

Abstract
Systemically delivered adeno-associated viral (AAV) vectors are now in early-phase clinical trials for a variety of diseases. While there is a general consensus on inclusion and exclusion criteria for each of these trials, the conditions under which vectors are infused vary significantly. In this study, we evaluated the impact of intravenous infusion rate of AAV8 vector in cynomolgus macaques on transgene expression, vector clearance from the circulation, and potential activation of the innate immune system. The dose of AAV8 vector in terms of genome copies per kilogram body weight and its concentration were fixed, while the rate of infusion varied to deliver the entire dose over different time periods, including 1, 10, or 90 minutes. Analyses during the in-life phase of the experiment included sequential evaluation of whole blood for vector genomes and appearance of proinflammatory cytokines. Liver tissues were analyzed at the time of necropsy for enhanced green fluorescent protein (eGFP) expression and vector genomes. The data were remarkable with a relative absence of any statistically significant effect of infusion time on vector transduction, safety, and clearance. However, some interesting and unexpected trends did emerge.
Introduction
Adeno-associated viral (AAV) vectors are now in early-phase clinical trials for a variety of diseases following systemic vector administration.1–4 While there has been a consensus related to the inclusion and exclusion criteria for each of these trials, such as the impact of pre-existing AAV-neutralizing antibody (NAb) titers and concurrent liver disease, the method by which these vectors are administered by intravenous (IV) infusion varies.
Preclinical studies of liver-directed gene therapy have demonstrated similar biodistribution of AAV vectors injected via peripheral vein versus directly into the hepatic circulation. These data provided justification for most liver-directed clinical protocols to deliver vector through a peripheral vein.5,6 Patients enrolled in the St. Jude’s phase 1 clinical trial for the treatment of hemophilia B received their dose of AAV8 vector expressing human coagulation factor IX in a volume of 200 ml infused IV over 1 hour.2,3 In the phase 1 clinical trial for acute intermittent porphyria (AIP), the AAV5 vector dose was diluted to a final volume of 20 ml and infused IV over 20 minutes.4 In both cases, the volume and time of infusion was fixed with the concentration of vector varying based on dose and mass of the patient. In the open phase 1 clinical trial for the treatment of homozygous familial hypercholesterolemia (HoFH) using AAV8, the University of Pennsylvania team elected to keep the concentration and time of infusion constant (i.e., 20 minutes); the dose is varied by adjusting the total volume of infused vector (ClinicalTrials.gov Identifier: NCT02651675).
Others have recently tried to investigate the effect of IV infusion time on vector delivery to the liver in rodents.7 However due to the small size of a mouse or rat, only a limited range of infusion conditions can be tested and the relevance to infusion kinetics in humans is limited. By using cynomolgus macaques to model vector administration in humans, we evaluated the impact of infusion rate on transgene expression, vector clearance from the circulation, and potential activation of the innate immune system.
Results
Male cynomolgus macaques were infused via the saphenous vein with 7.5 × 1012 genome copies (GC) per kg of AAV8.TBG.eGFP vector at a concentration of 7.5 × 1012 GC per ml over intervals that varied from 1, 10, and 90 minutes (Table 1). Only animals with NAbs to AAV8 < 1:5 were enrolled in this study. The volume of vector was adjusted between 6.00 and 7.70 ml to account for differences in the size of the animals. The rate of infusion differed slightly within a fixed infusion rate group to account for the adjustments in vector doses due to variations in animal size (e.g., 6.00 to 7.15 ml/minute for the 1 minute infusion group). The infusion rates varied more significantly between groups to account for the 90-fold difference in total infusion time ranging between 1 minute and 90 minutes of infusion.
Table 1. Summary of infusion conditions for cynomolgus macaques administered IV with 7.5 × 1012 GC/kg of AAV8.TBG.eGFP vector.
| NHP ID | Weight (kg) | Vector dose (GC/kg)a | Injected vector volume (ml)b | Infusion concentration (GC/ml) | Infusion time (minutes) | Infusion rate (ml/minute) |
|---|---|---|---|---|---|---|
| M11297 | 6.00 | 7.5 × 1012 | 6.00 | 7.5 × 1012 | 1 | 6.000 |
| M11298 | 6.90 | 7.5 × 1012 | 6.90 | 7.5 × 1012 | 1 | 6.900 |
| M11356 | 7.15 | 7.5 × 1012 | 7.15 | 7.5 × 1012 | 1 | 7.150 |
| M11211 | 7.35 | 7.5 × 1012 | 7.35 | 7.5 × 1012 | 10 | 0.735 |
| M11327 | 7.70 | 7.5 × 1012 | 7.70 | 7.5 × 1012 | 10 | 0.770 |
| M11658 | 6.30 | 7.5 × 1012 | 6.30 | 7.5 × 1012 | 10 | 0.630 |
| M11215 | 7.10 | 7.5 × 1012 | 7.10 | 7.5 × 1012 | 90 | 0.079 |
| M11517 | 6.35 | 7.5 × 1012 | 6.35 | 7.5 × 1012 | 90 | 0.071 |
| M11528 | 6.80 | 7.5 × 1012 | 6.80 | 7.5 × 1012 | 90 | 0.076 |
The same dose of vector was administered as a bolus injection over the course of 1 minute, infusion of vector over 10 minutes, or vector infused over 90 minutes.
The injection volume was based on the weight of the animal and the infusion rate calculated to perform the vector infusion within the infusion time.
Prior to and following completion of vector infusion (i.e., 1 minute, 15 minutes, 30 minutes, 1 hour, 2 hours, 3 hours, 4 hours, 6 hours, 8 hours, 24 hours, 48 hours, 72 hours, and 7 days), whole blood was sampled for the detection and quantification of GC to determine the rate of vector clearance from the circulation (Figure 1a). Average peak vector levels in blood measured 1 minute after completion of the infusion were inversely proportional to the time of infusion: peak vector concentrations of 6.3 × 1011 ± 4.9 × 1011 GC/ml, 4.7 × 1011 ± 2.6 × 1011 GC/ml, and 1.1 × 1012 ± 8.9 × 1011 GC/ml were measured for animals infused over 1, 10, and 90 minutes, respectively (mean ± SEM, no statistical difference). Vector GC were essentially cleared from whole blood in most animals by 48–72 hours post-vector administration, becoming undetectable in all animals by day 7 post-vector administration. The half-life for vector clearance was calculated for each animal (Figure 1b) and for each group (Figure 1c). Animals infused with the AAV8 vector over 90 minutes demonstrated a significantly lower clearance of vector from the circulation (P < 0.05), with no significant difference in vector clearance between the 1- and 10-minute infusion times.
Figure 1.
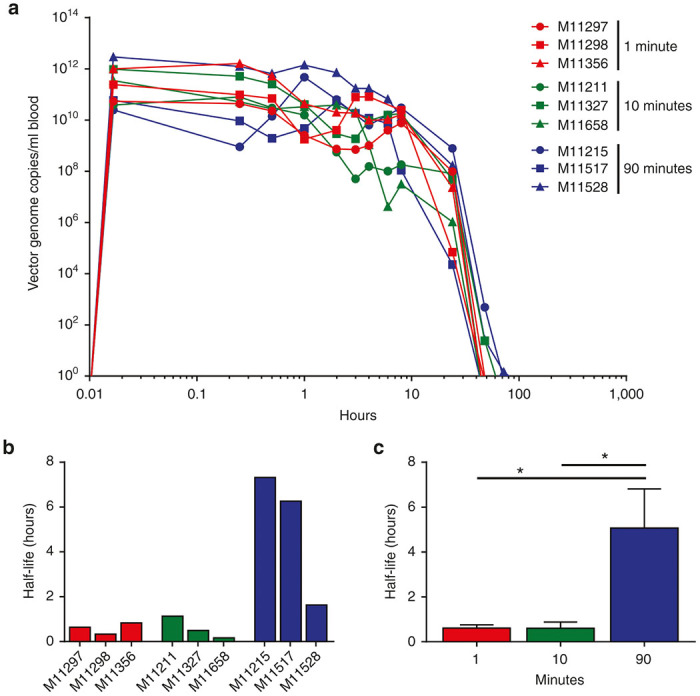
Vector clearance rates do not differ with infusion time. Cynomolgus macaques were administered IV with 7.5x1012 GC/kg of AAV8.TBG.eGFP vector. The same dose of vector was administered as a bolus injection over the course of 1 minute, infusion of vector over 10 minutes, or infusion of vector over 90 minutes. (a) Prior to and following completion of vector administration, whole blood samples were taken to evaluate vector clearance from the circulation over time. DNA was extracted from whole blood and vector GC were quantified against a standard curve produced from DNA extracted from whole blood and spiked with known quantities of vector. The half-life for vector clearance was quantified for each animal individually (b) and compared by infusion group (c). Mean ± SEM values shown (n = 3/group). *P < 0.05.
Prior to and following completion of the vector infusion, serum was sampled for cytokine analysis. A transient, low-level elevation in IL-6 was seen with the highest peaks noted to be 67.3 and 87.5 pg/ml in macaques M11297 and M11356, respectively. These peak IL-6 levels were minor and similar to those reported previously following IV administration of phosphate-buffered saline containing 10% glycerol.8 The area under the curve was calculated for each animal (Figure 2b) and compared with respect to infusion time (Figure 2c). While there was a trend toward higher IL-6 area under the curves with shorter vector infusion times, these differences did not reach statistical significance.
Figure 2.

Increased IL-6 levels are produced with bolus vector injections. Cynomolgus macaques were administered IV with 7.5 × 1012 GC/kg of AAV8.TBG.eGFP vector. The same dose of vector was administered as a bolus injection over the course of 1 minute, infusion of vector over 10 minutes, or infusion of vector over 90 minutes. (a) Prior to and following completion of vector administration serum samples were taken to evaluate cytokine levels over time by multiplexed cytokine gene expression analysis. The area under the curve for IL-6 levels was quantified for each animal individually (b) and compared by infusion group (c). Other cytokines tested included GM-CSF, G-CSF, IFNγ, IL-1β, IL-1ra, IL-2, IL-4, IL-5, IL-8, IL-10, IL-12/23(p40), IL-13, IL-15, IL-17a, IL-18, MCP-1, MIP-1α, MIP-1β, sCD40L, TGFα, TNFα, and VEGF. At baseline and following vector administration GM-CSF, IFNγ, IL-1β, IL-4, IL-8, IL-13, IL-18, MIP-1β, and TNFα were below the limit of detection. IL-2, IL-10, IL-12/23(p40), IL-15, IL-17a, MCP-1, MIP-1α, sCD40L, TGFα, and VEGF were detectable but no elevations were seen. Minor elevations in G-CSF, IL-1ra, and IL-5 were seen but were not consistent within a group. Mean ± SEM values shown (n = 3/group).
On day 7 post-vector administration, the macaques were euthanized and liver was harvested for evaluation of eGFP expression, first by quantification of transduction efficiency by morphometric analysis of histological sections, and second by measurement of total eGFP protein expression by enzyme-linked immunosorbent assay. To assure representative sampling, eGFP expression was measured from 14 samples taken from across the four liver lobes of each animal. The standard reporter gene eGFP was used in this study to facilitate analyses of transduction efficiency. Like most other reporter genes, eGFP is immunogenic and could result in the activation of cytotoxic T lymphocytes, which would target transduced hepatocytes leading to a decline in eGFP expression, confounding the assessment of transduction efficiency.9 Therefore, we terminated the study at the peak of expression for the eGFP transgene and prior to any potential cytotoxic T lymphocyte responses.
There were no regional differences in expression as determined by visual inspection of eGFP histological sections; representative fluorescent images from each animal are shown in Figure 3. The previously described periportal pattern of AAV8 transduction seen in macaques was evident in each group.10 High levels of transduction were observed in all animals in all groups, with some variation observed between animals within each group. The variation in transduction noted within an infusion group appears to be similar to that previously observed in NAb-negative animals.11 However, one macaque (M11517) appeared to have significantly higher eGFP expression than any other animal in the study, regardless of infusion time. Morphometric analysis of representative tissues sections was performed for individual animals (Figure 4a), which were also compared in aggregate between different groups (Figure 4b). There was a trend toward higher transduction efficiency with longer vector infusion time but there were no significant differences (Figure 4b). There were no significant differences in eGFP expression as determined by enzyme-linked immunosorbent assay between infusion groups (Figure 4d); the level of eGFP protein present was similar to that determined previously in a similar study.11
Figure 3.

eGFP transgene expression in liver following different vector infusion times. Cynomolgus macaques were administered IV with 7.5x1012 GC/kg of AAV8.TBG.eGFP vector. The same dose of vector was administered as a bolus injection over the course of 1 minute, infusion of vector over 10 minutes, or infusion of vector over 90 minutes. Animals were necropsied on day 7 post-vector administration and livers harvested for eGFP expression. Representative images of each animal are shown. Scale bar = 500 µm.
Figure 4.

Quantification of eGFP expression and vector GC in liver following different vector infusion times. Cynomolgus macaques were administered IV with 7.5x1012 GC/kg of AAV8.TBG.eGFP vector. The same dose of vector was administered as a bolus injection over the course of 1 minute, infusion of vector over 10 minutes, or infusion of vector over 90 minutes. Animals were necropsied on day 7 post-vector administration and livers harvested for eGFP expression. eGFP expression was quantified from images of eGFP expression taken on 14 samples from throughout the liver (a, b). eGFP expression was also quantified by enzyme-linked immunosorbent assay on samples taken from three sites throughout the liver (c, d). DNA was extracted from three sites throughout the liver and vector GC per diploid genome were quantified (e, f). Data are presented for each animal individually (a, c, and e) and compared by infusion group (b, d, and f). Mean ± SEM values shown (n = 3/group). *P < 0.05, **P < 0.01.
Tissues were also harvested at necropsy for biodistribution analysis. Vector infusion over 10 minutes resulted in 62.8 GC/diploid genome in the liver, which was higher than 18.9 and 32.6 GC/diploid genome present in macaques infused with vector over 1 and 90 minutes, respectively (P < 0.05, 10 versus 90 minutes; P < 0.01, 1 versus 10 minutes). This level of gene transfer following vector administration over 10 minutes is similar to that previously reported for nonhuman primates (NHPs) with undetectable pre-existing NAbs to the vector capsid.11
Deposition of vector in brain, heart, kidney, lung, muscle, and spleen was also evaluated (Figure 5). Consistent with previous studies, the highest levels of extra-hepatic vector genomes was in spleen, with heart and lung showing intermediate levels. There was an interesting trend in which the 10-minute infusion group yielded a different profile than the 1 and 90 minute groups (e.g., higher in muscle and heart but lower in brain and lung) but the differences were not significant.
Figure 5.

Quantification of vector GC in brain, heart, kidney, lung, muscle, and spleen following different vector infusion times. DNA was extracted from brain (a), heart (b), kidney (c), lung (d), muscle (e), and spleen (f) and vector GC per diploid genome were quantified. Data are presented as the average of each infusion group. Mean ± SEM values shown (n = 3/group).
Discussion
In this study, we sought to evaluate the impact of infusion rate on transgene expression, vector clearance from the circulation, and potential activation of the innate immune system. Our original hypothesis was that infusion times would indeed influence key measures of efficacy and safety with the bolus injection leading to greater transduction but more acute toxicity. The clearance of vector resembled standard first order decay with a half-life that varied from approximately 0.6 hours for 1- and 10-minute infusion groups to 5 hours for the 90-minute infusion group. However, we were surprised that infusion times spanning 1 to 90 minutes of the same dose and volume of vector had remarkably very little impact on outcomes of efficacy and safety, although there were some trends. As expected, the shorter infusion times resulted in higher peak levels when measured 1 minute after the infusion was stopped. There was remarkably little evidence of acute activation of innate immunity, with the only trend being higher levels of transient elevations of IL-6 associated with shorter infusion times. These elevations in IL-6 were similar to that observed with infusion of vehicle and are logs lower than what was observed following IV infusion of adenoviral vectors in macaques and humans.8,12
The real surprise, however, was that infusion time was not associated with significant differences in transduction efficiency. There was a trend toward higher transduction with longer infusion times that would suggest the time of exposure to the vector, which was higher in the 90-minute infusion where the half-life was longer, may be more important than the peak vector concentration, which was higher in the shorter infusion times. However, we concede that these are only trends and not sufficiently robust to confirm or refute the original hypotheses.
Several other aspects of in vivo transduction emerged from our studies that may be relevant to human applications. Preliminary results emerging from the AAV hemophilia A and B clinical trials are indeed promising, although they have been notable for significant subject-to-subject variation in transgene expression that is independent of pre-existing immunity to the capsid, as all research subjects were negative for NAbs to the vector capsid. We also observed variation in transgene expression between animals within a group that appears greater than the variation we observe with similar vectors injected IV in mice.10,13–15 While the mechanism that underlies this variability remains unknown, it does suggest that NHPs may be useful in elucidating it. The other interesting trend related to the impact of vector infusion times was on biodistribution. First, there appeared to be no relationship between transduction efficiency/transgene expression and vector genomes. Second, there was an interesting relationship between vector infusion time and vector deposition, with the 10-minute infusion group producing results different than the 1- and 90-minute group. These differences were not substantial for most tissues relevant to in vivo gene therapy, except for heart and skeletal muscle where the 10-minute group yielded higher transduction. Optimization of the conditions for AAV8 infusion may be useful in improving the outcome of gene therapy in conditions such as Pompe disease and Duchenne muscular dystrophy, where heart and skeletal muscle are target tissues.16,17
In conclusion, the emphasis of gene therapy pharmaceutical development has been on the actual composition of the product such as the capsid, promoter, codon usage, and genome structure. This study begins to address one key aspect of the successful development of this pharmaceutical, which is the time interval of its IV administration. While we were unable to demonstrate a robust impact of vector infusion time on AAV8 vector performance, some interesting and unanticipated trends did emerge. The development of gene therapy as a true pharmaceutical will require more attention to other aspects of its route of administration and issues related to formulation. These studies are critical for successful and optimal development of effective therapies.
Materials and Methods
AAV vector production
All AAV vectors were produced by the Penn Vector Core at the University of Pennsylvania as described previously.18 Briefly, plasmids expressing eGFP from the liver-specific thyroxine binding globulin (TBG) promoter were packaged with the AAV8 viral capsid.
NHP
Male cynomolgus macaques were housed at the Nonhuman Primate Research Program facility of the Gene Therapy Program of the University of Pennsylvania (Philadelphia, PA) during the studies. Studies were performed according to a study protocol approved by the IACUC, the Environmental Health and Radiation Safety Office and the Institutional Biosafety Committee of the University of Pennsylvania. All animals were housed in stainless steel caging with perches and maintained on a 12-hour light/dark cycle controlled via an Edstrom Watchdog system. Temperature was maintained within the range of 18–26 °C with 50% (±10%) humidity. Animals were fed Certified Primate Diet 5048 (PMI Feeds, Brentwood, MO) two times per day (morning and evening). Water was available ad libitum from an automatic watering system. Food enrichment such as fruits, vegetables, nuts, and cereals were provided daily. Manipulanda such as kongs, mirrors, puzzle feeder, and raisin balls were provided daily. Animals also received visual enrichment along with human interaction on a daily basis.
All macaques had NAb titers of <1:5 at the start of the studies, determined as described previously.19 A dose of 7.5 × 1012 GC/kg of AAV8.TBG.eGFP.BGH vector was infused IV, via the saphenous vein, over 1, 10, or 90 minutes. Blood samples were taken prior to the initiation of the study and during the study via venipuncture of the cephalic, saphenous, or femoral veins. Whole blood was collected in lavender topped K2 tubes containing potassium ethylenediaminetetraacetic acid (EDTA) prior to vector administration and 1 minute, 15 minutes, 30 minutes, 1 hour, 2 hours, 3 hours, 4 hours, 6 hours, 8 hours, 24 hours, 48 hours, 72 hours, and 7 days following the completion of the vector infusion for vector clearance analysis. Blood was collected in serum separator tubes and serum subsequently isolated prior to vector administration and 1 hour, 2 hours, 6 hours, 8 hours, 24 hours, 72 hours, and 7 days following the completion of the vector infusion for cytokine analysis. All clinical pathology tests on blood samples were conducted by Antech Diagnostics (Irvine, CA), including complete blood counts and differentials, and complete clinical chemistries.
On day 7 post-vector administration, all macaques were euthanized. The animal was first anesthetized with a mixture of ketamine (10–15 mg/kg) and dexmedetomidine (0.05–0.10 mg/kg) injected intramuscularly and euthanized using sodium pentobarbital (80 mg/kg) injected IV. Death was confirmed by absence of heartbeat and respiration.
Vector clearance
Detection and quantification of vector GC in extracted DNA from whole blood was performed to determine the rate of vector clearance from the circulation. DNA was isolated from 200 µl of whole blood collected in lavender topped K2 tubes containing potassium EDTA using the Qiagen QIAamp DNA mini kit and following the manufacturer’s instructions. Vector GCs were quantified against a standard curve generated following extraction of DNA from whole blood from a naive male cynomolgus macaque that was spiked with a known vector concentration and using primers/probe designed against the BGH polyA sequence of the vector as described previously.20
Cytokine analysis
Serum samples taken prior to the initiation of the study and during the study were analyzed for cytokine content by Luminex using a cytokine Non-Human Primate Cytokine Magnetic Bead Panel - Premixed 23 Plex (Thermo Fisher Scientific, Waltham, MA) as performed by the Human Immunology Core (University of Pennsylvania, Philadelphia, PA). Cytokines tested included GM-CSF, G-CSF, IFNγ, IL-1β, IL-1ra, IL-2, IL-4, IL-5, IL-6, IL-8, IL-10, IL-12/23(p40), IL-13, IL-15, IL-17a, IL-18, MCP-1, MIP-1α, MIP-1β, sCD40L, TGFα, TNFα, and VEGF.
eGFP expression and quantification
At necropsy, samples of liver were harvested from all four liver lobes for evaluation of eGFP expression; four samples taken from across the middle lobe (from the hilus to the edge of the liver), four samples from the left lobe, five samples from the middle lobe, and one sample from the caudate lobe. Tissues were processed as described previously to visualize eGFP expression.11,21 Images were taken for all 14 samples of liver from each macaque and eGFP expression in liver was quantified as the percentage of area expressing eGFP as described previously.11,21
eGFP enzyme-linked immunosorbent assay
eGFP enzyme-linked immunosorbent assay was performed as described previously on homogenized liver samples.11
Vector biodistribution
Tissues samples were snap frozen at the time of necropsy and DNA was extracted using the QIAamp DNA Mini Kit (Qiagen, Valencia, CA). Detection and quantification of vector GC in extracted DNA were performed by real-time PCR as described previously.20 Briefly, genomic DNA was isolated and vector GCs were quantified using primers/probe designed against the BGH polyA sequence of the vector.
Data and statistical analysis
All analyses were performed in Prism (GraphPad Software, San Diego, CA). Half-life was determined by one-phase decay modeling. A P value of <0.05 was considered significant. Comparisons between multiple groups were performed using one-way analysis of variance (Tukey’s Multiple Comparison post-test). All values expressed as mean ± standard error of the mean. Where the mean was calculated by evaluating multiple samples from a single animal it is represented as mean ± SEM values for biological replicates. Where the mean was measured per treatment group the values are presented as mean ± SEM values for the means of the three animals per treatment group.
Acknowledgments
We would like to thank Cassandra Brown, Mohamad Nayal, Deirdre McMenamin, and Christine Draper (University of Pennsylvania Perelman School of Medicine, Gene Therapy Program) for invaluable technical assistance and the Human Immunology Core (University of Pennsylvania, Philadelphia, PA). Funding for this research was provided by a grant from Dimension Therapeutics.
The authors declare having potential competing financial interests. J.M.W. is an advisor to REGENXBIO, Dimension Therapeutics, and Solid Gene Therapy, and is a founder of, holds equity in, and has a sponsored research agreement with REGENXBIO and Dimension Therapeutics; in addition, he is a consultant to several biopharmaceutical companies and is an inventor on patents licensed to various biopharmaceutical companies. L.M.K. is an employee of Dimension Therapeutics. The other authors declared no conflict of interest.
References
- Manno, CS, Pierce, GF, Arruda, VR, Glader, B, Ragni, M, Rasko, JJ et al. (2006). Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat Med 12: 342–347. [DOI] [PubMed] [Google Scholar]
- Nathwani, AC, Tuddenham, EG, Rangarajan, S, Rosales, C, McIntosh, J, Linch, DC et al. (2011). Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N Engl J Med 365: 2357–2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathwani, AC, Reiss, UM, Tuddenham, EG, Rosales, C, Chowdary, P, McIntosh, J et al. (2014). Long-term safety and efficacy of factor IX gene therapy in hemophilia B. N Engl J Med 371: 1994–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Avola, D, López-Franco, E, Sangro, B, Pañeda, A, Grossios, N, Gil-Farina, I et al. (2016). Phase I open label liver-directed gene therapy clinical trial for acute intermittent porphyria. J Hepatol 65: 776–783. [DOI] [PubMed] [Google Scholar]
- Harding, TC, Koprivnikar, KE, Tu, GH, Zayek, N, Lew, S, Subramanian, A et al. (2004). Intravenous administration of an AAV-2 vector for the expression of factor IX in mice and a dog model of hemophilia B. Gene Ther 11: 204–213. [DOI] [PubMed] [Google Scholar]
- Nathwani, AC, Gray, JT, McIntosh, J, Ng, CY, Zhou, J, Spence, Y et al. (2007). Safe and efficient transduction of the liver after peripheral vein infusion of self-complementary AAV vector results in stable therapeutic expression of human FIX in nonhuman primates. Blood 109: 1414–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg, JB, De, BP, Quach, L, Russo, C, Kaminsky, SM, Sondhi, D et al. (2016). Consequences of infusion time on efficiency of intravenous delivery of vector genomes to the liver. Mol Ther 24: S187. [Google Scholar]
- Varnavski, AN, Zhang, Y, Schnell, M, Tazelaar, J, Louboutin, JP, Yu, QC et al. (2002). Preexisting immunity to adenovirus in rhesus monkeys fails to prevent vector-induced toxicity. J Virol 76: 5711–5719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, G, Wang, Q, Calcedo, R, Mays, L, Bell, P, Wang, L et al. (2009). Adeno-associated virus-mediated gene transfer to nonhuman primate liver can elicit destructive transgene-specific T cell responses. Hum Gene Ther 20: 930–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell, P, Wang, L, Gao, G, Haskins, ME, Tarantal, AF, McCarter, RJ et al. (2011). Inverse zonation of hepatocyte transduction with AAV vectors between mice and non-human primates. Mol Genet Metab 104: 395–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, L, Calcedo, R, Bell, P, Lin, J, Grant, RL, Siegel, DL et al. (2011). Impact of pre-existing immunity on gene transfer to nonhuman primate liver with adeno-associated virus 8 vectors. Hum Gene Ther 22: 1389–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raper, SE, Chirmule, N, Lee, FS, Wivel, NA, Bagg, A, Gao, GP et al. (2003). Fatal systemic inflammatory response syndrome in a ornithine transcarbamylase deficient patient following adenoviral gene transfer. Mol Genet Metab 80: 148–158. [DOI] [PubMed] [Google Scholar]
- Kassim, SH, Li, H, Vandenberghe, LH, Hinderer, C, Bell, P, Marchadier, D et al. (2010). Gene therapy in a humanized mouse model of familial hypercholesterolemia leads to marked regression of atherosclerosis. PLoS One 5: e13424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, L, Wang, H, Bell, P, McCarter, RJ, He, J, Calcedo, R et al. (2010). Systematic evaluation of AAV vectors for liver directed gene transfer in murine models. Mol Ther 18: 118–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassim, SH, Li, H, Bell, P, Somanathan, S, Lagor, W, Jacobs, F et al. (2013). Adeno-associated virus serotype 8 gene therapy leads to significant lowering of plasma cholesterol levels in humanized mouse models of homozygous and heterozygous familial hypercholesterolemia. Hum Gene Ther 24: 19–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain, JS (2002). Gene therapy of muscular dystrophy. Hum Mol Genet 11: 2355–2362. [DOI] [PubMed] [Google Scholar]
- Byrne, BJ, Falk, DJ, Pacak, CA, Nayak, S, Herzog, RW, Elder, ME et al. (2011). Pompe disease gene therapy. Hum Mol Genet 20(R1): R61–R68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, G, Lu, Y, Calcedo, R, Grant, RL, Bell, P, Wang, L et al. (2006). Biology of AAV serotype vectors in liver-directed gene transfer to nonhuman primates. Mol Ther 13: 77–87. [DOI] [PubMed] [Google Scholar]
- Calcedo, R, Vandenberghe, LH, Gao, G, Lin, J and Wilson, JM (2009). Worldwide epidemiology of neutralizing antibodies to adeno-associated viruses. J Infect Dis 199: 381–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell, P, Moscioni, AD, McCarter, RJ, Wu, D, Gao, G, Hoang, A et al. (2006). Analysis of tumors arising in male B6C3F1 mice with and without AAV vector delivery to liver. Mol Ther 14: 34–44. [DOI] [PubMed] [Google Scholar]
- Wang, L, Calcedo, R, Wang, H, Bell, P, Grant, R, Vandenberghe, LH et al. (2010). The pleiotropic effects of natural AAV infections on liver-directed gene transfer in macaques. Mol Ther 18: 126–134. [DOI] [PMC free article] [PubMed] [Google Scholar]