

Abstract
Brain-derived neurotrophic factor (BDNF), a neurotrophin produced by airway smooth muscle (ASM), enhances inflammation effects on airway contractility, supporting the idea that locally produced growth factors influence airway diseases such as asthma. We endeavored to dissect intrinsic mechanisms regulating endogenous, as well as inflammation (TNF-α)-induced BDNF secretion in ASM of nonasthmatic vs. asthmatic humans. We focused on specific Ca2+ regulation- and inflammation-related signaling cascades and quantified BDNF secretion. We find that TNF-α enhances BDNF release by ASM cells, via several mechanisms relevant to asthma, including transient receptor potential channels TRPC3 and TRPC6 (but not TRPC1), ERK 1/2, PI3K, PLC, and PKC cascades, Rho kinase, and transcription factors cAMP response element binding protein and nuclear factor of activated T cells. Basal BDNF expression and secretion are elevated in asthmatic ASM and increase further with TNF-α exposure, involving many of these regulatory mechanisms. We conclude that airway BDNF secretion is regulated at multiple levels, providing a basis for autocrine effects of BDNF under conditions of inflammation and disease, with potential downstream influences on contractility and remodeling.
Keywords: neurotrophin, brain-derived neurotrophic factor, asthma, airway, secretion, inflammation
airway smooth muscle (asm) is important not just for regulation of airway tone and contractility, but also in airway structure, especially in the context of allergy, inflammation, and fibrosis (2, 33). In addition to being the recipient of factors that influence contractility, relaxation, proliferation, and migration, which determine structure and function of the smooth muscle layer of bronchial airways, ASM cells are increasingly recognized as active modulators of the local environment via production of inflammatory cytokines and other growth factors with autocrine/paracrine effects, overall contributing to the pathophysiology of diseases such as asthma (10, 11, 14, 18, 19, 33, 35). Accordingly, understanding the mechanisms that regulate growth factor expression and secretion represents a novel, unmet investigative area toward alleviating the effects of inflammation, with downstream consequences for airway structure and function.
There is increasing interest in the role of neurotrophins, particularly BDNF (brain-derived neurotrophic factor) in mediating and modulating inflammation effects on airway structure and function (33, 35). BDNF has been previously observed in lungs of different species (30, 35, 38) and has been reported to be increased in asthma (30, 38, 41). Recently, enhanced BDNF expression in epithelium of severe asthmatics has garnered interest in the idea that local BDNF expression and effects may be important in asthma phenotypes (49). We have shown that ASM responds to exogenous BDNF with elevated intracellular Ca2+ ([Ca2+]i) and cell proliferation, particularly in the presence of cytokines such as TNF-α and IL-13 (1, 7, 34, 37). However, ASM appears to be not only a target, but also a source of BDNF (1, 7, 44, 45, 49). What is less understood are the mechanisms by which local (airway) BDNF is regulated.
Local BDNF levels in the airway may be regulated via expression as well as active secretion into the extracellular milieu. Unlike the fast, highly regulated synaptic vesicular exocytosis of BDNF observed in neurons [where neurotrophins are more well known to be involved in modulation of synaptic transmission (24)], BDNF release in smooth muscle is probably slower. Nonetheless, BDNF secretion is likely modulated by [Ca2+]i (44), which, on a slower time scale, could also regulate BDNF protein expression. As elevated [Ca2+]i is a hallmark of asthmatic ASM, it is possible that higher [Ca2+]i also enhances BDNF expression and/or secretion, but this aspect has not been previously explored. Further, elevated [Ca2+]i in ASM involves Ca2+ regulatory proteins such as transient receptor potential (TRPC) and Orai1 channels, which have been implicated in asthma (40, 48), and initial studies from our group have shown that TRPC3 channel activation is important for ASM BDNF secretion (44). Along similar lines, in fetal ASM cells, cAMP and Epac2 appear to be important in driving BDNF expression and secretion (43). However, the roles for these secretion-regulatory pathways in asthmatic ASM are unknown.
An important aspect of local BDNF secretion is the likely autocrine effect on ASM. BDNF exerts its effects by binding specifically to tropomyosin-related kinase B (TrkB), activating a variety of signaling events, including extracellular signal regulated kinase (ERK), phosphoinositide 3-kinase (PI3K) and phospholipase C (PLC) pathways overall leading to an increase in [Ca2+]i (35). Interestingly, many of these pathways are not only important in asthma pathophysiology (15, 32, 33, 51), but are also important for regulation of BDNF expression per se, thus potentially leading to a positive feedback mechanism for elevated BDNF levels in the context of the asthmatic airway. Therefore, in the present study, we explored the mechanisms that control the expression and the secretion of BDNF by ASM cells, focusing on Ca2+-dependent or Ca2+-responsive pathways relevant to asthma. Experiments were performed in ASM cells derived from both nonasthmatic and asthmatic adult individuals, with the hypothesis that BDNF levels are intrinsically higher in asthmatic ASM, reflecting a role for regulatory pathways also relevant to asthma.
MATERIALS AND METHODS
Materials.
PD98059 (ERK/MAPK inhibitor), wortmannin (PI3K inhibitor), BIS (bisindolylmaleimide; PKC inhibitor), Y27632 (ROCK inhibitor), SN-50 (NF-κB inhibitor), SKF-96365 (TRPC inhibitor), and the cell-permeable nuclear factor of activated T cells (NFAT) inhibitor were purchased from EMD/Calbiochem (Culver City, CA). U73122 (PLCγ inhibitor) was obtained from Sigma-Aldrich (St. Louis, MO). G-protein antagonist 2A (GPA-2A) was purchased from Enzo Life Sciences (Farmingdale, NY). Small interfering RNAs (siRNAs) against TRPC1, TRPC3, and TRPC6 were purchased from Dharmacon (Lafayette, CO). BDNF antibody was obtained from Santa Cruz Biotechnologies (Santa Cruz, CA; cat. no. sc20981; dilution: 1:100), β-actin antibodies were obtained from Sigma-Aldrich (cat. no. A5316; dilution: 1:500), and TRPC3 (cat. no. ab188802; dilution: 1:500) and TRPC6 (cat. no. ab62461; dilution: 1:500) antibodies were obtained from Abcam (Cambridge, MA). Quantikine ELISA kit to estimate human BDNF was purchased from R&D Systems (Minneapolis, MN).
ASM cells and tissue.
Human ASM cells were isolated using previously described protocols (1, 7, 44, 45). Briefly, lung samples were resected incidental to patient thoracic surgery at Mayo Clinic (Rochester, MN), and 3rd–6th level bronchi were dissected from normal-appearing areas (identified with the aid of the surgical pathologist). Patient records were reviewed to identify age, sex, asthma history, medications, tobacco use, and other relevant comorbidities, but patient identifiers were not retained (approved by Mayo Institutional Review Board as a minimal risk protocol). Epithelial layers were removed by blunt dissection, and ASM tissue either used directly for pharmacological and/or TNF-α treatments, or enzymatically digested to isolate ASM cells. Cells were maintained in Complete-DMEM (DMEM supplemented with 10% FBS and 1% antibiotic-antimycotic). For all experiments, cells that had been passaged (subcultured) 3 times or less were used and were serum-deprived at least for 24 h. ASM phenotype was frequently verified by expression of smooth muscle markers (actin and myosin, Ca2+ channel regulatory proteins such TRPC3, CD38, and Orai1), and by the lack of expression of epithelial and fibroblast markers.
Transfections.
ASM cells were transfected with 200 pM siRNA [against TRPC1, TRPC3, TRPC6, or cAMP response element binding protein (CREB)] under serum- and antibiotic-free conditions using Lipofectamine (Invitrogen, Carlsbad, CA) (6, 44). Vehicle-alone treatment and transfection with scrambled nontargeting RNA served as controls. Cells were treated with 20 ng/ml TNF-α 2 days posttransfection and were analyzed 48 h later.
Pharmacological treatments.
ASM tissue and cells were exposed to drugs that inhibit various signaling pathway components for 2 h prior to TNF-α, 20 ng/ml) treatment. Concentrations of inhibitors used are listed are given in the figure captions.
ELISA.
BDNF in the cell culture medium was measured using the Quantikine ELISA kit from R&D Systems. After appropriate treatments, supernatants were collected and concentrated to about 1/6th of the original volume, using Amicon Ultracel-3K Centrifugal filters (Millipore, Billerica, MA). ELISA was performed in a 96-well format, following manufacturer's protocol, and using kit-provided BDNF for standards, which was used for absolute quantification.
Immunoblotting.
Whole cell lysates were isolated and separated on denaturing PAGE, using standard techniques. Approximately 20 μg of total protein was loaded. Protein expression was detected, and densities of bands were quantified on a Li-Cor Odyssey IR scanning system (Lincoln, NE). Band intensities were normalized against β-actin.
Statistical analysis.
All experiments were performed in quadruplicate using different sets of ASM cells isolated from at least four different individuals for each group (represented as n). Controls represent cells that were not transfected, not exposed to drugs (inhibitors) or TNF-α. For immunoblotting, extracts were obtained from four nonasthmatic ASM populations, and for ASM tissue, samples were isolated from at least three nonasthmatic individuals. All experiments using any specific individual's cells were repeated three times. For all experiments, comparisons were performed across groups with independent two-way ANOVA, and Bonferroni corrections were used for repeated comparisons. Statistical significance was tested at the P < 0.05 level. Values are reported as means ± SE; n values representing numbers of individuals (i.e., patients) are provided in the figure captions.
RESULTS
BDNF secretion in asthmatic vs. nonasthmatic ASM.
Endogenous BDNF secretion by ASM cells of nonasthmatic vs. asthmatic individuals under unstimulated vs. TNF-α-stimulated (inflammation) conditions was examined using ELISA. ASM cells from asthmatic individuals showed a significantly higher amount of secreted BDNF (approximately twofold) even under basal conditions (Fig. 1). Treatment with 20 ng/ml TNF-α for 48 h upregulated BDNF release in both nonasthmatic and asthmatic cells (Fig. 1), although the increase in asthmatic cells was not disproportionately higher than that in nonasthmatic cells. Values of BDNF were normalized to total cell protein. One potential concern with TNF-α or BDNF exposure is that both can individually increase ASM proliferation (6), which would be accounted for by normalization for total protein. However, to ensure that only proliferation needs to be considered, and not cell viability per se, trypan blue exclusion assay was used (data not shown). The ratio of live to dead cells was consistent between untreated and TNF-α- or BDNF-treated groups, suggesting that TNF-α and BDNF increase ASM proliferation rather than just enhance cell survival.
Fig. 1.
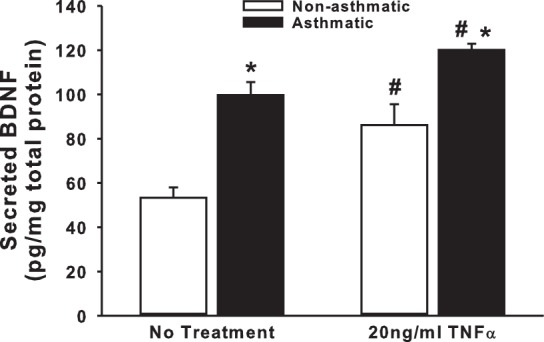
Inflammatory stimulus upregulates brain-derived neurotrophic factor (BDNF) secretion by human airway smooth muscle (ASM) cells. Nonasthmatic and asthmatic ASM cells, under serum-free conditions, were exposed to 20 ng/ml TNF-α for 48 h before BDNF was quantified in the extracellular medium, by ELISA. Secretion of BDNF at baseline is also higher in asthmatic ASM than in nonasthmatic cells; n = 4 patients. *Significant difference from “No Treatment” control, P < 0.05. #Significant difference between nonasthmatic and asthmatic samples, P < 0.05.
BDNF secretion is regulated by the TRPC channels.
We previously demonstrated that BDNF secretion is regulated by TRPC3 (44). Given the importance of TRPC channels in asthma pathophysiology, and the expression of multiple TRPC isoforms in ASM, particularly TRPC3 and TRPC6 (48, 50), we tested the role of TRPC3 and TRPC6 channels on BDNF release by nonasthmatic and asthmatic ASM cells, using siRNAs (efficacy of siRNA inhibition shown in Fig. 2A). As shown in Fig. 2B, both TRPC3 and TRPC6 positively regulate BDNF release, and reduced expression of these proteins substantially blunts both endogenous and TNF-α-enhanced secretion of BDNF. Compared with nonasthmatic ASM cells, where blocking TRPC3 or TRPC6 attenuates BDNF secretion by ∼25%, asthmatic cells exposed to TNF-α secreted substantially less BDNF secretion (Fig. 2). It should be noted here that we also inhibited TRPC1 with a specific siRNA, but did not see a significant change in BDNF release (results not shown). These data were further supported by pharmacological data obtained by inhibiting TRPC channels with 50 μM SKF-96365, a blocker of TRPC-mediated Ca2+ entry (13) (Fig. 2C).
Fig. 2.
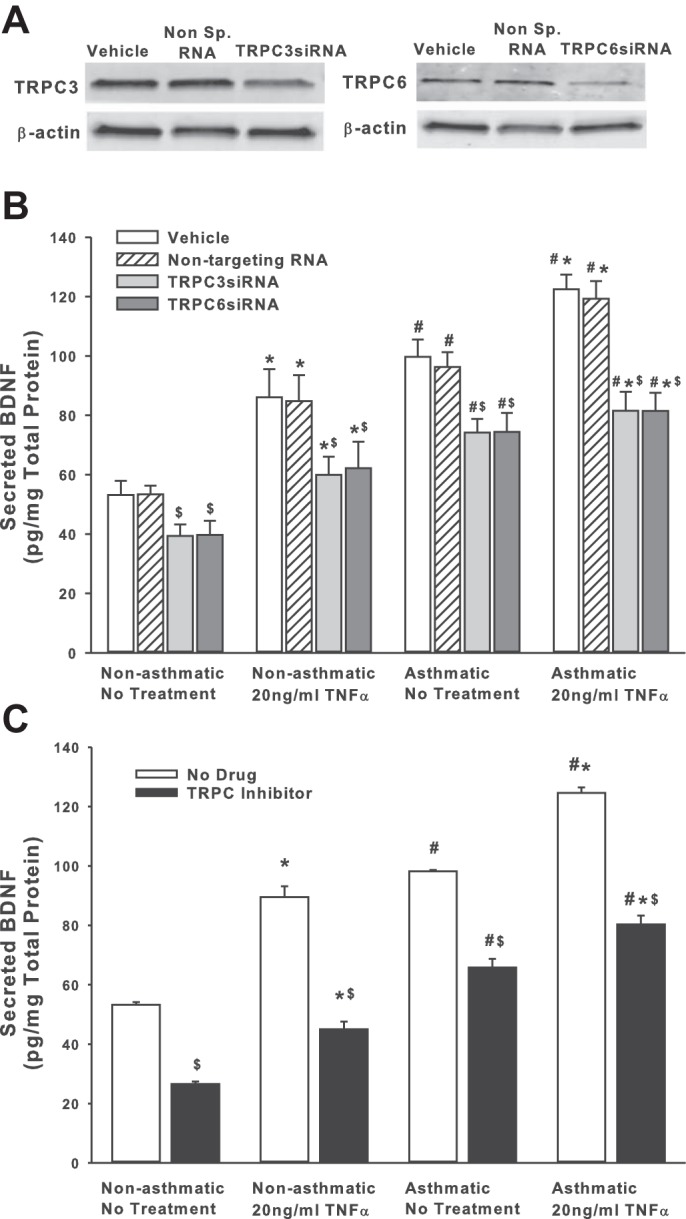
Transient receptor potential TRPC3 and TRPC channels are involved in regulating BDNF secretion by ASM cells. Nonasthmatic and asthmatic ASM cells were transfected with siRNA against TRPC3 or TRPC6 and were either exposed to medium or to 20 ng/ml TNF-α for 48 h, before cell supernatants and lysates were collected. A: immunoblot on the lysates showing inhibition efficiency of TRPC3 and TRPC6 siRNAs. These siRNAs specifically reduce the expression of their target proteins. Expression of TRPC3 and TRPC6 is not affected in “Vehicle” control and the “Nontargeting RNA” systems. β-actin was used as a loading control. B: BDNF was quantified in the extracellular medium by ELISA. Inhibition of either TRPC3 or TRPC6 resulted in decreased BDNF in the supernatant. Exposure to TNF-α did not elevate BDNF levels much higher. A similar effect was observed in asthmatic ASM cells as well. Vehicle refers to “No Transfection” control, where the transfection reagent Lipofectamine, with no DNA or RNA, was added to the cells. A nonspecific RNA that did not target any gene was used to control for siRNA specificity; n = 4 patients. *Significant difference from Vehicle-No TNF-α control, P < 0.05. #Significant difference between nonasthmatic and asthmatic cells, P < 0.05. $Significant difference between Vehicle and siRNA-transfected systems, P < 0.05. C: ASM cells were treated with 50 μM SKF-96365 (a TRPC inhibitor) for 2 h before being exposed to 20 ng/ml TNF-α for 48 h, before cell supernatants and lysates were collected; n = 4 patients. *Significant difference from No Drug-No TNF-α control, P < 0.05. #Significant difference between nonasthmatic and asthmatic cells, P < 0.05. $Significant difference between No Drug and TRPC Inhibitor-treated systems, P < 0.05.
Regulation of BDNF secretion by multiple signaling pathways.
We explored mechanisms additional to TRPC channels that are involved in controlling BDNF secretion by ASM cells. First, we examined Rho-kinase (ROCK), based on its relevance to modulation of intracellular Ca2+ levels as well as activation of TRPC3 and TRPC6, both of which are expressed by ASM cells. ROCK activation is also thought to play a crucial role in the development of allergic sensitization and asthma (39). We treated nonasthmatic and asthmatic cells with 10 μM Y27632 (ROCK inhibitor) under control (unexposed) or 20 ng/ml TNF-α conditions. Y27632 treatment dramatically decreased BDNF secretion in both nonasthmatic and asthmatic cells (Fig. 3), although the extent of reduction in BDNF secretion was greater in nonasthmatic ASM (50% vs. 20% in asthmatic cells). ROCK inhibition blunted TNF-α enhancement of BDNF secretion in nonasthmatic and asthmatic ASM cells. These results strongly suggest that ROCK activity is important for BDNF secretion by airway cells.
Fig. 3.
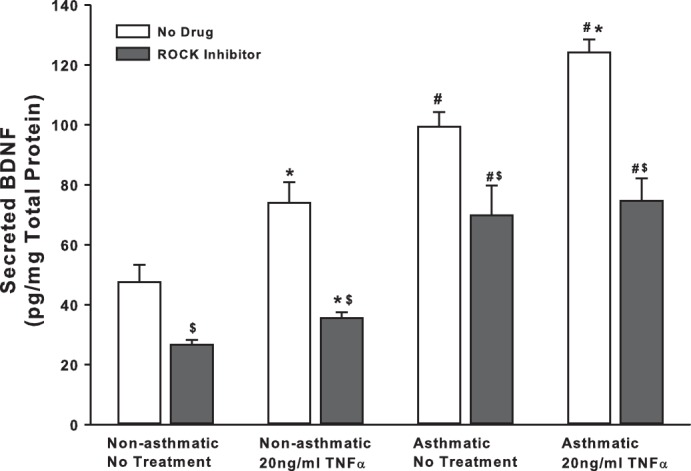
Rho kinase (ROCK) regulates basal BDNF secretion by ASM cells. Nonasthmatic and asthmatic ASM cells were treated, under serum-free conditions, with 10 μM Y27632 (a specific ROCK inhibitor). Two hours later, cells were either exposed to medium or to 20 ng/ml TNF-α for 48 h, before cell supernatants were collected for quantifying BDNF secretion by ELISA. Inhibition of ROCK caused a substantial reduction in BDNF in the supernatant. ROCK inhibition blocked BDNF secretion in asthmatic ASM cells as well, although to a lesser extent than in nonasthmatic cells. Exposure to TNF-α did not enhance BDNF secretion in either of the cell types treated with ROCK inhibitor; n = 4 patients. *Significant difference from No Drug-No TNF-α control, P < 005. #Significant difference between nonasthmatic and asthmatic cells, P < 0.05. $Significant difference between No Drug and ROCK Inhibitor systems, P < 0.05.
Next, we examined signaling pathways that are activated in inflammation and asthma, and whose role in BDNF secretion has been suggested in neurons and other systems, namely, ERK/MAPK, and PI3K pathways (52). We used pharmacological agents (PD98059 at 2 μM and wortmannin at 50 nM) to perturb ERK or PI3K signals, respectively, before exposing the cells to TNF-α. Furthermore, we expected that under conditions of bronchoconstrictor agonism (e.g., by ACh), activation of GPCRs will promote the PLC pathway via Gq. Therefore, we tested the G protein antagonist 2A (GPA-2A). All three compounds strongly suppressed BDNF secretion, in both the presence and absence of TNF-α (Fig. 4). Their action was equally potent in asthmatic ASM, irrespective of the addition of an inflammatory stimulus. These data imply that ERK/MAPK and PI3K pathways are essential for enhancing BDNF secretion.
Fig. 4.

ERK/MAPK and PI3K signaling cascades modulate BDNF secretion by ASM cells. Nonasthmatic and asthmatic ASM cells were treated, under serum-free conditions, with 10 μM GPA-2A (Gq inhibitor), 2 μM PD98059 (ERK/MAPK inhibitor), or 50 nM wortmannin (PI3K inhibitor). Two hours later, cells were either exposed to medium or to 20 ng/ml TNF-α for 48 h, before cell supernatants were collected for quantifying BDNF secretion by ELISA. Inhibition of ERK or PI3K signals attenuated BDNF secretion substantially, in both nonasthmatic and asthmatic ASM cells. In the presence of these inhibitors, TNF-α did not enhance BDNF secretion in either of the cell types; n = 4 patients. *Significant difference from No Treatment-No TNF-α control, P < 0.05. #Significant difference between nonasthmatic and asthmatic cells, P < 0.05. $Significant difference between No Drug and Inhibitor systems, P < 0.05.
Given its importance for synaptic regulation, several other pathways have been implicated for BDNF secretion in neurons, including PLC and PKC (8, 20, 29). These pathways are well recognized in ASM in the context of Ca2+ regulation. We inhibited PLC with 5 μM U73122 and PKC with 10 ng/ml BIS (bisindolylmaleimide) and quantified resultant changes in BDNF secretion. We found that inhibition of either PLC or PKC signaling causes a remarkable reduction in BDNF secretion in both asthmatic and nonasthmatic ASM cells, even under baseline conditions. Further, BDNF secretion is suppressed in TNF-α-exposed cells (Fig. 5). These results suggest that PLC and PKC pathways also play a vital role in regulating BDNF release and, thus, its downstream effects in the airway.
Fig. 5.
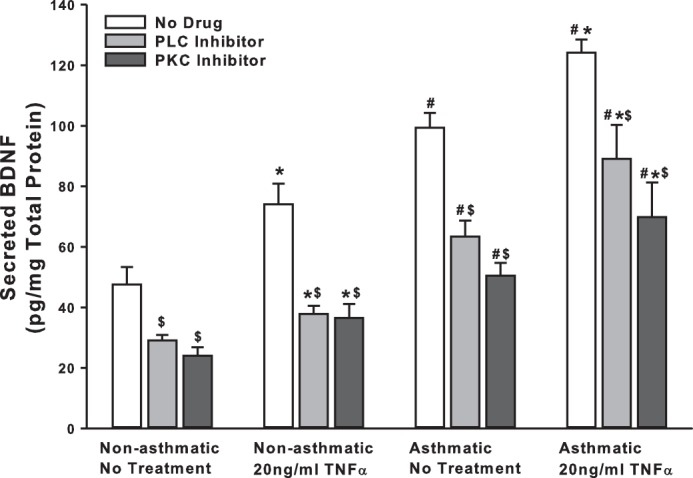
PLC and PKC signaling plays a role in basal BDNF secretion by ASM cells. Nonasthmatic and asthmatic ASM cells were treated, under serum-free conditions, with 5 μM U73122 (PLCγ inhibitor) or 10 ng/ml BIS (bisindolylmaleimide, a PKC inhibitor). Two hours later, cells were either exposed to medium or to 20 ng/ml TNF-α for 48 h, before cell supernatants were collected for quantifying BDNF secretion by ELISA. Inhibition of PLC or PKC signals attenuated BDNF secretion substantially, in both nonasthmatic and asthmatic ASM cells. In the presence of these inhibitors, TNF-α did not restore BDNF secretion in either of the cell types; n = 4 patients. *Significant difference from No Treatment-No TNF-α control, P < 0.05. #Significant difference between Nonasthmatic and Asthmatic cells, P < 0.05. $Significant difference between No Drug and Inhibitor systems, P < 0.05.
Role of transcription factors on BDNF secretion.
One of the expected outcomes of TRPC channel activation and increased PLC activity is increased [Ca2+]i, which, in turn, can trigger the MAPK cascade and/or activate Ca2+-sensitive proteins, such as calcineurin. MAPK and calcineurin converge on the transcriptional factors CREB and NFAT (16), respectively. Interestingly, promoter analysis on genes known to affect BDNF secretion (CAPS2, SYT4, and SNAP25, for example) shows that promoter regions of these genes do, in fact, have putative binding sites for transcription factors CREB and NFAT (Table 1), suggesting that BDNF secretion may be controlled at a transcriptional level. To determine the role of these transcription factors in BDNF secretion by ASM cells, we inhibited CREB (via siRNA), and NFAT (via a cell-permeable inhibitor). There was a significant drop in BDNF secretion due to either treatment (Fig. 6) in both asthmatic and nonasthmatic cells. Exposure to TNF-α did not seem to eliminate the inhibitor effects.
Table 1.
Promoter analysis on genes that may regulate BDNF secretion by ASM cells
| Gene | CREB | NFAT |
|---|---|---|
| BDNF | −88 to −80 | −284 to −277 |
| −366 to −359 | ||
| CAPS2 | −7782 to −7775 | −957 to −950 |
| −1348 to −1341 | ||
| −1509 to −1502 | ||
| SNAP25 | −3107 to −3100 | −569 to −562 |
| −3793 to −3786 | ||
| −3802 to −3795 | ||
| SYT4 | −1150 to −1143 | −765 to −758 |
| −5629 to −5622 | −1581 to −1574 | |
| −5776 to −5769 |
Approximately 10–12-kb region, upstream of start codon (ATG), was analyzed for putative cAMP response element binding protein (CREB) or nuclear factor of activated T cells (NFAT) binding sites, based on published consensus sequences (5′TGACGTCA3′ for CREB and 5′AGGAAATT3′ or 5′AGGAAACA3′ for NFAT).
BDNF, brain-derived neurotrophic factor; ASM, airway smooth muscle. Binding site location is presented as distance from the start codon.
Fig. 6.

BDNF secretion by ASM cells is controlled by Ca2+-responsive transcription factors. A: nonasthmatic and asthmatic ASM cells were transfected with siRNA against CREB and were either exposed to medium or to 20 ng/ml TNF-α for 48 h, before cell supernatants were collected for BDNF quantification by ELISA. Inhibition of either CREB significantly decreases BDNF secretion. Exposure to TNF-α did not elevate BDNF levels much higher. A similar effect was observed in asthmatic ASM cells as well. Vehicle refers to No Transfection control, where the transfection reagent Lipofectamine, with no DNA or RNA, was added to the cells. A nonspecific RNA that did not target any gene was used to control for siRNA specificity; n = 4 patients. *Significant difference from No Transfection-No TNFα control, P < 0.05. #Significant difference between Nonasthmatic and Asthmatic populations, P < 0.05. $Significant difference between Vehicle and siRNA-transfected systems, P < 0.05. B: nonasthmatic and asthmatic ASM cells were treated, under serum-free conditions, with a cell-permeable NFAT inhibitor (1 μM). Two hours later, cells were either exposed to medium or to 20 ng/ml TNF-α for 48 h, before cell supernatants were collected for quantifying BDNF secretion by ELISA. Inhibition of NFAT downregulated BDNF secretion significantly, in both nonasthmatic and asthmatic ASM cells. In the presence of NFAT inhibitor, TNF-α did not restore BDNF secretion in either of the cell types; n = 4 patients. *Significant difference from No Drug-No Treatment control, P < 0.05. #Significant difference between Nonasthmatic and Asthmatic populations, P < 0.05. $Significant difference between No Drug and Inhibitor systems, P < 0.05.
BDNF secretion is affected by cellular BDNF expression.
From the data reported above, it is clear that several factors and pathways influence the amount of BDNF secreted by ASM cells. In addition to nongenomic mechanisms, it is possible for BDNF to regulate its own expression, given that many of the signaling intermediates involved in BDNF signaling converge on the BDNF gene. We collected lysates from ASM cells after each of the treatments described above and assayed BDNF protein expression via immunoblotting. We found that while TNF-α treatment increased BDNF expression, blocking TRPC3/6, Gq, ERK/MAPK, PI3K, PLC or PKC signaling results in reduced BDNF expression (Fig. 7, A–D). Similarly, inhibition of ROCK or transcription factors CREB and NFAT also attenuates BDNF expression (Fig. 7, E–G). In asthmatic cells, BDNF expression is higher (than in nonasthmatic cells) under basal conditions, and it is further augmented following exposure to TNF-α (Fig. 7H). Together, these data suggest that transcriptional and/or posttranscriptional mechanisms also contribute toward regulating ASM secretion of BDNF.
Fig. 7.

Regulation of BDNF secretion is partially contingent on BDNF expression by ASM cells. Nonasthmatic ASM cells were subjected to treatments mentioned above (i.e., siRNA transfections or drug treatments), to perturb various signaling mechanisms, before being exposed to medium or to 20 ng/ml TNF-α for 48 h. Whole cell lysates were collected and analyzed for BDNF expression. A: transfection with siRNA against TRPC3 or TRPC6 inhibits BDNF expression, both in the presence and absence of TNF-α. Similarly, inhibition of TRPC3 (B), Gq, ERK/MAPK, or PI3K (C), PLC or PKC (D), ROCK (E), CREB (F), or NFAT (G) downregulate BDNF expression to appreciable levels, irrespective of TNF-α treatment. H: expression of BDNF in asthmatic ASM cells is higher at control conditions than in nonasthmatic cells, and TNF-α treatment enhances it further. β-Actin was used as loading control in all cases; n = 4 patients. *Significant difference from No Treatment-No TNF-α control, P < 0.05. #Significant difference between Nonasthmatic and Asthmatic populations, P < 0.05. $Significant difference between No Drug and Inhibitor systems, P < 0.05.
Verification of BDNF regulation in ASM tissues.
To ensure that the cellular in vitro data were not a feature of cellular isolation and culturing, we verified results in epithelium-denuded ASM tissue from nonasthmatic human subjects. After removing the epithelium, we incubated the ASM tissue (2 h) with pharmacological compounds used in our cellular studies above, before treating with TNF-α. Forty-eight hours later, ELISA was performed to quantify BDNF secreted into the medium and supernatant. Our results indicate that multiple signaling pathways, including TRPC, Gq, ERK/MAPK, PI3K, PLC, and PKC do regulate BDNF secretion by ASM tissue as well (Fig. 8). As with cells, inhibiting any of these signals has a substantial impact on BDNF release, and exposure to TNF-α is only partially restorative (TRPC, PI3K, PLC, and PKC; Fig. 8, A and B). Similarly, ROCK activity and NFAT activation seem to be essential for BDNF release, as blocking either of these results in decreased BDNF secretion (Fig. 8, C and D).
Fig. 8.

BDNF secretion in ASM tissue also involves several regulatory mechanisms. Epithelium-denuded ASM tissue from nonasthmatic donors was subjected to pharmacological treatments (similar to ASM cells, as described above), to perturb various signaling mechanisms, before being exposed to medium or to 20 ng/ml TNF-α for 48 h. BDNF secretion was quantified by ELISA on the media and supernatants. Drugs that inhibit TRPC, ERK/MAPK, or PI3K had substantial effects on BDNF secretion by ASM tissue (A). A similar effect was observed when PLC or PKC (B), ROCK (C), or NFAT (D) was inhibited, supporting the cellular data; n = 3 patients. *Significant difference from No Treatment-No TNF-α control, P < 0.05. $Significant difference between No Drug and Inhibitor systems, P < 0.05.
DISCUSSION
There is increasing interest in exploring the role of locally produced factors in the airway, particularly those enhanced by inflammation, in asthma pathophysiology. Here, factors produced by resident airway cells especially under chronic conditions have the potential to substantially mediate and modulate inflammation effects on multiple aspects of asthma, including airway hyperreactivity and remodeling. We previously showed that BDNF acts like a growth factor, working in conjunction with inflammatory mediators relevant to asthma, such as TNF-α and IL-13, to modulate [Ca2+]i (37) and ASM proliferation (7). In the airway, BDNF can be secreted by a variety of cell types, including immune cells, neurons, epithelial cells, and smooth muscle cells, and recently, BDNF expression and regulation, as well as its action via TrkB in these cells, have gained attention as a potentially significant mechanism in the pathogenesis of asthma (33, 35, 44, 49). The data presented in the current study present a picture of complex regulation of endogenous BDNF expression and secretion by human ASM in the context of inflammation (represented by TNF-α) and highlight a potential autocrine role for this neurotrophin.
In the nervous system, BDNF is thought to control its expression and activity in an autocrine fashion, and BDNF/TrkB activation initiates a variety of downstream signaling cascades, including the Ras/ERK, PI3K, and PLC pathways (23). We previously showed that in ASM cells, exogenous BDNF works via TrkB to activate ERK and PI3K pathways (7) and that BDNF is expressed in and secreted by human ASM (44). However, the mechanisms that regulate BDNF expression and secretion, particularly in the context of airway inflammation and asthma, are only beginning to be elucidated. Recent reports from our group showed that TRPC3 activation and the associated elevation in [Ca2+]i, seen during inflammation (44) and hyperoxic conditions (in fetal ASM) (43), can be potent modulators of BDNF secretion. In the current study, we have examined several additional modes via which BDNF regulation can be achieved. Our data not only provide a broad insight into the regulation of secreted BDNF in the airway, but they also highlight the complexity of this process, particularly, in the context of pathways that are common to both BDNF regulation and, interestingly, BDNF activity, as well as proinflammatory pathophysiology in asthma.
The interdependent connection between BDNF and TRPC3/6 channels is established in the nervous system. Here, BDNF has been shown to induce Ca2+ elevation via TRPC3, and BDNF activity (i.e., excitatory postsynaptic neurotransmission and synaptic vesicle fusion), which is mediated by TrkB, seems to require intracellular and extracellular Ca2+ (3, 28). TRPC3 is also necessary for activity-dependent release of BDNF, and for BDNF function, via a nonselective cationic current, during development (4, 25). TRPC6 is involved in BDNF processing (from its precursor to mature BDNF) in retinal ganglion cells (47). In nonneuronal tissues, TRPC3 has been shown to affect BDNF release by playing a significant role in Ca2+ influx regulation in the airway (44), and the evidence for the reverse relationship has been reported in cardiomyocytes, where BDNF regulates TRPC3/6 (17). Regardless, it is clear that TRPC3/6 activation and the resultant increase in [Ca2+]i are vital for BDNF expression and signaling, with data from the present study showing that TRPC3/6 expression is also essential for BDNF secretion in the airway.
The BDNF-TRPC axis may have additional implications: it has been shown in neurons that BDNF activation of CaMKK, which phosphorylates Akt1, requires TRPC6 (13) and that signaling pathways that are switched on in response to growth factor-induced TRPC activation include ERK and CREB (53). Thus, the roles that ERK and CREB that we see in the airway play in BDNF secretion could be attributed mainly to TRPC3/6-mediated increase in [Ca2+]i.
Regulation of [Ca2+]i in ASM involves release of intracellular Ca2+ stores. The key role that BDNF plays in the release of Ca2+ from intracellular stores via PLCγ-associated IP3/IP3R pathway has long been recognized (26, 46). Given that PLC activation by TrkB in neurons (23) and increased Ca2+ release from the sarcoplasmic reticulum (SR) during airway inflammation have been reported (34, 37), one could infer from our current data that increased PLC activity and IP3-induced SR Ca2+ outflow following TNF-α treatment, may be due to the autocrine signaling by BDNF. Furthermore, our results in ASM cells are consistent with what is observed in neurons: PLC activation is a requisite for BDNF release (20).
Another interesting aspect explored in this study is the plausible role of the transcription factors NFAT and CREB in modulating BDNF secretion, suggesting that BDNF expression, at least partially, controls its release. Studies on neurons have confirmed that BDNF is a target for CREB (42) and that BDNF expression is dependent on the Ca2+/CREB pathway (12). It is likely that in the ASM, inflammatory signals trigger pathways and second messengers converging at CREB, which eventually targets and upregulates the expression of BDNF. As is well documented, the regulation of [Ca2+]i homeostasis and changes in gene expression go hand in hand. Therefore, it is not entirely surprising that an increase in [Ca2+]i activates other Ca2+-sensitive factors, such as NFAT, a calcineurin effector. In addition to being an effector of BDNF and activities, NFAT is also thought to regulate TRPC3/6 expression (21). Thus, it is possible that both NFAT and CREB are involved in the regulation of BDNF expression. Our identification of near-consensus CREB and NFAT binding sites on BDNF promoter supports this idea. Further, while our ASM cellular data by themselves shed light on the mechanisms underlying BDNF production in the airway, results from our ASM tissue studies lend credence to the notion that the complex regulation of BDNF can occur in situ in the airway.
We previously showed vesicular release of BDNF by human ASM cells (44). Although somewhat unprecedented, such a mechanism does offer an additional and efficient tier of regulation for BDNF secretion. The involvement of the Soluble NSF Attachment Protein Receptor (SNARE) complex proteins and the regulation of the exocytotic machinery in releasing BDNF from secretory vesicles is better understood in neurons (31). Drawing from these studies, we looked at the promoter regions of SNAP25 (a SNARE protein), CAPS2 (a secretory granule associated protein), and SYT4 (a membrane-trafficking vesicle protein). We found putative binding sites for CREB and NFAT on the promoters of genes coding for all three of these proteins. It is tempting to speculate that in addition to directly controlling BDNF expression, CREB and NFAT may also target genes that are involved in the vesicular trafficking and release of BDNF, especially in view of our recent finding that secretion of BDNF is altered concurrently with SNAP25 expression in ASM cells treated with sex steroids (45). It should be mentioned here that although the vesicular release of BDNF by neurons is tightly regulated and rapid (22), we expect a slightly dissimilar mechanism in the ASM, especially during the much slower remodeling process. The exact mechanism(s) via which these vesicle proteins regulate BDNF secretion by the ASM remains to be examined.
Finally, the work presented here is the first to directly compare nonasthmatic ASM to asthmatic ASM, in the context of inflammation-induced BDNF secretion, and the mechanisms thereof, which bear significant physiological and pathophysiological relevance to asthma. For example, ROCK signaling is a vital determinant of structural modifications in the airway during asthma, as it promotes MLCK (myosin light chain or MLC phosphorylation) and inhibits the MLCP (MLC phosphatase), thus being directly involved in smooth muscle contractility (9). While RhoA signaling is regulated by [Ca2+]i under normal conditions (27), both TNF-α- and BDNF-induced MLC phosphorylation events have been shown to require RhoA and ROCK signaling (5). ROCK regulation of BDNF function at the secretion level, as our study suggests, thus, underscores the potential of RhoA signaling as a therapeutic target for asthma.
On the basis of our current findings, we envision a number of events that occur, perhaps concurrently, during inflammation, culminating in BDNF release, as depicted in Fig. 9. In this model, TNF-α and any agonists (e.g., ACh, which is known to increase BDNF) promote the PLC/IP3 and DAG/PKC pathways, enhancing [Ca2+]i, which may already be on the increase due to augmented TRPC3/6 activity. Subsequently, Ca2+- sensitive pathways, such as ERK and calcineurin, are turned on, resulting in the translocation of CREB and NFAT to the nucleus, to regulate the expression of BDNF and other proteins involved in its release. Activation of ROCK and the autocrine function of BDNF may also aid in regulating BDNF secretion. Secreted BDNF can then have autocrine effects of regulating a multitude to genes (via ERK and other pathways) to influence ASM structure and function. Here, asthmatic ASM cells may have a baseline upregulated level of a number of upstream and downstream mechanisms, allowing for higher basal BDNF levels (and effects).
Fig. 9.

Schematic model of the complexity of BDNF regulation in ASM, particularly with inflammation. BDNF secretion may be controlled at multiple levels: 1) Inflammatory stimuli such as TNF-α, particularly with ongoing bronchoconstrictor agonist stimulation involving GPCRs, can enhance the PLC/IP3 and DAG/PKC pathways. DAG can modulate TRPC3 and TRPC6 channels, while a number of these regulatory components can increase Ca2+. The net effects of enhanced [Ca2+]i and other upstream pathways are activation of the Ca2+-sensitive MAPK signaling cascade, and calcineurin (CaN), both of which activate the transcription factor CREB (2). CaN also turns on NFAT (3). CREB and NFAT translocate to the nucleus and regulate the expression of BDNF, TRPC3/6, and the genes for vesicle proteins, such as SNAP25, CAPS2, and SYT4. These proteins, thus, play a role in vesicle release of BDNF (4). Alternatively, RhoA/ROCK (which also responds to TrkB-ligand binding, possibly via RasGRF1) can promote exocytosis of BDNF-containing vesicles (5). Finally, BDNF, in an autocrine manner (6), binds to and turns on TrkB receptor, which may result in an autocrine loop of sustained BDNF expression, secretion, and signaling.
In conclusion, we show that asthmatic cells secrete more BDNF, even in the absence of an inflammation trigger, suggesting that the endogenous production and release of BDNF may “prime” these cells to be hyperresponsive to subsequent inflammatory conditions. This is an important factor to consider, as the autocrine activity of BDNF, combined with additional episodes of inflammation, likely gives rise to a “snowball effect,” which, in turn, heightens the severity of asthma as it progresses.
GRANTS
This article was supported by grants from the National Institutes of Health (R01 HL-088029 and R01 HL-056470) to Y. S. Prakash, and by a Young Clinical Scientist Award from the Flight Attendants Medical Research Institute to B. Aravamudan.
DISCLOSURES
No conflicts of interest, financial or otherwise are declared by the authors.
AUTHOR CONTRIBUTIONS
B.A. and Y.S.P. conception and design of research; B.A. performed experiments; B.A. analyzed data; B.A. interpreted results of experiments; B.A. prepared figures; B.A. drafted manuscript; B.A., M.A.T., and Y.S.P. edited and revised manuscript; B.A., C.M.P., and Y.S.P. approved final version of manuscript.
REFERENCES
- 1.Abcejo AJ, Sathish V, Smelter DF, Aravamudan B, Thompson MA, Hartman WR, Pabelick CM, Prakash YS. Brain-derived neurotrophic factor enhances calcium regulatory mechanisms in human airway smooth muscle. PLos One 7: e44343, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Al-Muhsen S, Johnson JR, Hamid Q. Remodeling in asthma. J Allergy Clin Immunol 128: 451–462; quiz 463-454, 2011. [DOI] [PubMed] [Google Scholar]
- 3.Amaral MD, Pozzo-Miller L. Intracellular Ca2+ stores and Ca2+ influx are both required for BDNF to rapidly increase quantal vesicular transmitter release. Neural Plast 2012: 203536, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Amaral MD, Pozzo-Miller L. TRPC3 channels are necessary for brain-derived neurotrophic factor to activate a nonselective cationic current and to induce dendritic spine formation. J Neurosci 27: 5179–5189, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Anastasia A, Deinhardt K, Wang S, Martin L, Nichol D, Irmady K, Trinh J, Parada L, Rafii S, Hempstead BL, Kermani P. Trkb signaling in pericytes is required for cardiac microvessel stabilization. PloS One 9: e87406, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aravamudan B, Kiel A, Freeman M, Delmotte P, Thompson M, Vassallo R, Sieck GC, Pabelick CM, Prakash YS. Cigarette smoke-induced mitochondrial fragmentation and dysfunction in human airway smooth muscle. Am J Physiol Lung Cell Mol Physiol 306: L840–L854, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aravamudan B, Thompson M, Pabelick C, Prakash YS. Brain-derived neurotrophic factor induces proliferation of human airway smooth muscle cells. J Cell Mol Med 16: 812–823, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bagayogo IP, Dreyfus CF. Regulated release of BDNF by cortical oligodendrocytes is mediated through metabotropic glutamate receptors and the PLC pathway. ASN Neuro 1: pii: e00001, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chiba Y, Nakazawa S, Todoroki M, Shinozaki K, Sakai H, Misawa M. Interleukin-13 augments bronchial smooth muscle contractility with an up-regulation of RhoA protein. Am J Respir Cell Mol Biol 40: 159–167, 2009. [DOI] [PubMed] [Google Scholar]
- 10.Dekkers BG, Maarsingh H, Meurs H, Gosens R. Airway structural components drive airway smooth muscle remodeling in asthma. Proc Am Thorac Soc 6: 683–692, 2009. [DOI] [PubMed] [Google Scholar]
- 11.Doherty T, Broide D. Cytokines and growth factors in airway remodeling in asthma. Curr Opin Immunol 19: 676–680, 2007. [DOI] [PubMed] [Google Scholar]
- 12.Finkbeiner S. Calcium regulation of the brain-derived neurotrophic factor gene. Cell Mol Life Sci 57: 394–401, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fortin DA, Srivastava T, Dwarakanath D, Pierre P, Nygaard S, Derkach VA, Soderling TR. Brain-derived neurotrophic factor activation of CaM-kinase kinase via transient receptor potential canonical channels induces the translation and synaptic incorporation of GluA1-containing calcium-permeable AMPA receptors. J Neurosci 32: 8127–8137, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gosens R, Grainge C. Bronchoconstriction and airway biology: potential impact and therapeutic opportunities. Chest 147: 798–803, 2015. [DOI] [PubMed] [Google Scholar]
- 15.Guedes AG, Deshpande DA, Dileepan M, Walseth TF, Panettieri RA Jr, Subramanian S, Kannan MS. CD38 and airway hyper-responsiveness: studies on human airway smooth muscle cells and mouse models. Can J Physiol Pharmacol 93: 145–153, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hai CM. Airway smooth muscle cell as therapeutic target of inflammation. Curr Med Chem 14: 67–76, 2007. [DOI] [PubMed] [Google Scholar]
- 17.Hang P, Zhao J, Cai B, Tian S, Huang W, Guo J, Sun C, Li Y, Du Z. Brain-derived neurotrophic factor regulates TRPC3/6 channels and protects against myocardial infarction in rodents. Int J Biol Sci 11: 536–545, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harkness LM, Ashton AW, Burgess JK. Asthma is not only an airway disease, but also a vascular disease. Pharmacol Ther 148: 17–33, 2015. [DOI] [PubMed] [Google Scholar]
- 19.Howarth PH, Knox AJ, Amrani Y, Tliba O, Panettieri RA Jr, Johnson M. Synthetic responses in airway smooth muscle. J Allergy Clin Immunol 114: S32–S50, 2004. [DOI] [PubMed] [Google Scholar]
- 20.Jean YY, Lercher LD, Dreyfus CF. Glutamate elicits release of BDNF from basal forebrain astrocytes in a process dependent on metabotropic receptors and the PLC pathway. Neuron Glia Biol 4: 35–42, 2008. [DOI] [PubMed] [Google Scholar]
- 21.Koitabashi N, Aiba T, Hesketh GG, Rowell J, Zhang M, Takimoto E, Tomaselli GF, Kass DA. Cyclic GMP/PKG-dependent inhibition of TRPC6 channel activity and expression negatively regulates cardiomyocyte NFAT activation. Novel mechanism of cardiac stress modulation by PDE5 inhibition. J Mol Cell Cardiol 48: 713–724, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuczewski N, Porcher C, Lessmann V, Medina I, Gaiarsa JL. Activity-dependent dendritic release of BDNF and biological consequences. Mol Neurobiol 39: 37–49, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leal G, Comprido D, Duarte CB. BDNF-induced local protein synthesis and synaptic plasticity. Neuropharmacology 76: 639–656, 2014. [DOI] [PubMed] [Google Scholar]
- 24.Li W, Calfa G, Larimore J, Pozzo-Miller L. Activity-dependent BDNF release and TRPC signaling is impaired in hippocampal neurons of Mecp2 mutant mice. Proc Natl Acad Sci USA 109: 17,087–17,092, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li Y, Calfa G, Inoue T, Amaral MD, Pozzo-Miller L. Activity-dependent release of endogenous BDNF from mossy fibers evokes a TRPC3 current and Ca2+ elevations in CA3 pyramidal neurons. J Neurophysiol 103: 2846–2856, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li YX, Xu Y, Ju D, Lester HA, Davidson N, Schuman EM. Expression of a dominant negative TrkB receptor, T1, reveals a requirement for presynaptic signaling in BDNF-induced synaptic potentiation in cultured hippocampal neurons. Proc Natl Acad Sci USA 95: 10,884–10,889, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mbikou P, Fajmut A, Brumen M, Roux E. Contribution of Rho kinase to the early phase of the calcium-contraction coupling in airway smooth muscle. Exp Physiol 96: 240–258, 2011. [DOI] [PubMed] [Google Scholar]
- 28.Mizoguchi Y, Kato TA, Seki Y, Ohgidani M, Sagata N, Horikawa H, Yamauchi Y, Sato-Kasai M, Hayakawa K, Inoue R, Kanba S, Monji A. Brain-derived neurotrophic factor (BDNF) induces sustained intracellular Ca2+ elevation through the up-regulation of surface transient receptor potential 3 (TRPC3) channels in rodent microglia. J Biol Chem 289: 18,549–18,555, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morioka N, Yoshida Y, Nakamura Y, Hidaka N, Hisaoka-Nakashima K, Nakata Y. The regulation of exon-specific brain-derived neurotrophic factor mRNA expression by protein kinase C in rat cultured dorsal root ganglion neurons. Brain Res 1509: 20–31, 2013. [DOI] [PubMed] [Google Scholar]
- 30.Nassenstein C, Kerzel S, Braun A. Neurotrophins and neurotrophin receptors in allergic asthma. Prog Brain Res 146: 347–367, 2004. [DOI] [PubMed] [Google Scholar]
- 31.O'Sullivan NC, McGettigan PA, Sheridan GK, Pickering M, Conboy L, O'Connor JJ, Moynagh PN, Higgins DG, Regan CM, Murphy KJ. Temporal change in gene expression in the rat dentate gyrus following passive avoidance learning. J Neurochem 101: 1085–1098, 2007. [DOI] [PubMed] [Google Scholar]
- 32.Perez-Zoghbi JF, Karner C, Ito S, Shepherd M, Alrashdan Y, Sanderson MJ. Ion channel regulation of intracellular calcium and airway smooth muscle function. Pulm Pharmacol Ther 22: 388–397, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Prakash YS. Airway smooth muscle in airway reactivity and remodeling: what have we learned? Am J Physiol Lung Cell Mol Physiol 305: L912–L933, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Prakash YS, Iyanoye A, Ay B, Mantilla CB, Pabelick CM. Neurotrophin effects on intracellular Ca2+ and force in airway smooth muscle. Am J Physiol Lung Cell Mol Physiol 291: L447–L456, 2006. [DOI] [PubMed] [Google Scholar]
- 35.Prakash YS, Martin RJ. Brain-derived neurotrophic factor in the airways. Pharmacol Ther 143: 74–86, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Prakash YS, Thompson MA, Pabelick CM. Brain-derived neurotrophic factor in TNF-α modulation of Ca2+ in human airway smooth muscle. Am J Respir Cell Mol Biol 41: 603–611, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Renz H. Neurotrophins in bronchial asthma. Respir Res 2: 265–268, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schaafsma D, Gosens R, Bos IS, Meurs H, Zaagsma J, Nelemans SA. Allergic sensitization enhances the contribution of Rho-kinase to airway smooth muscle contraction. Br J Pharmacol 143: 477–484, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Spinelli AM, Gonzalez-Cobos JC, Zhang X, Motiani RK, Rowan S, Zhang W, Garrett J, Vincent PA, Matrougui K, Singer HA, Trebak M. Airway smooth muscle STIM1 and Orai1 are upregulated in asthmatic mice and mediate PDGF-activated SOCE, CRAC currents, proliferation, and migration. Pflügers Arch 464: 481–492, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Szczepankiewicz A, Rachel M, Sobkowiak P, Kycler Z, Wojsyk-Banaszak I, Schoneich N, Szczawinska-Poplonyk A, Breborowicz A. Neurotrophin serum concentrations and polymorphisms of neurotrophins and their receptors in children with asthma. Respir Med 107: 30–36, 2013. [DOI] [PubMed] [Google Scholar]
- 42.Tang M, Shi S, Guo Y, Xu W, Wang L, Chen Y, Wang Z, Qiao Z. GSK-3/CREB pathway involved in the gx-50's effect on Alzheimer's disease. Neuropharmacology 81: 256–266, 2014. [DOI] [PubMed] [Google Scholar]
- 43.Thompson MA, Britt RD Jr, Kuipers I, Stewart A, Thu J, Pandya HC, MacFarlane P, Pabelick CM, Martin RJ, Prakash YS. cAMP-mediated secretion of brain-derived neurotrophic factor in developing airway smooth muscle. Biochim Biophys Acta 1853: 2506–2514, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vohra PK, Thompson MA, Sathish V, Kiel A, Jerde C, Pabelick CM, Singh BB, Prakash YS. TRPC3 regulates release of brain-derived neurotrophic factor from human airway smooth muscle. Biochim Biophys Acta 1833: 2953–2960, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang S, Freeman MR, Sathish V, Thompson MA, Pabelick CM, Prakash YS. Sex steroids influence brain-derived neurotropic factor secretion from human airway smooth muscle cells. J Cell Physiol 231: 1586–1592, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang X, Berninger B, Poo M. Localized synaptic actions of neurotrophin-4. J Neurosci 18: 4985–4992, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang X, Teng L, Li A, Ge J, Laties AM, Zhang X. TRPC6 channel protects retinal ganglion cells in a rat model of retinal ischemia/reperfusion-induced cell death. Invest Ophthalmol Vis Sci 51: 5751–5758, 2010. [DOI] [PubMed] [Google Scholar]
- 48.Wang YX, Zheng YM. Molecular expression and functional role of canonical transient receptor potential channels in airway smooth muscle cells. Adv Exp Med Biol 704: 731–747, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Watanabe T, Fajt ML, Trudeau JB, Voraphani N, Hu H, Zhou X, Holguin F, Wenzel SE. Brain-derived neurotrophic factor (BDNF) expression in asthma: association with severity and Type-2 inflammatory processes. Am J Respir Cell Mol Biol 53: 844–852, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.White TA, Xue A, Chini EN, Thompson M, Sieck GC, Wylam ME. Role of transient receptor potential C3 in TNF-α-enhanced calcium influx in human airway myocytes. Am J Respir Cell Mol Biol 35: 243–251, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wright DB, Tripathi S, Sikarwar A, Santosh KT, Perez-Zoghbi J, Ojo OO, Irechukwu N, Ward JP, Schaafsma D. Regulation of GPCR-mediated smooth muscle contraction: implications for asthma and pulmonary hypertension. Pulm Pharmacol Ther 26: 121–131, 2013. [DOI] [PubMed] [Google Scholar]
- 52.Yamada K, Nabeshima T. Brain-derived neurotrophic factor/TrkB signaling in memory processes. J Pharm Sci 91: 267–270, 2003. [DOI] [PubMed] [Google Scholar]
- 53.Yao H, Peng F, Fan Y, Zhu X, Hu G, Buch SJ. TRPC channel-mediated neuroprotection by PDGF involves Pyk2/ERK/CREB pathway. Cell Death Differ 16: 1681–1693, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]