride-channel gating. Mol Pharmacol 67: 1797–1807, 2005. [DOI] [PubMed] [Google Scholar]
- 29.Phuan PW, Yang B, Knapp JM, Wood AB, Lukacs GL, Kurth MJ, Verkman AS. Cyanoquinolines with independent corrector and potentiator activities restore DeltaPhe508-cystic fibrosis transmembrane conductance regulator chloride channel function in cystic fibrosis. Mol Pharmacol 80: 683–693, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Price MP, Ishihara H, Sheppard DN, Welsh MJ. Function of Xenopus cystic fibrosis transmembrane conductance regulator (CFTR) Cl channels and use of human-Xenopus chimeras to investigate the pore properties of CFTR. J Biol Chem 271: 25184–25191, 1996. [DOI] [PubMed] [Google Scholar]
- 31.Rahman KS, Cui G, Harvey SC, McCarty NA. Modeling the conformational changes underlying channel opening in CFTR. PLoS One 8: e74574, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ramsey BW, Davies J, McElvaney NG, Tullis E, Bell SC, Drevinek P, Griese M, McKone EF, Wainwright CE, Konstan MW, Moss R, Ratjen F, Sermet-Gaudelus I, Rowe SM, Dong Q, Rodriguez S, Yen K, Ordonez C, Elborn JS. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Eng J Med 365: 1663–1672, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Scott-Ward TS, Cai Z, Dawson ES, Doherty A, Da Paula AC, Davidson H, Porteous DJ, Wainwright BJ, Amaral MD, Sheppard DN, Boyd AC. Chimeric constructs endow the human CFTR Cl− channel with the gating behavior of murine CFTR. Proc Natl Acad Sci USA 104: 16365–16370, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thiagarajah JR, Verkman AS. CFTR inhibitors for treating diarrheal disease. Clin Pharmacol Ther 92: 287–290, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tucker SJ, Tannahill D, Higgins CF. Identification and developmental expression of the Xenopus laevis cystic fibrosis transmembrane conductance regulator gene. Hum Mol Genet 1: 77–82, 1992. [DOI] [PubMed] [Google Scholar]
- 36.Uhler MD, Abou-Chebl A. Cellular concentrations of protein kinase A modulate prostaglandin and cAMP induction of alkaline phosphatase. J Biol Chem 267: 8658–8665, 1992. [PubMed] [Google Scholar]
- 37.Van Goor F, Hadida S, Grootenhuis PD, Burton B, Cao D, Neuberger T, Turnbull A, Singh A, Joubran J, Hazlewood A, Zhou J, McCartney J, Arumugam V, Decker C, Yang J, Young C, Olson ER, Wine JJ, Frizzell RA, Ashlock M, Negulescu P. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc Natl Acad Sci USA 106: 18825–18830, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Van Goor F, Straley KS, Cao D, Gonzalez J, Hadida S, Hazlewood A, Joubran J, Knapp T, Makings LR, Miller M, Neuberger T, Olson E, Panchenko V, Rader J, Singh A, Stack JH, Tung R, Grootenhuis PD, Negulescu P. Rescue of ΔF508-CFTR trafficking and gating in human cystic fibrosis airway primary cultures by small molecules. Am J Physiol Lung Cell Mol Physiol 290: L1117–L1130, 2006. [DOI] [PubMed] [Google Scholar]
- 39.Van Goor F, Yu H, Burton B, Hoffman BJ. Effect of ivacaftor on CFTR forms with missense mutations associated with defects in protein processing or function. J Cyst Fibros 13: 29–36, 2014. [DOI] [PubMed] [Google Scholar]
- 40.Veit G, Avramescu RG, Perdomo D, Phuan PW, Bagdany M, Apaja PM, Borot F, Szollosi D, Wu YS, Finkbeiner WE, Hegedus T, Verkman AS, Lukacs GL. Some gating potentiators, including VX-770, diminish DeltaF508-CFTR functional expression. Sci Trans Med 6: 246ra297, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wainwright CE, Elborn JS, Ramsey BW, Marigowda G, Huang X, Cipolli M, Colombo C, Davies JC, De Boeck K, Flume PA, Konstan MW, McColley SA, McCoy K, McKone EF, Munck A, Ratjen F, Rowe SM, Waltz D, Boyle MP. Lumacaftor-Ivacaftor in patients with cystic fibrosis homozygous for Phe508del CFTR. N Eng J Med 373: 220–231, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Walters R, Welsh M. Mechanism by which calcium phosphate coprecipitation enhances adenovirus-mediated gene transfer. Gene Ther 6: 1845–1850, 1999. [DOI] [PubMed] [Google Scholar]
- 43.Wang W, Hong JS, Rab A, Sorscher EJ, Kirk KL. Robust stimulation of W1282X-CFTR channel activity by a combination of allosteric modulators. PLoS One 11: e0152232, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang W, Li G, Clancy JP, Kirk KL. Activating cystic fibrosis transmembrane conductance regulator channels with pore blocker analogs. J Biol Chem 280: 23622–23630, 2005. [DOI] [PubMed] [Google Scholar]
- 45.Wolber G, Langer T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J Chem Inf Model 45: 160–169, 2005. [DOI] [PubMed] [Google Scholar]
- 46.Yu K, Whitlock JM, Lee K, Ortlund EA, Cui YY, Hartzell HC. Identification of a lipid scrambling domain in ANO6/TMEM16F. eLife 4: e06901, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang ZR, Zeltwanger S, McCarty NA. Direct comparison of NPPB and DPC as probes of CFTR expressed in Xenopus oocytes. J Membr Biol 175: 35–52, 2000. [DOI] [PubMed] [Google Scholar]