

Abstract
Background:
Keloid is a fibroproliferative skin disorder that is characterized by collagen accumulation and blood vessel proliferation in the reticular layer of the dermis. It is caused by prolonged inflammation after cutaneous injury. Several studies suggested recently that epithelial mesenchymal transition (EMT) is involved in the development of fibrosis. This study assessed whether EMT also participates in keloid development and/or aggravation.
Methods:
Resected keloid (n = 19) and normal skin (n = 13) samples were subjected to immunohistochemical, immunofluorescent, and Western blot analyses of their expression of epidermal (E-cadherin) and mesenchymal (vimentin) proteins.
Results:
Immunohistochemical analysis showed that the keloid tissues had more vimentin-positive cells in the epidermis than the normal tissues. When normal primary keratinocytes were cultured with proinflammatory cytokines, the cobblestone-shaped cells changed to a spindle shape and many vimentin-positive cells were detected. When immortalized HaCaT keratinocytes were cocultured in split-well plates with normal or keloid-derived fibroblasts, they also underwent EMT, as indicated by their greater vimentin expression on Western blot analysis compared with HaCaT cells that were cultured alone.
Conclusions:
EMT was observed in keloid specimens. EMT was induced by inflammatory cytokines and fibroblasts. EMT may be involved in keloid generation and/or aggravation and may have potential as a keloid treatment target.
Keloid is a fibroproliferative disorder of the skin1 that is caused by abnormal healing of skin that has been injured or irritated. Common causes of injury and irritation are trauma, burn, surgery, vaccination, skin piercing, acne, and herpes zoster infection. Keloid scars are red and elevated, have an unappealing appearance, and associate with intermittent pain, persistent itching, and a sensation of contraction. The inflammation in the scars is continuous, localized, and particularly prominent in the reticular layer of the dermis of the skin.2 The reticular layer also exhibits enhanced angiogenesis and collagen accumulation. At the histological level, keloids often have thickened hyalinized collagen bundles (keloidal collagen) that are the result of excessive deposition of extracellular matrix components, including collagen, elastin, and glycosaminoglycan. These features suggest that keloids are caused by an aberrant wound healing process in the damaged reticular layer of the dermis.
Several recent studies suggest that the fibrosis in hypertrophic scars involves the epithelial mesenchymal transition (EMT).3–6 The EMT is characterized by the loss by the epidermis of its epithelial characteristics (in particular E-cadherin expression) and its adoption of mesenchymal features (in particular vimentin expression). A number of growth factors, in particular tumor necrosis factor-alpha (TNFα) and transforming growth factor-beta (TGFβ), are responsible for the transition.7,8 Numerous studies suggest that EMT also plays a significant role in the infiltration of tumors, metastasis, and wound healing.4,7,9–11 We speculated that EMT may also be involved in the development and/or aggravation of keloids. This notion is supported by the recent study of Ma et al, who showed that the keratinocytes in keloids exhibited characteristic EMT-related changes and that these changes could be induced in normal keratinocyte cultures by exposing them to hypoxia.3 In addition, Do et al suggested that interleukin 18, its receptor, and its antagonist play an important role in keloid pathogenesis by inducing EMT.12 However, the mechanisms by which EMT is induced in keloid keratinocytes remain unclear. Moreover, whether regulating EMT has therapeutic potential for keloids has also not been explored.
The present study was performed to assess whether EMT participates in the development and/or aggravation of keloids.
MATERIALS AND METHODS
Tissue Specimens
Resected tissues from 19 patients who underwent plastic surgery to remove keloids were obtained. Normal skin samples were also obtained from 13 patients. All samples were selected by experienced plastic surgeons and removed as part of reconstructive procedures. Informed consent was obtained from all patients, and the study was conducted in accordance with the guidelines of the institutional review board of Nippon Medical School. None of the keloid patients had received medication previously.
Antibodies Used for Immunohistochemical and Immunofluorescence Analyses
The following antibodies were purchased: rabbit monoclonal anti–E-cadherin antibody (Epitomics, Burlingame, Calif.), monoclonal mouse antivimentin clone V7 (DAKO Japan, Tokyo, Japan), anti–FSP-1 antibody (MERCK Millipore, Temecula, Calif.), and anticortactin antibody (G-20: sc-6544; Santa Cruz Biotech, Dallas, Tex.). FSP-1 is a mesenchymal marker, whereas cortactin is an epidermal marker.
Keloid and Normal Skin Analyses
All skin specimens were fixed with 4% paraformaldehyde, and paraffin sections were prepared according to the usual method. The sections were stained with hematoxylin and eosin and subjected to histological analysis to confirm that the tissues had keloid or normal skin characteristics. All samples were also subjected to immunofluorescence and immunohistochemical analyses of vimentin and/or E-cadherin expression. For immunofluorescence staining, frozen sections were cut into 5-μm-thick slices and incubated with anti–E-cadherin and antivimentin antibodies. To detect these primary antibodies, the sections were incubated with antirabbit (for E-cadherin) and antimouse (for vimentin) secondary antibodies at a 1:800 dilution for 2 hours at room temperature, then washed in phosphate-buffered saline, and mounted with or without 4′,6′-diamidino-2-phenylindole nuclear staining. For the immunohistochemistry analyses, the numbers of vimentin-positive cells in the epidermis and E-cadherin-positive cells in the dermis were counted in five randomly selected viewing fields (magnification ×40). The data were expressed as means. In the immunofluorescence analyses, the E-cadherin and vimentin images were merged to indicate the cells that expressed both markers.
Effect of Proinflammatory Cytokine Treatment on EMT in Normal Primary Keratinocytes
Full-thickness samples of normal skin were incubated at 37°C for 3 hours in 25 mmol/L ethylene diamine tetraacetic acid (EDTA) in Dulbecco’s modified Eagle’s medium (DMEM) with the epidermis facing down. Thereafter, the skin samples were immersed in 0.25% trypsin at 37°C for 30 minutes. The epithelial sheet was then separated using forceps and further digested in DMEM containing 0.25 mmol/L EDTA. The harvested keratinocytes were cultured in KGM-Gold medium with penicillin (100 IU/ml) for 96 hours with the proinflammatory cytokine TNFα (10 ng/mL) or TGFβ (1 ng/mL; both from R&D Systems, Minneapolis, Minn.). The cells were then subjected to immunofluorescence analysis with the antivimentin, anticortactin, and anti–FSP-1 antibodies.
Effect of Co-culture with Normal and Keloid-derived Fibroblasts on EMT in HaCaT Keratinocytes
In this experiment, primary cultures of normal fibroblasts (n = 3) and keloid fibroblasts (n = 3) obtained from excised skin samples were cultured in split wells with the immortalized keratinocyte cell line HaCaT, a spontaneously transformed aneuploidy immortal keratinocyte cell line from human skin. HaCaT is frequently used as a skin keratinocyte model in vitro because of its highly preserved differentiation.13 To generate the primary fibroblast cultures, full-thickness samples of normal and keloid skin were incubated as described above with 25 mmol/L EDTA and then 0.25% trypsin, after which the epithelial and dermal sheets were separated by using forceps and the dermal sheets were further digested in DMEM containing 0.25 mmol/L EDTA. The harvested fibroblasts were then cultured in DMEM with penicillin (100 IU/mL) and streptomycin (100 μg/mL) for 72 hours. Similarly, HaCaT cells were cultured in DMEM for 3–5 days. Thereafter, the fibroblasts and HaCaT cells were placed in plates with six split wells, namely, wells with two chambers that are separated by a permeable membrane: the upper chamber contained 1 × 104 HaCaT cells/mL whereas the lower chamber contained a monolayer of 1 × 105 fibroblasts per milliliter. The cells were cocultured in 3 mL per well of DMEM containing 10% fetal bovine serum for 72 hours, after which the HaCaT cells were collected and lysed on ice with cell lysis buffer (10 mM Tris–HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 10 mM Na4P2O7·10 H2O, 100 mM NaF, 1% Triton-X, 0.5% NP-40, 10 mM Na3VO4, 100 KIU aprotinin, PI cocktail). After centrifugation at 15,000 rpm for 30 minutes, the filtrates were collected and subjected to Western blot analysis. Thus, equal amounts of protein (30 μg per lane) were size-fractionated on SDS-polyacrylamide gels and blotted onto Immobilon polyvinylidene difluoride membranes (Bio-Rad Laboratories, Hercules, Calif.). These membranes were then incubated with antivimentin and antiactin antibodies overnight at 4°C in transfer buffer containing 192 mM glycine, 25 mM Tris–HCl, pH 8.3, 20% v/v methanol, and 0.02% SDS. The membranes were washed with 0.1% Tween buffer (50 mM Tris–HCl, pH 7.5, 150 mM NaCl, 0.1% Tween-20) and treated with the corresponding alkaline phosphatase-conjugated secondary antibodies. The reactions were developed by using the ProtoBlor NBT and BICP Color Development System (Promega, Madison, Wis.). Western blot bands were quantitated by densitometric measurements using the Image J software.
Statistical Analysis
The vimentin-positive cells in the epidermis of the normal skin and keloid samples were counted. The normal and keloid skin samples were compared in terms of mean numbers of epidermal vimentin-positive cells by using the Kruskal–Wallis H test followed by the Mann-Whitney U test with the Bonferroni correction. HaCaT cells cultured on their own or with normal or keloid-derived fibroblasts were compared in terms of vimentin expression levels by using the Mann-Whitney U test. All statistical analyses were performed by using StatMate IV software version 4.01 (Advanced Technology for Medicine and Science, Tokyo, Japan). P values that were <0.05 were considered to indicate statistical significance.
RESULTS
Keloids, but Not Normal skin, Exhibit EMT
E-cadherin served as an epithelial marker whereas vimentin served as a mesenchymal marker.14 Thirteen normal skin samples from 8 male and 5 female patients with a mean (range) age of 43.6 (23–65) years were obtained from the back (n = 5), chest (n = 5), and abdomen (n = 5). Immunohistochemical and immunofluorescence analyses revealed the presence of E-cadherin and vimentin in the epidermis and the dermis, respectively. There were very few vimentin-positive cells in the epidermis (1.36 cells per view in a high-power field). When 19 keloid specimens from 10 men and 9 women with a mean (range) age of 41.8 (28–66) years (17 and 3 were from the chest and shoulder, respectively) were similarly analyzed, E-cadherin and vimentin were also mainly expressed in the epidermis and dermis, respectively. However, there were also numerous vimentin-positive cells in the epidermis (Figs. 1, 2). This was more pronounced in the strongly inflamed areas (the reddish areas at the edge) of the keloid (12.98 vimentin-positive cells per view) than in the less inflamed areas (the less reddish areas near the center of the keloid; 8.10 cells per view) (Fig. 3). Figure 4 shows clearly that the strongly inflamed area of a keloid expresses vimentin more strongly in the epidermis than in the less inflamed area. This is consistent with the finding by Lu et al, who reported that fibroblasts from the peripheral regions of keloids were largely in the proliferative period of the cell cycle (G2 and S phases) whereas fibroblasts from the centers were mostly in the G0 or G1 phases.15 Moreover, the latter cells were resistant to apoptosis. The differences between the normal skin, less inflamed keloid areas, and strongly inflamed keloid areas in terms of vimentin-positive cells in the epidermis were statistically significant (all P < 0.05; Fig. 3).
Fig. 1.
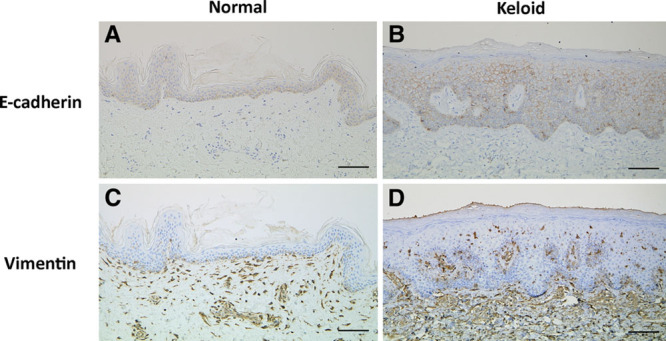
Immunohistochemical localization of E-cadherin and vimentin in representative samples of normal skin (A and C) and keloid tissue (B and D). The keloid keratinocytes exhibited greater vimentin expression (brown stain) than the normal keratinocytes. Bar, 100 μm.
Fig. 2.
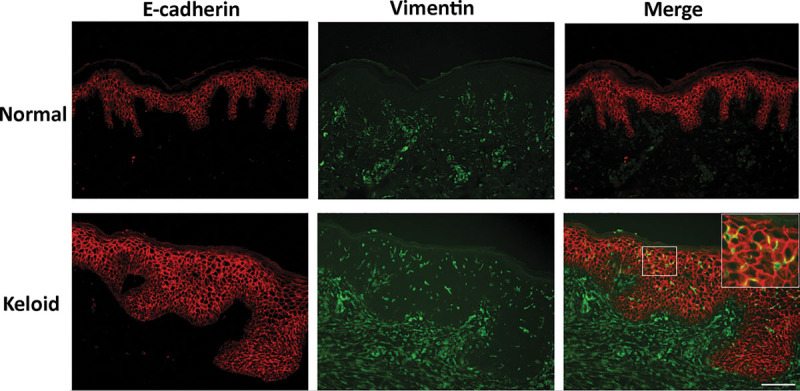
Immunofluorescence staining of representative samples of normal skin and keloid tissue for E-cadherin (red) and vimentin (green). The images were merged in the right-hand panels. Although the keloid epidermal cells did not exhibit any loss of E-cadherin expression, several did coexpress vimentin. Bar, 100 μm.
Fig. 3.
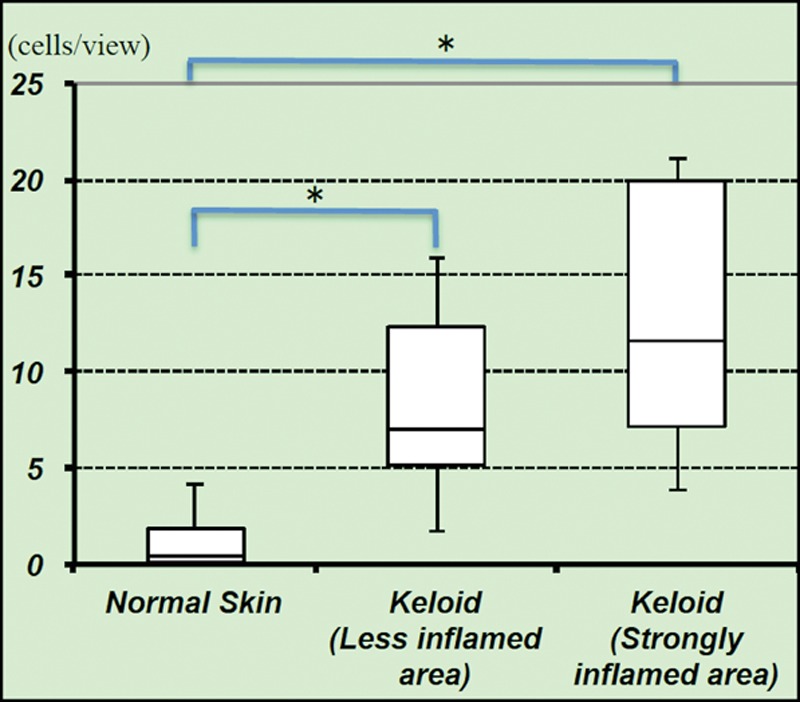
Number of vimentin-positive cells in the epidermis of resected normal skin and the weakly and strongly inflammatory areas of resected keloids. Under a high-power field, the mean numbers of vimentin-positive cells in the epidermis of normal skin, weakly inflammatory keloid areas, and strongly inflammatory keloid areas were 1.36, 8.10, and 12.98 cells per view, respectively. P < 0.01 as determined by using the Kruskal–Wallis H test. χ2 value = 9.1028, degrees of freedom = 2, asymptopic significance = 0.001.
Fig. 4.

Gross (A) and corresponding microscopic (B) images of a keloid. The peripheral (strongly inflamed) area (A) of the keloid expressed more vimentin (brown stain) in the epidermis than the more central (less inflamed) area (B).
EMT Can Be Induced in Normal Primary Keratinocytes by Proinflammatory Cytokines
Keloids express high levels of the proinflammatory cytokines TNFα and TGFβ. To test whether these cytokines contribute to the emergence of EMT in keloids, normal primary keratinocytes were cultured with or without each cytokine and then subjected to double-fluorescence immunohistochemistry for E-cadherin and vimentin. FSP-1 also served as a mesenchymal marker in these analyses. After 72 hours of culture, the inflammatory cytokine treatments induced mesenchymal morphology: there was little adhesion between the cells and the cells changed from a cobblestone shape to a spindle shape (Fig. 5A). When the cells were stained for vimentin and cortactin after 5 days of culture, immunofluorescence staining showed that the spindle-shaped cells were vimentin-positive and cortactin-negative (Fig. 5B). Moreover, staining for vimentin and FSP-1 showed that after 5 days of inflammatory cytokine treatment, most of the keratinocytes had converted into vimentin-positive cells: these cells consisted of 2 populations of mesenchymal-positive cells, namely, vimentin-positive FSP-1-positive cells and vimentin-positive FSP-1-negative cells (Fig. 5C). These phenomena were observed with epidermis cells not becoming fibroblasts themselves but acquiring a fibroblast-like appearance.
Fig. 5.
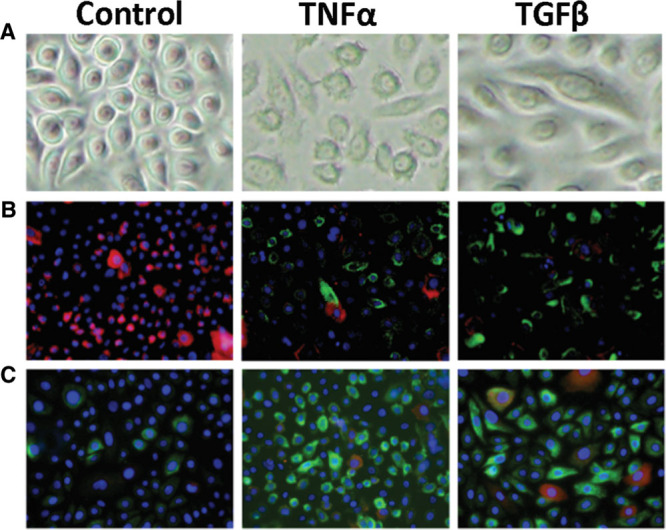
Proinflammatory cytokine treatment induces mesenchymal features in normal primary keratinocytes. Primary keratinocytes were cultured with TNFα (10 ng/mL) or TGFβ (1 ng/mL). A, After 72 hours, the keratinocytes exhibited mesenchymal morphology, as shown by phase contrast microscopy. B, After 5 days, the cells were immunofluorescence-stained for vimentin and cortactin expression. The cells predominantly expressed vimentin (green) and cortactin (red stain). The nuclei were stained by DAPI (blue). C, After 5 days, the cells were immunofluorescence-stained for vimentin and FSP-1 expression. TGFβ treatment converted many of the epithelial cells to vimentin-positive (green) and FSP-1-positive (red) double-positive cells (yellow). Thus, TGF-1 generated 2 populations of mesenchymal-positive cells, namely, the double-positive cells and the vimentin-positive FSP-1-negative cells.
Normal and Keloid-derived Fibroblasts Induce EMT in a Keratinocyte Cell Line
The preceding experiment indicated that the proinflammatory cytokines that characterize keloids can induce EMT in keratinocytes. Thus, EMT in the epidermis may cause the aberrant fibroblast proliferation in keloids. To test whether factors from fibroblasts can induce EMT in keratinocytes, we cocultured normal and keloid-derived fibroblasts with the immortalized keratinocyte cell line HaCaT. We hypothesized that both normal and keloid-derived fibroblasts would induce EMT in the HaCaT cells whereas the HaCaT cells would not exhibit any signs of EMT when cultured on their own. Indeed, Western blot analysis showed that both normal and keloid-derived fibroblasts induced HaCaT cells to express significantly more vimentin than when the HaCaT cells were cultured on their own. Notably, the keloid-derived fibroblasts induced significantly more HaCaT vimentin expression than normal fibroblasts (Fig. 6).
Fig. 6.
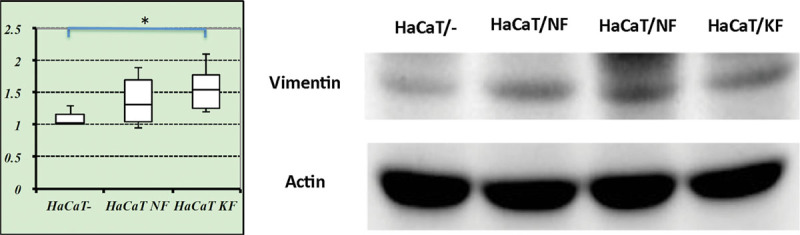
HaCaT keratinocytes cocultured with normal and keloid-derived fibroblasts develop the epidermal mesenchymal transition. Normal and keloid-derived fibroblasts were obtained from 3 normal skin and 3 keloid samples, respectively. HaCaT cells were cultured on their own (HaCaT-) or in split wells with the fibroblasts. Vimentin expression of the HaCaT cells was determined by Western blot analysis. *P < 0.05, as determined by using the Mann–Whitney U test. NF, normal fibroblasts; KF, keloid-derived fibroblasts.
DISCUSSION
EMT and Inflammatory Cytokines
EMT is characterized by the loss in epithelia of cell adhesion and polarity and changes in cell plasticity that cause them to acquire mesenchymal features. Kalluri and Weinberg suggest that EMT is encountered in 3 distinct biological settings. The type 1 settings are implantation and embryonic gastrulation, where EMT gives rise to the mesoderm, endoderm, and mobile neural crest cells. The type 2 settings are inflammation and fibrosis. The type 3 settings arise when the secondary epithelia associated with many organs transform into cancer cells; these cells later undergo EMT, which facilitates their invasion and metastasis.7 Because the acquisition of EMT associates with increased cellular movement and the accumulation of extracellular matrix, the process has gained attention in recent years: it has now been shown that it is involved in cancer cell infiltration and metastasis,8,9 fibrosis of organs,16–20 wound healing,4,5 scarring,21 and the development of keloids.6
Funayama et al22 reported that the overlying keratinocytes of keloid lesions play an important role in keloidogenesis because they promote the proliferation and reduce the apoptosis of the underlying fibroblasts through paracrine and double paracrine effects. Cytokines are also involved in the EMT in wound repair and tissue fibrosis.8,9 For example, TGFβ associates with fibrosis and the expression by epithelial cells of mesenchymal markers such as fibronectin. We believe that keloid generation and/or aggravation is related to imbalances in the extracellular matrix that are caused by inflammatory cytokine-mediated interactions between fibroblasts and epithelial cells.
EMT and Fibrosis
Fibrosis is characterized by excessive collagen production. Collagen is part of the extracellular matrix, and several lines of evidence suggest that it plays a key role in EMT.23 Patients with skin fibrosis have also been reported to exhibit EMT: when Takahashi et al analyzed the skin lesions of 6 patients with morphea and 11 control skin samples, the dermal eccrine glands in the morphea samples displayed EMT. In particular, the morphea samples had elevated levels of TGFβ1, Snail1, and fibronectin and reduced levels of E-cadherin and α-smooth muscle actin, particularly in the eccrine glands.24 Nakamura and Tokura11 also suggested that EMT may occur in systemic sclerosis: they reported enhanced expression of Snail1 and TWIST1 in the eccrine glands of 3 patients with diffuse cutaneous systemic sclerosis. They also noted that some pericytes in the eccrine glands of patients with systemic sclerosis had differentiated into myofibroblasts.25
Evidence of EMT in Keloid Tissues
The present study showed that the keloid epidermis contains significantly more vimentin-positive cells than the epidermis of normal skin. This phenomenon was especially marked in the marginal regions of the keloids, where inflammation was particularly pronounced. This suggests that EMT may be involved in the development and/or aggravation of keloids. Moreover, when a normal primary keratinocyte cell line was cultured with TNFα or TGFβ, many of the cells started expressing vimentin; in addition, many of the vimentin-positive cells also expressed FSP-1. FSP-1 is a widely used specific marker of fibroblasts.26 This suggests that the proinflammatory cytokines that are expressed at high levels in keloids promote EMT. Furthermore, we found that an immortalized keratinocyte cell line started expressing significantly higher levels of vimentin when it was cocultured with normal and keloid-derived fibroblasts; this effect was particularly pronounced when the fibroblasts were derived from keloids. These observations suggest that at least some of the overabundant fibroblasts seen in keloids are derived from the epidermis.
Potential of EMT Regulation in Keloid Therapy
The mechanisms underlying keloid generation remain to be completely elucidated. The lack of animal models for this pathology has contributed to this lack of understanding. Nevertheless, we speculate that several drugs that are currently being developed or used could be useful therapies for keloids because they have the capacity to inhibit EMT.
Potential anti-EMT drugs may include short cyclic His-Ala-Val peptides. Williams et al27 suggested that these peptides can inhibit the function of the mesenchymal marker N-cadherin. Importantly, the peptides induced apoptosis in N-cadherin-expressing vascular endothelial cells, which suggests that they may suppress angiogenesis.27 Shintani et al also reported that the cyclic peptide ADH-1 (N-Ac-CHAVC-NH2) specifically inhibits N-cadherin and suppresses the cell scattering and migration of pancreatic adenocarcinoma cell lines. Moreover, when mice that were transplanted with pancreatic cancer cells received this peptide by orthotopic injection (50 mg/kg in 100 μL of phosphate-buffered saline once a day 5 times a week for 4 weeks), it suppressed tumor progression and peritoneal N-cadherin-controlled tumor metastasis.28 Another possible anti-EMT drug is sorafenib, which has tyrosine multikinase inhibitory effects and suppresses the angiogenesis and proliferation of tumor cells. It has been approved as an oral therapy for some cancers by the American Food and Drug Administration.29,30 Chen et al suggested that sorafenib may also be useful as a therapy for fibrosis because it not only profoundly inhibits TGFβ-induced EMT in alveolar epithelial cells, it also reduces the proliferation and collagen synthesis of fibroblasts.31 Another anti-EMT drug is erubulin, which inhibits mitosis and thereby exerts anticancer activity. Yoshida et al32 suggested that erubulin significantly inhibits EMT and may decrease the migration and invasiveness of tumors. Several studies have also suggested that a secreted frizzled-related protein may also be useful as an anti-EMT therapy that inhibits keloid development and/or aggravation.33,34 Finally, it has been proposed that 5-fluorouracil, which is a pyrimidine analog that is widely used as a cancer chemotherapy, and interferon may also be useful keloid therapies.35,36 Thus, therapies that regulate EMT may have potential as a therapeutic or prophylactic keloid treatment. For example, sorafenib and His-Ala-Val peptides could be useful because they decrease collagen production and control angiogenesis, respectively. Further research on the usefulness of these drugs for keloid prevention or amelioration is warranted.
Footnotes
Disclosure: The authors have no financial interest to declare in relation to the content of this article. The Article Processing Charge was paid for by the authors.
REFERENCES
- 1.Tredget EE, Nedelec B, Scott PG, et al. Hypertrophic scars, keloids, and contractures. The cellular and molecular basis for therapy. Surg Clin North Am. 1997;77:701–730. doi: 10.1016/s0039-6109(05)70576-4. [DOI] [PubMed] [Google Scholar]
- 2.Huang C, Murphy GF, Akaishi S, et al. Keloids and hypertrophic scars: update and future directions. Plast Reconstr Surg Glob Open. 2013;1:e25. doi: 10.1097/GOX.0b013e31829c4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ma X, Chen J, Xu B, et al. Keloid-derived keratinocytes acquire a fibroblast-like appearance and an enhanced invasive capacity in a hypoxic microenvironment in vitro. Int J Mol Med. 2015;35:1246–1256. doi: 10.3892/ijmm.2015.2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yan C, Grimm WA, Garner WL, et al. Epithelial to mesenchymal transition in human skin wound healing is induced by tumor necrosis factor-alpha through bone morphogenic protein-2. Am J Pathol. 2010;176:2247–2258. doi: 10.2353/ajpath.2010.090048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ong CT, Khoo YT, Tan EK, et al. Epithelial-mesenchymal interactions in keloid pathogenesis modulate vascular endothelial growth factor expression and secretion. J Pathol. 2007;211:95–108. doi: 10.1002/path.2081. [DOI] [PubMed] [Google Scholar]
- 6.Yan L, Cao R, Wang L, et al. Epithelial-mesenchymal transition in keloid tissues and TGF-β1-induced hair follicle outer root sheath keratinocytes. Wound Repair Regen. 2015;23:601–610. doi: 10.1111/wrr.12320. [DOI] [PubMed] [Google Scholar]
- 7.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2:442–454. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- 9.Miyazono K. Transforming growth factor-beta signaling in epithelial-mesenchymal transition and progression of cancer. Proc Jpn Acad Ser B Phys Biol Sci. 2009;85:314–323. doi: 10.2183/pjab.85.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Quaggin SE, Kapus A. Scar wars: mapping the fate of epithelial-mesenchymal-myofibroblast transition. Kidney Int. 2011;80:41–50. doi: 10.1038/ki.2011.77. [DOI] [PubMed] [Google Scholar]
- 11.Nakamura M, Tokura Y. Epithelial-mesenchymal transition in the skin. J Dermatol Sci. 2011;61:7–13. doi: 10.1016/j.jdermsci.2010.11.015. [DOI] [PubMed] [Google Scholar]
- 12.Do DV, Ong CT, Khoo YT, et al. Interleukin-18 system plays an important role in keloid pathogenesis via epithelial-mesenchymal interactions. Br J Dermatol. 2012;166:1275–1288. doi: 10.1111/j.1365-2133.2011.10721.x. [DOI] [PubMed] [Google Scholar]
- 13.Schoop VM, Mirancea N, Fusenig NE. Epidermal organization and differentiation of HaCaT keratinocytes in organotypic coculture with human dermal fibroblasts. J Invest Dermatol. 1999;112:343–353. doi: 10.1046/j.1523-1747.1999.00524.x. [DOI] [PubMed] [Google Scholar]
- 14.Moreno-Bueno G, Peinado H, Molina P, et al. The morphological and molecular features of the epithelial-to-mesenchymal transition. Nat Protoc. 2009;4:1591–1613. doi: 10.1038/nprot.2009.152. [DOI] [PubMed] [Google Scholar]
- 15.Lu F, Gao J, Ogawa R. Biological differences between fibroblasts derived from peripheral and central area of keloid tissues. Plast Reconstr Surg. 2007;120:625–630. doi: 10.1097/01.prs.0000270293.93612.7b. [DOI] [PubMed] [Google Scholar]
- 16.Guarino M, Tosoni A, Nebuloni M. Direct contribution of epithelium to organ fibrosis: epithelial-mesenchymal transition. Hum Pathol. 2009;40:1365–1376. doi: 10.1016/j.humpath.2009.02.020. [DOI] [PubMed] [Google Scholar]
- 17.Kim KK, Kugler MC, Wolters PJ, et al. Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc Natl Acad Sci USA. 2006;103:13180–13185. doi: 10.1073/pnas.0605669103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zeisberg M, Yang C, Martino M, et al. Fibroblasts derive from hepatocytes in liver fibrosis via epithelial to mesenchymal transition. J Biol Chem. 2007;282:23337–23347. doi: 10.1074/jbc.M700194200. [DOI] [PubMed] [Google Scholar]
- 19.Zeisberg M, Kalluri R. Fibroblasts emerge via epithelial-mesenchymal transition in chronic kidney fibrosis. Front Biosci. 2008;13:6991–6998. doi: 10.2741/3204. [DOI] [PubMed] [Google Scholar]
- 20.Willis BC, Borok Z. TGF-beta-induced EMT: mechanisms and implications for fibrotic lung disease. Am J Physiol Lung Cell Mol Physiol. 2007;293:L525–L534. doi: 10.1152/ajplung.00163.2007. [DOI] [PubMed] [Google Scholar]
- 21.Mukhopadhyay A, Tan EK, Khoo YT, et al. Conditioned medium from keloid keratinocyte/keloid fibroblast coculture induces contraction of fibroblast-populated collagen lattices. Br J Dermatol. 2005;152:639–645. doi: 10.1111/j.1365-2133.2005.06545.x. [DOI] [PubMed] [Google Scholar]
- 22.Funayama E, Chodon T, Oyama A, et al. Keratinocytes promote proliferation and inhibit apoptosis of the underlying fibroblasts: an important role in the pathogenesis of keloid. J Invest Dermatol. 2003;121:1326–1331. doi: 10.1111/j.1523-1747.2003.12572.x. [DOI] [PubMed] [Google Scholar]
- 23.Baum B, Settleman J, Quinlan MP. Transitions between epithelial and mesenchymal states in development and disease. Semin Cell Dev Biol. 2008;19:294–308. doi: 10.1016/j.semcdb.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 24.Takahashi M, Akamatsu H, Yagami A, et al. Epithelial-mesenchymal transition of the eccrine glands is involved in skin fibrosis in morphea. J Dermatol. 2013;40:720–725. doi: 10.1111/1346-8138.12235. [DOI] [PubMed] [Google Scholar]
- 25.Nakamura M, Tokura Y. Expression of SNAI1 and TWIST1 in the eccrine glands of patients with systemic sclerosis: possible involvement of epithelial-mesenchymal transition in the pathogenesis. Br J Dermatol. 2011;164:204–205. doi: 10.1111/j.1365-2133.2010.10021.x. [DOI] [PubMed] [Google Scholar]
- 26.Strutz F, Okada H, Lo CW, et al. Identification and characterization of a fibroblast marker: FSP1. J Cell Biol. 1995;130:393–405. doi: 10.1083/jcb.130.2.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Williams E, Williams G, Gour BJ, et al. A novel family of cyclic peptide antagonists suggests that N-cadherin specificity is determined by amino acids that flank the HAV motif. J Biol Chem. 2000;275:4007–4012. doi: 10.1074/jbc.275.6.4007. [DOI] [PubMed] [Google Scholar]
- 28.Shintani Y, Fukumoto Y, Chaika N, et al. ADH-1 suppresses N-cadherin-dependent pancreatic cancer progression. Int J Cancer. 2008;122:71–77. doi: 10.1002/ijc.23027. [DOI] [PubMed] [Google Scholar]
- 29.Escudier B, Eisen T, Stadler WM, et al. TARGET Study Group. Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med. 2007;356:125–134. doi: 10.1056/NEJMoa060655. [DOI] [PubMed] [Google Scholar]
- 30.Kane RC, Farrell AT, Saber H, et al. Sorafenib for the treatment of advanced renal cell carcinoma. Clin Cancer Res. 2006;12:7271–7278. doi: 10.1158/1078-0432.CCR-06-1249. [DOI] [PubMed] [Google Scholar]
- 31.Chen YL, Zhang X, Bai J, et al. Sorafenib ameliorates bleomycin-induced pulmonary fibrosis: potential roles in the inhibition of epithelial-mesenchymal transition and fibroblast activation. Cell Death Dis. 2013;4:e665. doi: 10.1038/cddis.2013.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yoshida T, Ozawa Y, Kimura T, et al. Eribulin mesilate suppresses experimental metastasis of breast cancer cells by reversing phenotype from epithelial-mesenchymal transition (EMT) to mesenchymal-epithelial transition (MET) states. Br J Cancer. 2014;110:1497–1505. doi: 10.1038/bjc.2014.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Igota S, Tosa M, Murakami M, et al. Identification and characterization of Wnt signaling pathway in keloid pathogenesis. Int J Med Sci. 2013;10:344–354. doi: 10.7150/ijms.5349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ren J, Wang R, Song H, et al. Secreted frizzled related protein 1 modulates taxane resistance of human lung adenocarcinoma. Mol Med. 2014;20:164–178. doi: 10.2119/molmed.2013.00149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Naylor MC, Brissett AE. Current concepts in the etiology and treatment of keloids. Facial Plast Surg. 2012;28:504–512. doi: 10.1055/s-0032-1325644. [DOI] [PubMed] [Google Scholar]
- 36.Al-Attar A, Mess S, Thomassen JM, et al. Keloid pathogenesis and treatment. Plast Reconstr Surg. 2006;117:286–300. doi: 10.1097/01.prs.0000195073.73580.46. [DOI] [PubMed] [Google Scholar]