

Abstract
Multiple myeloma is an incurable malignancy of plasma B-cells. Traditional chemotherapeutic regimes often induce initial tumor regression; however, virtually all patients eventually succumb to relapse caused by either reintroduction of disease during autologous transplant or expansion of chemotherapy resistant minimal residual disease. It has been previously demonstrated that an oncolytic virus known as myxoma can completely prevent myeloma relapse caused by reintroduction of malignant cells during autologous transplant. The ability of this virus to treat established residual disease in vivo, however, remained unknown. Here we demonstrate that intravenous administration of myxoma virus into mice bearing disseminated myeloma results in the elimination of 70–90% of malignant cells within 24 hours. This rapid debulking was dependent on direct contact of myxoma virus with residual myeloma and did not occur through destruction of the hematopoietic bone marrow niche. Importantly, systemic myxoma therapy also induced potent antimyeloma CD8+ T cell responses which localized to the bone marrow and were capable of completely eradicating established myeloma in some animals. These results demonstrate that oncolytic myxoma virus is not only effective at preventing relapse caused by reinfusion of tumor cells during stem cell transplant, but is also potentially curative for patients bearing established minimal residual disease.
Introduction
Multiple myeloma (MM) is a clonal malignancy of plasma B cells.1 Unfortunately, despite its clonal nature, MM displays a high degree of genetic heterogeneity, most likely due to the genetic instability inherent in immunoglobulin rearrangement. Thus, even with the introduction of new classes of chemotherapeutic agents,2 the disease remains extremely difficult to completely eradicate and virtually all patients eventually succumb to relapse.3,4 Novel treatment options capable of producing durable complete remissions or cures are therefore urgently needed.
One such novel treatment is the use of live viruses which preferentially infect and kill malignant cells.5 This strategy, known as oncolytic virotherapy (OV) is attractive in the setting of MM since the multipronged mechanisms through which oncolytic viruses kill infected cells renders the development of cancer resistance unlikely. Indeed, it has recently been demonstrated that OV using a recombinant measles virus can induce complete remissions even in multiply relapsed MM patients.6 Unfortunately, while this trial demonstrates that OV has tremendous promise for relapsed MM patients, application of the particular oncolytic measles virus tested is heavily restricted due to the high sero-prevalence of antimeasles immunoglobulin. In order to overcome this obstacle, our lab had previously investigated the antiMM potential of a nonhuman oncolytic agent, known as myxoma virus (MYXV),5,7,8 whose natural tropism is tightly restricted to lagomorphs (rabbits).9,10 This work demonstrated that, despite its nonhuman tropism, MYXV infection efficiently killed human MM cells by inducing a rapid apoptotic response.7,8 This response was not observed in normal hematopoietic cells allowing ex vivo treatment with MYXV to prevent MM relapse in the context of autologous stem cell transplant (auto-SCT).7 Unfortunately, while reinfusion of malignant cells is thought to play some role in MM recurrence following auto-SCT,11,12 the primary cause of relapse in most patients is likely expansion of minimal residual disease (MRD) which escapes chemotherapeutic eradication within protected bone marrow (BM) niches.13,14 Therefore we, set out to investigate the therapeutic potential of MYXV against MRD which was already established in vivo.
Results
Murine MOPC-315 cells accurately recapitulate the response of human MM cells to MYXV treatment
The efficacy of OV is mediated both by direct viral lysis as well as the induction of secondary antitumor immunotherapy.15 Complete analysis of oncolytic potential therefore requires experiments to be conducted in immune competent tumor models which accurately mimic the responses of human disease. To facilitate studies into how MYXV treatment impacts MRD, we therefore sought to identify a murine MM cell which recapitulated the previously described response of human MM to MYXV treatment.7,8 Four established murine MM cell lines: P3.6.2.8.1, MOPC-31C, MOPC-315, and MOPC-315.BM were either mock treated or infected with vMYX-GFP. The rate of infection (Figure 1a) and lytic potential (Figure 1b) of MYXV in each cell line was then compared with that seen in the human U266 MM cell line using flowcytometry and MTT assay. Consistent with our previous results,7,8 virtually all human U266 cells (95.0%) displayed evidence of GFP expression 6 hours after infection and this infection resulted in a significant loss of cellular viability (55% remaining viability) by 24 hours. In the murine MM lines, both MOPC-315 and P3.6.2.8.1 cells displayed rates of infection similar to that observed in U266 cells (MOPC-315 = 93.8%, P3.6.2.8.1 = 99.0%). However, in P3.6.2.8.1 cells, this infection resulted in a significantly higher reduction in cellular viability then was typically observed in U266 cells (24% versus 55%, P = 0.03). In contrast, the reduction in viability observed in MOPC-315 cells was statistically identical to that seen in U266 cells (45% versus 55%). Infection of either MOPC-31C or MOPC-315. BM cells resulted in only limited infection and reduction in cellular viability. These results suggested that the MOPC-315 cell line might display susceptibility to MYXV treatment similar to human myeloma in vivo.
Figure 1.

MYXV infects murine MM cell lines. The indicated cell lines were mock infected or infected with vMYX-GFP at an MOI = 10. (a) Six hours postinfection, the number of GFP expressing cells was determined using flow cytometry. (b) Twenty-four hours postinfection, the effects of viral treatment on cellular viability was determined using MTT assay. Significance was determined using student’s t-test (*P < 0.05, ***P < 0.0001). GFP, green fluorescent protein; MM, multiple myeloma; MYXV, myxoma virus; MOI, multiplicity of infection.
We have previously demonstrated that MYXV-mediated killing of both primary and established human MM cells is independent of viral replication and occurs through the rapid induction of a lethal apoptotic response.7,8 To further determine whether MOPC-315 cells accurately recapitulated the response of human MM to MYXV treatment, we therefore compared both MYXV replication as well as the induction of lethal apoptosis in these cells to that observed in human U266 MM cells. To assay viral replication, we performed a single step growth curve to determine the numbers of new infectious MYXV progeny created in either U266 or MOPC-315 cells. The numbers of progeny virus produced in each MM cell line was then compared with the numbers of progeny virus produced in previously reported “permissive” cell lines such as A549, BSC40, LLC, or RL5. Consistent with previous reports,7 MYXV infection produced only a limited number of new infectious progeny following infection of human U266 cells (Figure 2a). Slightly higher numbers of infectious progeny were observed following infection of MOPC-315 cells; however, the numbers of these progeny were still 50–100 fold less what was observed in the other permissive cell lines suggesting that viral replication in MOPC-315 cells was possible but highly inefficient. To further assay whether viral infection resulted in a lethal apoptotic response, we next compared the initiation and progression of apoptosis in infected MOPC-315 cells to that observed in human U266 cells. The results indicated that, 2 hours after viral treatment both U266 cultures and MOPC-315 cultures displayed increased numbers of early apoptotic cells (as measured by AnnexinV/Propidium Iodide staining) (Figure 2b) and that magnitude of this increase was similar in both cell lines. Additionally, western blot analysis demonstrated that viral treatment of either MOPC-315 or U266 cells induced cleavage of both caspase-3 and PARP in both cell lines with similar kinetics (Figure 2c). Importantly, inhibition of viral replication, through the addition of cytarabine, did not prevent MYXV from inducing a loss of cellular viability in either MOPC-315 or U266 cells suggesting that the primary mechanism responsible for killing both cells was independent of viral replication (Figure 2d). Taken together, these data suggest that murine MOPC-315 MM cells are killed by MYXV-treatment with a similar efficacy and through a similar mechanism as is seen in human primary MM cells and therefore represent an appropriate model for analysis of treatment of MRD in vivo.
Figure 2.

MYXV induces apoptosis in MOPC-315 cells (a) The indicated cell lines were infected with MYXV at an MOI = 5. At various times postinfection, cells were harvested and the numbers of new infectious progeny were determined using foci forming assays. Data is displayed as the fold increase in progeny over that observed at 6 hours postinfection. MOPC-315 cells were mock-infected or infected with MYXV at an MOI = 10. (b) Six hours postinfection, cells were stained with annexinV (AnV) and propidium iodide (PI). (c) At the indicated times postinfection, cells were harvested and the cleavage of the indicated apoptotic proteins was analyzed using western blot. (d) MOPC-315 cells or U266 cells were mock-infected or infected with MYXV at an MOI = 10 in either the presence of absence of cytarabine (AraC). Cellular viability was then determined 24 hours after infection using MTT assay. Significance was determined using student’s t-test (***P < 0.0001). MM, multiple myeloma; MYXV, myxoma virus; MOI, multiplicity of infection.
Systemic MYXV treatment can cure established MRD
Having identified a murine MM cell line which appeared to accurately mimic the response of human MM to MYXV infection, next we asked whether viral treatment could be used to treat MRD already established in vivo. Syngeneic Balb/C mice were sublethally irradiated (450 cGy) and then injected intravenously (i.v.) with 5 × 106 MOPC-315 cells. Disease was allowed to establish until the mice began to show symptoms of hind limb paralysis (typically around 21 days postinjection) to model patients presenting with systemically disseminated, and symptomatic MM. Mice were then randomly separated into two cohorts and treated with three sequential i.v. injections of either saline or 1 × 108 foci forming units (FFU) of MYXV over a 5-day period. To determine the acute effects of viral treatment on MRD, animals were killed 24 hours after the final treatment and tumor burden was directly measured using flowcytometry.16 The results indicated that, in saline treated animals (n = 10), CD138hi/CD4+ MOPC-315 cells made up ~18% of the viable cells in the BM (Figure 3a). In contrast, all mice treated with MYXV (n = 12) displayed a significantly reduced number of MOPC-315 cells in their BM (6.4%, P < 0.0001) indicating that viral treatment could acutely debulk MRD in vivo. To further determine whether viral treatment would provide any significant survival benefit, Balb/C mice were seeded with MOPC-315 cells and treated as above. Animals were then monitored for signs of worsening clinical MM and killed when they displayed complete paralysis of both hind limbs. The results indicated that 100% of saline treated animals (n = 33) displayed symptoms of worsening MM which required euthanasia by day 67 postinjection (Figure 3b). In contrast, animals treated with MYXV (n = 39) displayed significantly improved overall survival (P = 0.001) which presented in two distinct forms. The first form, which presented as a modest (~6 day) delay in disease progression, provided only minimal survival benefit but occurred in a high percentage of animals (26 out of 39, 66%). In contrast, the second form, which presented as an apparent eradication of disease with no remaining symptoms at 80 days posttransplant, provided a much more significant survival benefit but occurred in only 25% of animals (10 out of 39). Taken together, these data indicate that systemic MYXV therapy can acutely debulk MRD in vivo and induce complete clinical regression in some animals.
Figure 3.
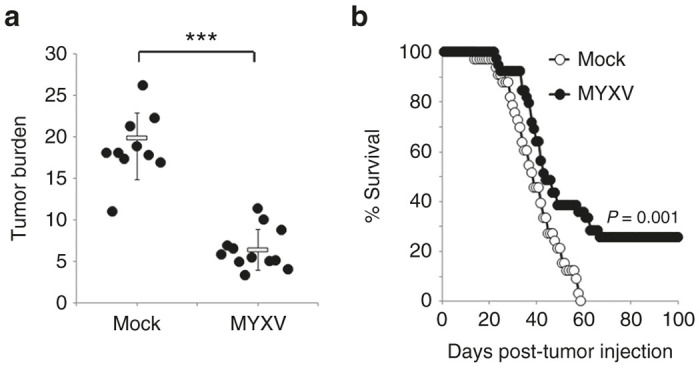
MYXV treatment can eradicate established MRD in vivo. Balb/C mice were sublethally irradiated (450 cGy) and then injected i.v. with 5 × 106 MOPC-315 cells. Disease was allowed to establish and then animals were given three i.v. injections of either saline or 1 × 108 FFU of MYXV over 5 days. (a) Twenty-four hours after the last treatment tumor burden in the BM of either saline (n = 10) or MYXV treated (n = 12) animals was determined using flowcytometry. Significance was determined using student’s t-test (***P < 0.0001). (b) Saline (n = 33) or MYXV treated (n = 39) animals were monitored for disease progression and killed when they displayed complete paralysis of both hind-limbs. Significance was determined using log-rank test. BM, bone marrow; MRD, minimal residual disease; MYXV, myxoma virus; FFU, foci forming units.
MYXV treatment does not functionally compromise the BM niche
In vivo, MM cells reside within protected BM niches and disruption of these niches can reduce tumor burden and slow disease progression.17–19 While MYXV is unable to replicate in normal human or mouse tissues, flooding the blood stream with billions of viral particles might significantly affect cells contiguous with the vasculature, such as the cells which make up the BM niche. Therefore, we wished to determine whether i.v. injection of MYXV might impact the course of established MM by altering either the make-up or functionality of the BM niche in vivo. To address this, nonirradiated, tumor-free, Balb/C mice were given three i.v. injections of either saline (n = 10) or 1 × 108 FFU of vMYX-GFP (n = 10) over a 5-day period. Twenty-four hours after the final injection, BM was harvested and the numbers, composition, and infection of viable cells were analyzed. The results indicated that mice injected with either saline or MYXV displayed identical numbers of viable cells in their BM (1.08 × 108 versus 7.58 × 107, P = 0.96) (Figure 4a). Additionally, BM from both groups of mice displayed similar FFS/SSC profiles and no GFP+ cells could be identified in virally inoculated mice (Figure 4b) suggesting that viral injection did not grossly alter the make-up of the murine BM. To determine whether injection of MYXV might compromise the functionality of the BM niche, we next asked whether i.v. injection of MYXV would impair engraftment of normal hematopoietic stem cells. Balb/C mice were lethally irradiated (700 cGy) and then rescued using i.v. injection of 5 × 106 total BM cells from non-irradiated syngeneic animals. A limited number of lethally irradiated animals which were not given BM transplant (n = 2) were included as controls. The following day, rescued animals were randomly separated into two cohorts and given three sequential i.v. injections of either saline (n = 10) or 1 × 108 FFU of MYXV (n = 12) on days 1, 3, and 5 post-transplant. Functional engraftment of hematopoietic stem cells to the BM niche was then determined by analyzing recovery of body weight after lethal irradiation with animals being killed if they fell below 75% of their pretreatment weight. The results indicated that BM transplanted mice displayed initial weight loss followed by rapid weight recovery with animals in both cohorts reaching their full starting weight by day 20 (Figure 4c). No statistical differences in either initial weight loss or subsequent recovery between the saline and MYXV treated cohorts were observed at any time and all transplanted animals in both groups survived >40 days (Figure 4d). In contrast, lethally irradiated animals which were not given BM transplant displayed rapid, secondary weight loss beginning around day 17 and requiring euthanasia by day 21.
Figure 4.
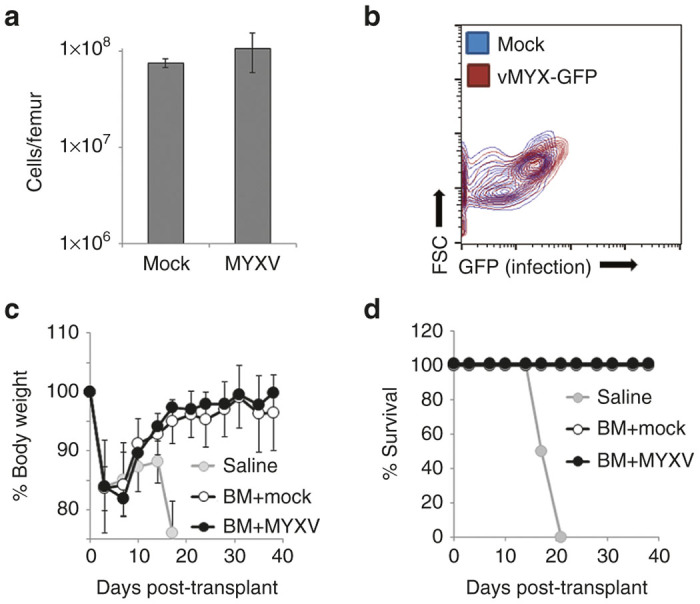
MYXV injection does not inhibit the functionality of the BM niche. Balb/C mice were given three injections of either saline (n = 10) or 1 × 108 FFU of vMYX-GFP (n = 10) over 5 days. Twenty-four hours after the final infection, animals were killed and the (a) numbers of cells in the BM of each mouse was determined using a hemocytometer while the (b) infection of these cells was determined using flowcytometry. Significance was determined using student’s t-test. Balb/C mice were lethally irradiated (700 cGy) and then injected with either saline (n = 2) or 5 × 106 syngeneic BM cells form a nonirradiated Balb/C donor. Transplanted animals were then separated into two groups and given three injections of either saline (n = 10) or 1 × 108 FFU of MYXV (n = 12) over 5 days. (c) Body weight was monitored twice weekly and (d) animals were killed when they dropped below 75% of their starting weight. Significance was determined using student’s t-test at each time point. BM, bone marrow; GFP, green fluorescent protein; FFU, foci forming units; MYXV, myxoma virus.
MYXV treatment requires direct contact of viral particles with residual MM cells
To further determine whether systemic MYXV injection debulked established MRD through a direct interaction with malignant cells or by altering the functionality of the tumor microenvironment, next we sought to identify a murine MM cell line which was immune to MYXV treatment due to a defect in viral absorption. Permissive MOPC-315 cells or nonpermissive MOPC-31C or MOPC-315.BM cells were incubated with MYXV virions chemically labeled with Cy5. Binding of MYXV virions to the cell surface was then analyzed immediately after viral adsorption. The results indicated that high levels of Cy5 fluorescence could be seen on virtually all (96%) control MOPC-315 cells, indicating that Cy5 labeling did not compromise binding of MYXV to the cell surface. In contrast, virtually no Cy5+ cells (3.6%) could be observed following incubation of labeled virus with MOPC-315.BM cells (Figure 5a). MOPC-31C cells displayed an intermediate phenotype in which viral binding was possible but inefficient (our unpublished observations). To further confirm that MOPC-315.BM cells inability to bind virus was maintained in vivo, we next asked whether virus could bind to these cells following their establishment into the BM. Balb/C mice were sublethally irradiated (450 cGy) and then injected i.v. with either 5 × 106 MOPC-315 cells or 1 × 106 MOPC-315.BM cells. Twenty days after tumor implantation, mice were killed and BM extracted. The ability of Cy5 labeled MYXV to bind to the surface of CD138hi engrafted MM cells was then analyzed. The results indicated that, while CD138hi cells in MOPC-315 engrafted mice displayed high levels of Cy5 fluorescence after ex vivo viral adsorption, this fluorescent shift was not seen on CD138hi cells from MOPC-315.BM engrafted mice (Figure 5b) indicating that the inability of these cells to support MYXV binding was maintained in vivo. Consistent with previous reports,20,21 a significant fluorescent shift was also observed on CD138lo normal plasma cells indicating that the virus is also capable of binding certain nonmalignant B cell populations. To determine whether MYXV treatment might impact MM engraftment indirectly, we next asked whether viral injection would have any impact on MRD established with binding deficient MOPC-315.BM cells. Balb/C mice were sublethally irradiated (450 cGy) and then injected i.v. with 1 × 106 MOPC-315.BM cells. Disease was allowed to establish until the mice began to show symptoms of hind limb paralysis (typically around 18–20 days postinjection) and then treated with three sequential i.v. injections of either saline or 1 × 108 FFU of MYXV over a 5-day period. Separate experiments were conducted to analyze both the acute effects of viral treatment on residual disease burden as well as whether viral treatment provided any significant survival benefit. In contrast to our previous observations (Figure 3), the results indicated that both saline (n = 7) and MYXV (n = 8) treated mice bearing MOPC-315.BM MRD displayed identical levels of tumor burden (0.49% versus 0.38%, P = 0.71) 24 hours after the final treatment (Figure 5c). Additionally, viral treatment failed to provide any significant survival benefit and all mice in both saline and MYXV-treated cohorts succumb to hind-limb paralysis by day 50 post-transplant (Figure 5d). Taken together, these data suggest that MYXV requires direct contact with MRD to impact the course of MM disease in vivo.
Figure 5.

MYXV treatment is dependent on direct contact with MRD. (a) MOPC-315 or MOPC-315.BM cells were incubated with Cy5 labeled MYXV virions. Binding of virus to the cell surface was then determined using flowcytometry. (b) Balb/C mice were sublethally irradiated (450 cGy) and then injected i.v. with 5 × 106 MOPC-315 or 1 × 106 MOPC-315.BM cells 20 days after tumor injection, BM was harvested and binding of Cy5 labeled MYXV virions to the surface of CD138hi MM cells was analyzed using flowcytometry. Balb/C mice were sublethally irradiated (450 cGy) and then injected i.v. with 1 × 106 MOPC-315.BM cells. Disease was allowed to establish and then animals were given three i.v. injections of either saline or 1 × 108 FFU of MYXV over 5 days. (c) Twenty-four hours after the last treatment tumor burden in the BM of either saline (n = 7) or MYXV treated (n = 8) animals was determined using flowcytometry. Significance was determined using student’s t-test (***P < 0.0001). (d) Saline (n = 11) or MYXV treated (n = 14) animals were monitored for disease progression and killed when they displayed complete paralysis of both hind-limbs. Significance was determined using log-rank test. BM, bone marrow; FFU, foci forming units; MYXV, myxoma virus; MM, multiple myeloma; MRD, minimal residual disease.
MYXV eliminates residual MM by inducing antitumor CD8+ T cell responses
Our previous findings indicated that MYXV eliminated MRD through direct binding of virions to malignant cells (Figure 5). MRD, however, was still readily observable in all animals following the completion of viral treatment (Figure 3a). Since MYXV replication is highly inefficient in MM cells (Figure 2 and refs. 8,9) it seemed unlikely that the complete elimination of MRD observed in 25% of our mice could be caused by newly generated progeny virus. Therefore, we hypothesized that eradication of MRD in mice displaying a complete response might be mediated through virally induced antitumor immunity. To test this, we asked whether MYXV treatment increased the abundance or functionality of anti-MM T cells. Balb/C mice were sublethally irradiated and then injected i.v. with either 5 × 106 MOPC-315 or 1 × 106 MOPC-315.BM cells. Three weeks after injection of tumor cells, mice were treated with three injections of either saline or 1 × 108 FFU of MYXV over a 5-day period. Animals were killed 24 hours after the final viral treatment and the numbers of both CD4+ and CD8+ T cells in the BM were analyzed using flowcytometry. The results indicated that MYXV treated mice bearing MOPC-315 tumors had significantly (P < 0.0001) increased numbers of CD8+ T cells within the BM compared with untreated saline treated animals (Figure 6a). This increase was not observed in MYXV-treated mice bearing MOPC-315.BM tumors suggesting it was dependent on a viral interaction with MRD. To confirm that the CD8+ T cells induced by MYXV treatment were tumor reactive, we further analyzed their ability to secrete IFNγ following ex vivo stimulation. Spleens from the previously treated mice, as well as tuor-naive controls were stimulated overnight with either saline, uninfected MOPC-315 cells, or free MYXV virions. The following day, secretion of IFNγ was measured using enzyme-linked immunosorbent assay (ELISA). The results indicated that splenocytes from either tumor-naive or saline-treated MOPC-315 bearing mice secreted virtually no IFNγ without stimulation (Figure 6b). Low levels of IFNγ was secreted from these cells following stimulation with MOPC-315 cells suggesting a small antitumor immune response might be present in these animals even in the absence of viral treatment. In contrast, splenocytes from MOPC-315 bearing animals treated with MYXV secreted significantly increased amounts of IFNγ following simulation with either MOPC-315 cells or free MYXV virions suggesting that viral treatment induced both antiviral and antitumor immune responses. To confirm that the eradication of MRD observed in our previous experiments was caused by the antitumor T cell responses, next we asked whether MYXV-treatment was effective against residual MM in the absence of an adaptive immune system. About 5 × 106 MOPC-315 cells were injected i.v. into highly immune deficient NOD/Scid/IL2Rγ−/− (NSG) mice. Ten days after injection animals were given three i.v. injections of either saline (n = 10) or 1 × 108 FFU of MYXV (n = 10) over a 5-day period. Separate experiments were conducted to analyze both the acute effects of viral treatment on residual disease burden as well as whether viral treatment provided any significant survival benefit. Consistent with our results in immune replete animals, tumor bearing mice injected with MYXV displayed significantly reduced tumor burden (6.4% versus 0.2%, P = 0.0003) 24 hours after the final injection compared with saline treated mice (Figure 6c). Also consistent with our results in immune replete animals, a high percentage (90%, 9/10) of MYXV-treated animals (n = 14) displayed a statistically significant (P = 0.008) but relatively minor (~3 days) increase in time to euthanasia compared with saline treated animals (n = 10) (Figure 6d). In striking contrast to our previous findings, however, MYXV-treatment in NSG mice failed to eradicate MRD in any mice and all virally treated animals rapidly developed hind-limb paralysis requiring euthanasia by day 18.
Figure 6.
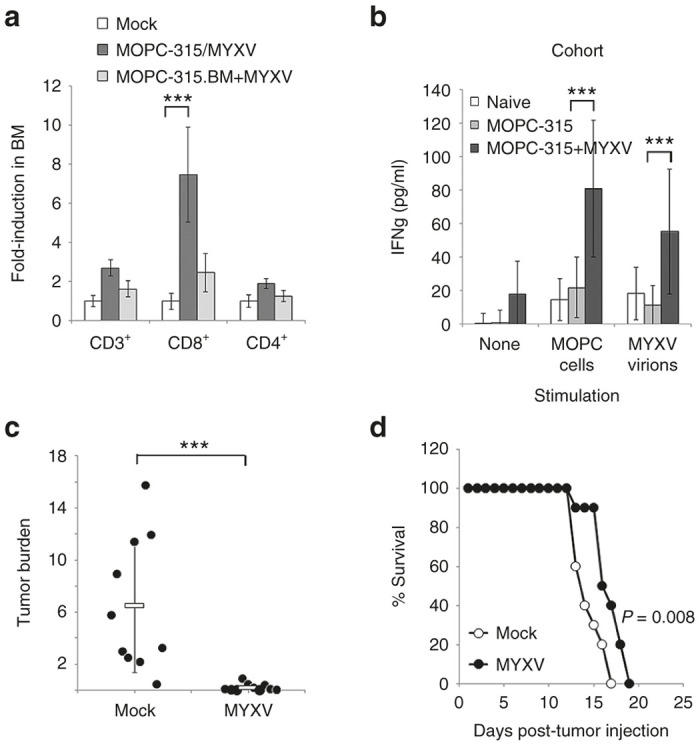
MYXV treatment induces anti-MM T cell responses. Balb/C mice were sublethally irradiated (450 cGy) and then injected i.v. with saline (naive), 5 × 106 MOPC-315 or 1 × 106 MOPC-315.BM cells. Disease was allowed to establish and then animals were given three i.v. injections of either saline or 1 × 108 FFU of MYXV over 5 days. (a) Twenty-four hours after the last injection, the numbers of various T cell subsets in the BM was determined using flowcytometry. Significance was determined using student’s t-test (***P < 0.0001). (b) Splenocytes from naive or MOPC-315 bearing mice above were stimulated as indicated for 24 hours and secretion of IFNγ analyzed using ELISA. Significance was determined using student’s t-test (*** P < 0.0001). Nonirradiated NSG mice were injected i.v. with 5 × 106 MOPC-315 cells and disease allowed to establish. Animals were then given three i.v. injections of either saline (n = 10) or 1 × 108 FFU of MYXV (n = 10) over 5 days. (c) Animals were killed and tumor burden in the BM was determined using flowcytometry. Significance was determined using student’s t-test (***P < 0.0001). (d) Animals were monitored for disease progression and killed when they displayed complete paralysis of both hind-limbs. Significance was determined using log-rank test. BM, bone marrow; ELISA, enzyme-linked immunosorbent assay; FFU, foci forming units; IFNγ, interferon γ; MYXV, myxoma virus; MM, multiple myeloma; MRD, minimal residual disease.
Discussion
Our data demonstrates that systemic treatment with oncolytic MYXV can effectively treat established residual MM in vivo. Interestingly, our results appear to uncover two distinct benefits associated with MYXV treatment. The first benefit is the acute elimination of the majority of residual MM cells from the BM. This benefit was seen rapidly after treatment (within 72 hours after initiation of treatment) was not dependent on the presence of an adaptive immune system (Figure 6) and was observed in virtually all treated animals (Figures 3 and 6) suggesting it likely represents direct oncolytic killing of residual MM cells. Unfortunately, while this effect was readily observable and extremely rapid, it resulted in only minimal clinical benefit (3–8 days increased survival in our models). This unimpressive survival benefit is most likely due to the poorly-replicative nature of MYXV in MM cells (Figure 2 and refs. 8,9) which severely restricts the potential expansion of the therapeutic agent in vivo. Never-the-less, the ability of viral treatment to rapidly eliminate a high-percentage of residual MM could have significant therapeutic benefits when combined with other treatment regimes.
In contrast to the benefits associated with direct viral killing of MRD cells, the second benefit associated with MYXV treatment occurred in a much smaller percentage of animals (10/39); however, it resulted in apparent eradication of clinical disease suggesting it might have more significant clinical impact. This result was observed only during treatment of immune competent animals and likely represents the viral induction of antitumor CD8+ T cell responses. Interestingly, the low response rate for this immune therapy suggests a rate limiting step exists between viral treatment and effective viro-immunotherapy. This rate limiting step did not appear to be killing of residual MM as debulking of acute disease was observed in 100% of treated animals. Similarly, increased numbers of CD8+ T cells in the BM was also observed in all treated animals suggesting that initiation of a T cell response was not limiting. In contrast, while stimulation with MM cells induced IFNγ secretion from splenic T cells of all MYXV treated animals, the levels of this secretion were significantly higher in ~25% (3/12) of animals suggesting a possible inhibition of anti-MM T cell function. This inhibition did not appear to be mediated by the PD1/PDL1 checkpoint axis since cotreatment of MOPC-bearing mice with both MYXV and anti-PD1 blocking antibody failed to significantly increase the response rate of viral treatment (our unpublished observations). Investigation of other potential immune-regulatory pathways, such as Tregs or other T cell checkpoints is therefore required.
Importantly, both the direct and immune-based benefits of viral treatment appeared to require direct binding of MYXV to residual MM in vivo (Figure 5). Similarly, systemic injection of large quantities of MYXV did not have any apparent deleterious effects on the make-up of the BM niche and did not prevent functional engraftment of normal hematopoietic stem cells (Figure 4). This is likely due to the failure of MYXV to bind to nonmalignant hematopoietic cells7,20–22 as well as the inability of the virus to productively infect normal murine tissues.23,24 These results are critical for several reasons. First, MYXV treatment has been proposed as a novel method to eliminate contaminating MM cells from auto-SCT samples prior to transplant.7 Previous work had demonstrated that MYXV did not alter engraftment of human hematopoietic stem cells into NSG mice due to an inability of the virus to adhere to these cells.7,20,25 However, this work had not addressed whether systemically injected virus might have any deleterious effects on the BM niche itself. Our data clearly indicates that systemic injection of MYXV does significantly alter the BM make-up and does not impair reconstitution of a functional hematopoietic system following lethal irradiation. This conclusion is supported by our observation that viral treatment does not significantly impact residual MRD in the absence of a direct interaction with the malignant cells. Our work therefore supports the use of MYXV, either as a systemic injection or as an ex vivo treatment of auto-SCT samples, during SCT rescue therapy. Second, immunotherapy strategies are often associated with high rates of auto-immune like complications. While MYXV is unable to infect the majority of normal human hematopoietic cells, one exception to this rule is CD19+ B cells.21 Thus, one possible explanation for our results was that virally induced anti-MM immune responses were actually generated through killing of normal resident B cells, a mechanism which would likely also produce autoimmune like toxicities. Interestingly, while our data indicates that MYXV is able to bind CD138lo normal plasma cells in vivo (Figure 5b) the induction of anti-MM immune responses requires direct interaction of MYXV with CD138hi malignant MM cells. This suggests that either the responses of normal plasma cells and MM cells to MYXV infection are fundamentally different or that clinically significant antigenic differences exist between the two cell populations which precludes the generation of a cross reactive T cell response. Either way, these data indicate that viral treatment is likely to generate highly tumor specific immune responses thus limiting potential autoimmune toxicities.
While the complete response rate to MYXV therapy observed in our experiments is relatively low, previous studies have suggested that MYXV is an effective oncolytic against the vast majority of primary human myeloma.7,8,26 This is likely due to the unique, virus specific mechanism through which MYXV kills infected MM cells8 and suggests that MYXV-treatment might be effective for patients who are undergoing relapse after failing previous therapies. Interestingly, our results with the MOPC-315.BM cell line indicate that it is possible for MM cells to become resistant to MYXV treatment through a loss of viral binding capacity. This could impact viral therapy in two ways. First, patients could initially present with a form of MM which was incapable of supporting MYXV binding. While this has not been observed in patient samples tested to date, such patients would likely not be amendable to MYXV therapy suggesting that prescreening myeloma samples for viral binding to CD138hi cells prior to the initiation of treatment might be warranted. Importantly, such screening could be readily accomplished using existing tools and methods and therefore should not present a significant barrier to clinical translation. Second, the high heterogeneity of primary MM suggests that small populations of resistant malignant cells might exist in otherwise MYXV-susceptible patients. Such cells would likely play some role in disease recurrence; however, our data indicates that direct killing of only 75% of MRD is capable to causing complete disease regression in some animals (Figure 3a,b). Therefore, the potential existence or development of small numbers of resistant MM clones within a patient would not appear to prevent the use of MYXV as a potential therapeutic.
Other oncolytic viruses have also recently been shown to be effective against MM. Compared with these viruses MYXV provides a variety of translational advantages. In particular, compared with other oncolytic viruses, MYXV processes an excellent safety profile, particularly for use in patients with potential immune dysfunction. The virus is not genetically modified, replicates exclusively in the cytoplasm, and fails to cause even mild disease in any known species except rabbits. This suggests that viral treatment could be conducted as a simple outpatient procedure with limited monitoring of viral shedding or transfers to other patients. Additionally, the virus is easy to engineer suggesting that future modifications to improve efficacy are feasible. Additional research to identify potential modifications, however, is obviously required. Unfortunately, in addition to its advantages, the translation of MYXV also presents several novel challenges, in particular the production of clinical grade virus. Unlike many other oncolytic agents, MYXV has never been used in humans as either a therapeutic or a vaccine; therefore no protocols exist for the production of GMP grade viral stocks. Similarly, while MYXV replicates to high titers in vitro in a variety of cells, only small amounts of virus are actually secreted requiring that virus be purified away from contaminating cell debris. Future translation of our findings therefore hinges of the development of better methods to generate high titer, high purify stocks of MYXV for use in patients.
Given that our therapy generates significant clinical benefits in a small percentage of treated animals, our results are somewhat similar to systemic oncolytic therapy with recombinant measles virus, which has recently been shown to be capable to inducing complete remissions in human patients who have relapsed from several previous therapies.6 However, while measles therapy has been highly effective for a limited number of late-stage patients, treatment appears to be mediated primarily by the direct oncolytic capacity of the measles virus which requires robust viral replication and spread. This limits effective treatment with this agent to highly immune suppressed individuals, those who are seronegative for antimeasles reactive immunoglobulin at the time of treatment. In contrast, therapy with MYXV actively relies on a functional, adaptive immune response and all patients should present as seronegative for antiviral immunoglobulin. Therefore, MYXV might represent an attractive treatment option for late-stage, or multiply relapsed patients who are not eligible for other oncolytic platforms.
Materials and Methods
Cell lines and reagents.
The MOPC-315 (TIB-23), MOPC-31C (CCL-130), P3.6.2.8.1 (TIB-8), U266 (TIB-196), and BSC40 (CRL-2761) cells were obtained from the American Type Culture Collection (Manassas, VA). MOPC-315.BM27 cells were a kind gift from Bjarne Bogen at the University of Oslo. MM cells were cultured between 0.2–0.8 × 106 cells/ml in Roswell Park Memorial Institute (RPMI)-1640 media supplemented with 20% fetal bovine serum (FBS) and 1× Penicillin/Streptomycin/Glutamine (Mediatech, Manassas, VA). IFNγ secretion was measured using the murine IFNγ ELISA kit (Cat# 551866, BD Biosciences, Franklin Lakes, NJ) per manufacturer’s recommendations. Cell viability was performed by CellTiter 96 Non-Radioactive Cell Proliferation Assay (MTT) assay (Promega, Madison, WI) per manufacturer’s recommendations. The following antibodies were used: αCD3 (clone 145-2c11), αCD4 (clone RM4-5), αCD8 (clone 53–6.7), αCD138 (clone 281–2), and Annexin-V (Cat# 550474) (BD Biosciences); αPARP (9542), αCasp3 (9662) (Cell Signaling Technology, Beverly, MA); αActin (sc1615) (Santa Cruz Biotechnology, Dallas, TX).
Virus purification and infection.
Parental MYXV (strain Lausanne) as well as vMYX-GFP28 were kind gifts from Grant McFadden at the University of Florida. Virus was amplified in BSC40 cells as previously described.29 Briefly, BSC40 cells were infected with virus and harvested after 72 hours. Cells were then mechanically lysed, and the resulting supernatant clarified through a 36% sucrose pad. Pellets were then further purified through discontinuous (40/36/32/28/24) percent sucrose gradient and viral virions extracted from the 40%/36% interface. Unless otherwise noted, experiments were carried out by infecting cells for 60 minutes at a multiplicity of infection (MOI) = 10. Fluorescently tagged MYXV virions (MYXV-Cy5) were created by incubating 1 × 109 purified MYXV particles with 50 μg NHS-Cy5 (GE Healthcare, Pittsburgh, PA) for 30 minutes at room temperature and then removing unbound NHS-Cy5 by purifying labelled virions through two 36% sucrose cushions.
In vivo models of MM.
A 6–8 week old, female Balb/C mice (Charles River Laboratories, Raleigh, NC) were irradiated using 450 cGy in an Xray-source irradiator. Twenty-four hours after irradiation, animals were injected i.v. with either 5 × 106 MOPC-315 cells or 1 × 106 MOPC-315.BM cells through the lateral tail vein. Viral treatments were initiated when the first animal displayed evidence of disease and consisted of three i.v. injections (every other day for 5 days) of 1 × 108 FFU of MYXV. MRD (tumor burden) was measured postmortem by harvesting cells from both the left and right hind femurs, and quantitating the percentage of single, viable cells which stained both CD138hi and CD4+ using flow cytometry.7,27 Disease progression was monitored by visually assessing hind-limb function and euthanizing animals when they displayed complete paralysis in both hind-limbs. All animal experiments were reviewed and approved by the Medical University of South Carolina IACUC.
Author Contributions
E.B. designed research, performed research, collected data, analyzed and interpreted data, performed statistical analysis, wrote the manuscript. M.B. performed research, collected data. B.B. contributed vital new reagents. X-Z.Y. contributed analytical tools.
Acknowledgments
This work was supported by grants to Eric Bartee from NIH-NIAID (1K22AI095372-01A1), NIH-NCI (1R01CA194090-01A1), the American Cancer Society (#IRG-97-219-14), and startup funding from the Medical University of South Carolina. This work was also supported in part by the Hollings Cancer Center’s Cancer Center Support Grant P30 CA138313, the South Carolina Clinical and Translational Research (SCTR) Institute NIH grant UL1TR000062.
The authors declared no conflict of interest.
References
- Palumbo, A and Anderson, K (2011). Multiple myeloma. N Engl J Med 364: 1046–1060. [DOI] [PubMed] [Google Scholar]
- Kumar, SK, Rajkumar, SV, Dispenzieri, A, Lacy, MQ, Hayman, SR, Buadi, FK et al. (2008). Improved survival in multiple myeloma and the impact of novel therapies. Blood 111: 2516–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexanian, R, Haut, A, Khan, AU, Lane, M, McKelvey, EM, Migliore, PJ et al. (1969). Treatment for multiple myeloma. Combination chemotherapy with different melphalan dose regimens. JAMA 208: 1680–1685. [DOI] [PubMed] [Google Scholar]
- Brenner, H, Gondos, A and Pulte, D (2008). Recent major improvement in long-term survival of younger patients with multiple myeloma. Blood 111: 2521–2526. [DOI] [PubMed] [Google Scholar]
- Bais, S, Bartee, E, Rahman, MM, McFadden, G and Cogle, CR (2012). Oncolytic virotherapy for hematological malignancies. Adv Virol 2012: 186512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell, SJ, Federspiel, MJ, Peng, KW, Tong, C, Dingli, D, Morice, WG, et al. (2014). Remission of disseminated cancer after systemic oncolytic virotherapy. Mayo Clinic Proc 89: 926–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartee, E, Chan, WM, Moreb, JS, Cogle, CR and McFadden, G (2012). Selective purging of human multiple myeloma cells from autologous stem cell transplantation grafts using oncolytic myxoma virus. Biol Blood Marrow Transplant 18: 1540–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartee, MY, Dunlap, KM and Bartee, E (2016). Myxoma virus induces ligand independent extrinsic apoptosis in human myeloma cells. Clin Lymphoma Myeloma Leuk 16: 203–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenner, F and Ratcliffe, FN (1965). Myxomatosis, Cambridge University Press: Cambridge, UK 1–394. [Google Scholar]
- Kerr, PJ and Best, SM (1998). Myxoma virus in rabbits. Rev Sci Tech 17: 256–268. [DOI] [PubMed] [Google Scholar]
- Gulati, SC and Acaba, L (1993). Rationale for purging in autologous stem cell transplantation. J Hematother 2: 467–471. [DOI] [PubMed] [Google Scholar]
- Melillo, L, Cascavilla, N, Lerma, E, Corsetti, MT and Carella, AM (2005). The significance of minimal residual disease in stem cell grafts and the role of purging: is it better to purge in vivo or in vitro? Acta Haematol 114: 206–213. [DOI] [PubMed] [Google Scholar]
- Bourhis, JH, Bouko, Y, Koscielny, S, Bakkus, M, Greinix, H, Derigs, G et al. European Group for Blood and Marrow Transplantation. (2007). Relapse risk after autologous transplantation in patients with newly diagnosed myeloma is not related with infused tumor cell load and the outcome is not improved by CD34+ cell selection: long term follow-up of an EBMT phase III randomized study. Haematologica 92: 1083–1090. [DOI] [PubMed] [Google Scholar]
- Hamadani, M (2014). Autologous hematopoietic cell transplantation: an update for clinicians. Ann Med 46: 619–632. [DOI] [PubMed] [Google Scholar]
- Melcher, A, Parato, K, Rooney, CM and Bell, JC (2011). Thunder and lightning: immunotherapy and oncolytic viruses collide. Mol Ther 19: 1008–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedel, SS, Mottok, A, Brede, C, Bäuerlein, CA, Jordán Garrote, AL, Ritz, M et al. (2012). Non-invasive imaging provides spatiotemporal information on disease progression and response to therapy in a murine model of multiple myeloma. PLoS One 7: e52398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirandola, L, Apicella, L, Colombo, M, Yu, Y, Berta, DG, Platonova, N et al. (2013). Anti-Notch treatment prevents multiple myeloma cells localization to the bone marrow via the chemokine system CXCR4/SDF-1. Leukemia 27: 1558–1566. [DOI] [PubMed] [Google Scholar]
- Iyer, SP, Beck, JT, Stewart, AK, Shah, J, Kelly, KR, Isaacs, R et al. (2014). A Phase IB multicentre dose-determination study of BHQ880 in combination with antimyeloma therapy and zoledronic acid in patients with relapsed or refractory multiple myeloma and prior skeletal-related events. Br J Haematol 167: 366–375. [DOI] [PubMed] [Google Scholar]
- Gooding, S and Edwards, CM (2016). New approaches to targeting the bone marrow microenvironment in multiple myeloma. Curr Opin Pharmacol 28: 43–49. [DOI] [PubMed] [Google Scholar]
- Kim, M, Madlambayan, GJ, Rahman, MM, Smallwood, SE, Meacham, AM, Hosaka, K et al. (2009). Myxoma virus targets primary human leukemic stem and progenitor cells while sparing normal hematopoietic stem and progenitor cells. Leukemia 23: 2313–2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villa, NY, Bais, S, Chan, WM, Meacham, AM, Wise, E, Rahman, MM et al. (2016). Ex vivo virotherapy with myxoma virus does not impair hematopoietic stem and progenitor cells. Cytotherapy 18: 465–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madlambayan, GJ, Bartee, E, Kim, M, Rahman, MM, Meacham, A, Scott, EW et al. (2012). Acute myeloid leukemia targeting by myxoma virus in vivo depends on cell binding but not permissiveness to infection in vitro. Leuk Res 36: 619–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, F, Ma, Y, Barrett, JW, Gao, X, Loh, J, Barton, E et al. (2004). Disruption of Erk-dependent type I interferon induction breaks the myxoma virus species barrier. Nat Immunol 5: 1266–1274. [DOI] [PubMed] [Google Scholar]
- Bartee, E, Mohamed, MR, Lopez, MC, Baker, HV and McFadden, G (2009). The addition of tumor necrosis factor plus beta interferon induces a novel synergistic antiviral state against poxviruses in primary human fibroblasts. J Virol 83: 498–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman, MM, Madlambayan, GJ, Cogle, CR and McFadden, G (2010). Oncolytic viral purging of leukemic hematopoietic stem and progenitor cells with Myxoma virus. Cytokine Growth Factor Rev 21: 169–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan, WM, Bartee, EC, Moreb, JS, Dower, K, Connor, JH and McFadden, G (2013). Myxoma and vaccinia viruses bind differentially to human leukocytes. J Virol 87: 4445–4460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofgaard, PO, Jodal, HC, Bommert, K, Huard, B, Caers, J, Carlsen, H et al. (2012). A novel mouse model for multiple myeloma (MOPC315.BM) that allows noninvasive spatiotemporal detection of osteolytic disease. PLoS One 7: e51892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lun, X, Yang, W, Alain, T, Shi, ZQ, Muzik, H, Barrett, JW et al. (2005). Myxoma virus is a novel oncolytic virus with significant antitumor activity against experimental human gliomas. Cancer Res 65: 9982–9990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smallwood, SE, Rahman, MM, Smith, DW and McFadden, G (2010). Myxoma virus: propagation, purification, quantification, and storage. Curr Protoc Microbiol Chapter 14: Unit 14A.1. [DOI] [PMC free article] [PubMed] [Google Scholar]