

Abstract
In the growing field of proteomics, tools for the in silico analysis of proteins and even of whole proteomes are of crucial importance to make best use of the accumulating amount of data. To utilise this data for healthcare and drug development, first the characteristics of proteomes of entire species—mainly the human—have to be understood, before secondly differentiation between individuals can be surveyed. Specialised databases about nucleic acid sequences, protein sequences, protein tertiary structure, genome analysis, and proteome analysis represent useful resources for analysis, characterisation, and classification of protein sequences. Different from most proteomics tools focusing on similarity searches, structure analysis and prediction, detection of specific regions, alignments, data mining, 2D PAGE analysis, or protein modelling, respectively, comprehensive databases like the proteome analysis database benefit from the information stored in different databases and make use of different protein analysis tools to provide computational analysis of whole proteomes.
INTRODUCTION
Continual advancement in proteome research has led to an influx of protein sequences from a wide range of species, representing a challenge in the field of Bioinformatics. Genome sequencing is also proceeding at an increasingly rapid rate, and this has led to an equally rapid increase in predicted protein sequences. All these sequences, both experimentally derived and predicted, need to be stored in comprehensive, nonredundant protein sequence databases. Moreover, they need to be assembled and analysed to represent a solid basis for further comparisons and investigations. Especially the human sequences, but also those of the mouse and other model organisms, are of interest for the efforts towards a better understanding of health and disease. An important instrument is the in silico proteome analysis.
The term “proteome” is used to describe the protein equivalent of the genome. Most of the predicted protein sequences lack a documented functional characterisation. The challenge is to provide statistical and comparative analysis and structural and other information for these sequences as an essential step towards the integrated analysis of organisms at the gene, transcript, protein, and functional levels.
Especially whole proteomes represent an important source for meaningful comparisons between species and furthermore between individuals of different health states. To fully exploit the potential of this vast quantity of data, tools for in silico proteome analysis are necessary. In the following, some important sources for proteome analysis like sequence databases and analysis tools will be described, which represent highly useful proteomics tools for the discovery of protein function and protein characterisation.
RESOURCES
Important tools for genome and proteome analysis are databases that store the huge amount of biological data, which is often no longer published in conventional publications. These databases, especially in combination with database search tools and tools for the computational analysis of the data, are necessary resources for biological and medical research.
Sequence databases
Sequence databases are of special importance for different fields of research because they are comprehensive sources of information on nucleotide sequences and proteins. There are basically three types of sequence-related databases, collecting nucleic acid sequences, protein sequences, and protein tertiary structures, respectively.
Nucleotide sequence databases
In nucleotide sequence databases, data on nucleic acid sequences as it results from the genome sequencing projects, and also from smaller sequencing efforts, is stored. The vast majority of the nucleotide sequence data produced is collected, organized, and distributed by the International Nucleotide Sequence Database Collaboration [1], which is a joint effort of the nucleotide sequence databases EMBL-EBI (European Bioinformatics Institute, http://www.ebi.ac.uk), DDBJ (DNA Data Bank of Japan, http://www.ddbj.nig.ac.jp), and GenBank (National Center for Biotechnology Information, http://www.ncbi.nlm.nih.gov). The nucleotide sequence databases are data repositories, accepting nucleic acid sequence data from the community and making it freely available. The databases strive for completeness, with the aim of recording and making available every publicly known nucleic acid sequence. EMBL, GenBank, and DDBJ automatically update each other every 24 hours with new or updated sequences. Since their conception in the 1980s, the nucleic acid sequence databases have experienced constant exponential growth. There is a tremendous increase of sequence data due to technological advances. At the time of writing, the DDBJ/EMBL/GenBank Nucleotide Sequence Database has more than 10 billion nucleotides in more than 10 million individual entries. In effect, these archives currently experience a doubling of their size every year. Today, electronic bulk submissions from the major sequencing centers overshadow all other input and it is not uncommon to add to the archives more than 7000 new entries, on average, per day.
Protein sequence databases
In protein sequence databases, information on proteins is stored. Here it has to be distinguished between universal databases covering proteins from all species and specialised data collections storing information about specific families or groups of proteins, or about the proteins of a specific organism. Two categories of universal protein sequence databases can be discerned: simple archives of sequence data and annotated databases where additional information has been added to the sequence record. Especially the latter are of interest for the needs of proteome analysis.
PIR, the protein information resource [2] (http://www-nbrf.georgetown.edu/) has been the first protein sequence database which was established in 1984 by the National Biomedical Research Foundation (NBRF) as a successor of the original NBRF Protein Sequence Database. Since 1988 it has been maintained by PIR-International, a collaboration between the NBRF, the Munich Information Center for Protein Sequences (MIPS), and the Japan International Protein Information Database (JIPID). The PIR release 71.04 (March 1, 2002) contains 283 153 entries. It presents sequences from a wide range of species, not especially focusing on human.
SWISS-PROT [3] is an annotated protein sequence database established in 1986 and maintained since 1988 collaboratively by the Swiss Institute of Bioinformatics (SIB) (http://www.expasy.org/) and the EMBL Outstation-The European Bioinformatics Institute (EBI) (http://www.ebi.ac.uk/swissprot/). It strives to provide a high level of annotation such as the description of the function of a protein, its domain structure, posttranslational modifications, variants, and so forth, and a minimal level of redundancy. More than 40 cross-references—about 4 000 000 individual links in total—to other biomolecular and medical databases, such as the EMBL/GenBank/DDBJ international nucleotide sequence database [1], the PDB tertiary structure database [4] or Medline, are providing a high level of integration. Human sequence entries are linked to MIM [5], the “Mendelian Inheritance in Man” database that represents an extensive catalogue of human genes and genetic disorders. SWISS-PROT contains data that originates from a wide variety of biological organisms. Release 40.22 (June 24, 2002) contains a total of 110 824 annotated sequence entries from 7459 different species; 8294 of them are human sequences. The annotation of the human sequences is part of the HPI project, the human proteomics initiative [6], which aims at the annotation of all known human proteins, their mammalian orthologues, polymorphisms at the protein sequence level, and posttranslational modifications, and at providing tight links to structural information and clustering and classification of all known vertebrate proteins. Seven hundred sixty-one human protein sequence entries in SWISS-PROT contain data relevant to genetic diseases. In these entries, the biochemical and medical basis of the diseases are outlined, as well as information on mutations linked with genetic diseases or polymorphisms, and specialised databases concerning specific genes or diseases are linked [7].
TrEMBL (translation of EMBL nucleotide sequence database) [3] is a computer-annotated supplement to SWISS-PROT, created in 1996 with the aim to make new sequences available as quickly as possible. It consists of entries in SWISS-PROT-like format derived from the translation of all coding sequences (CDSs) in the EMBL nucleotide sequence database, except the CDSs already included in SWISS-PROT. TrEMBL release 21.0 (June 21, 2002) contains 671 580 entries, which should be eventually incorporated into SWISS-PROT, 32 531 of them human. Before the manual annotation step, automated annotation [8, 9] is applied to TrEMBL entries where sensible.
SP_TR_NRDB (or abbreviated SPTR or SWALL) is a database created to overcome the problem of the lack of comprehensiveness of single-sequence databases: it comprises both the weekly updated SWISS-PROT work release and the weekly updated TrEMBL work release. So SPTR provides a very comprehensive collection of human sequence entries, currently 45 629.
The CluSTr (clusters of SWISS-PROT and TrEMBL proteins) database [10] (http://www.ebi.ac.uk/clustr) is a specialised protein sequence database, which offers an automatic classification of SWISS-PROT and TrEMBL proteins into groups of related proteins. The clustering is based on analysis of all pairwise comparisons between protein sequences. Analysis has been carried out for different levels of protein similarity, yielding a hierarchical organisation of clusters.
Protein tertiary structure databases
The number of known protein structures is increasing very rapidly and these are available through PDB, the protein data bank [4] (http://www.rcsb.org/pdb/). There is also a database of structures of “small” molecules of interest to biologists concerned with protein-ligand interactions, available from the Cambridge Crystallographic Data Centre (http://www.ccdc.cam.ac.uk/).
In addition, there are also a number of derived databases, which enable comparative studies of 3D structures as well as to gain insight on the relationships between sequence, secondary structure elements, and 3D structure. DSSP (dictionary of secondary structure in proteins, http://www.sander.ebi.ac.uk/dssp/) [11] contains the derived information on the secondary structure and solvent accessibility for the protein structures stored in PDB. HSSP (homology-derived secondary structure of proteins, http://www.sander.ebi.ac.uk/hssp/) [12] is a database of alignments of the sequences of proteins with known structure with all their close homologues. FSSP (families of structurally similar proteins, http://www.ebi.ac.uk/dali/fssp/) [13] is a database of structural alignments of proteins. It is based on an all-against-all comparison of the structures stored in PDB. Each database entry contains structural alignments of significantly similar proteins but excludes proteins with high sequence similarity since these are usually structurally very similar.
The SCOP (structural classification of proteins) database [14] (http://scop.mrc-lmb.cam.ac.uk/scop/) has been created by manual inspection and abetted by a battery of automated methods. This resource aims to provide a detailed and comprehensive description of the structural and evolutionary relationships between all proteins whose structure is known. As such, it provides a broad survey of all known protein folds and detailed information about the close relatives of any particular protein.
Another database, which attempts to classify protein structures in the PDB, is the CATH database [15] (http://www.biochem.ucl.ac.uk/bsm/cath_new/), a hierarchical domain classification of protein structures in the PDB.
Proteome analysis databases and tools
Tools and databases for proteome analysis are based on reliable algorithms and information about protein sequences and structures derived from comprehensive protein databases. It can be difficult to distinguish between “database” and “tool” since databases providing precomputed data and search algorithms can offer a high functionality towards protein analysis.
Proteome analysis databases
The classic proteomics databases are those of 2D gel electrophoresis data such as the SWISS-2DPAGE database (two-dimensional polyacrylamide gel electrophoresis database) [16] (http://www.expasy.ch/ch2d/). However, since the genome sequencing is proceeding at an increasingly rapid rate, this leads to an equally rapid increase in predicted protein sequences entering the protein sequence databases. Most of these predicted protein sequences are without a documented functional role. The challenge is to bridge the gap until functional data has been gathered through experimental research by providing statistical and comparative analysis and structural and other information for these sequences. This way of computational analysis can serve as an essential step towards the integrated analysis of organisms at the gene, transcript, protein, and functional levels.
Proteome analysis databases have been set up to provide comprehensive statistical and comparative analyses of the predicted proteomes of fully sequenced organisms.
The proteome analysis database [17] (http://www.ebi.ac.uk/proteome) has the more general aim of integrating information from a variety of sources that will together facilitate the classification of the proteins in complete proteome sets. The proteome sets are built from the SWISS-PROT and TrEMBL protein sequence databases that provide reliable, well-annotated data as the basis for the analysis. Proteome analysis data is available for all the completely sequenced organisms present in SWISS-PROT and TrEMBL, spanning archaea, bacteria, and eukaryotes. In the proteome analysis effort, the InterPro [18] (http://www.ebi.ac.uk/interpro/) and CluSTr resources have been used. Links to structural information databases like the HSSP and PDB are provided for individual proteins from each of the proteomes. A functional classification using gene ontology (GO; [19]) is also available. The proteome analysis database provides a broad view of the proteome data classified according to signatures describing particular sequence motifs or sequence similarities and at the same time affords the option of examining various specific details like structural or functional classification. It currently (June 2002) contains statistical and analytical data for the proteins from 77 complete genomes.
The international protein index (IPI) (http://www.ebi.ac.uk/IPI/IPIhelp.html) provides a top-level guide to the main databases that describe the human and mouse proteome, namely SWISS-PROT, TrEMBL, RefSeq [20], and EnsEMBL [21]. IPI maintains a database of cross-references between the primary data sources with the aim of providing a minimally redundant yet maximally complete set of human proteins (one sequence per transcript).
Proteome analysis tools
Traditional proteomics tools like those accessible from the ExPASy server (http://www.expasy.org) represent a variety of possibilities to analyse proteins. They help to identify and characterise proteins, to convert DNA sequences into amino acid sequences, and to perform similarity searches, pattern and profile searches, posttranslational modification prediction, primary structure analysis, secondary and tertiary structure prediction, detection of transmembrane regions, alignments, and biological text analysis. Moreover, there is a software available for 2D PAGE analysis, automated knowledge-based protein modelling, and structure display and analysis.
The analysis of whole proteomes represents an even bigger challenge. Large and comprehensive databases and knowledge bases are developed and used which provides large sets of precomputed data. To gather this comprehensive data, a vast amount of underlying information is necessary. The proteome analysis database, mentioned above, uses annotated information about proteins from the SWISS-PROT/TrEMBL database and automated protein classifications from InterPro, CluSTr, HSSP, TMHMM [22], and SignalP [23]. The precomputation permits comparisons of whole proteomes of completely sequenced organisms with those of others (Table 1). Users of the database can perform their own interactive proteome comparisons between any combinations of organisms in the database. Moreover, structural features of individual proteomes like the protein length distribution (Figure 1), amino acid composition (Figure 2), affiliation of the different proteins to protein families, and the number of sequences in total and those displayed by other databases can be requested. Users are also able to run a Fasta similarity search (Fasta3) on their own sequence against a complete proteome in the database with the help of a specific search form. It is possible to download a proteome set or a list of InterPro matches for a given organism, to see the current status of all complete proteomes in SWISS-PROT and TrEMBL, and to download GO annotation for the human proteome.
Table 1.
InterPro comparative analysis of Homo sapiens and Mus musculus proteomes—the first 17 of the top 30 hits are shown.
| H sapiens | M musculus | ||||
| InterPro | Proteins matched | Rank | Proteins matched | Rank | Description |
| (Proteome coverage) | (Proteome coverage) | ||||
| IPR000822 | 1165 (3.4%) | 1 | 341 (1.4%) | 5 | Zn-finger, C2H2 type |
| IPR003006 | 928 (2.7%) | 2 | 498 (2.1%) | 3 | Immunoglobulin/major histocompatibility complex |
| IPR000719 | 738 (2.2%) | 3 | 387 (1.6%) | 4 | Eukaryotic protein kinase |
| IPR000694 | 713 (2.1%) | 4 | 0 | Poline-rich region | |
| IPR000276 | 681 (2.0%) | 5 | 401 (1.7%) | 29 | Rhodopsin-like GPCR superfamily |
| IPR002290 | 515 (1.5%) | 6 | 275 (1.1%) | 7 | Serine/threonine protein kinase |
| IPR000561 | 417 (1.2%) | 7 | 212 (0.9%) | 13 | EGF-like domain |
| IPR001909 | 405 (1.2%) | 8 | 85 (0.4%) | 15 | KRAB box |
| IPR001680 | 386 (1.1%) | 9 | 168 (0.7%) | 17 | G-protein beta WD-40 repeat |
| IPR001245 | 375 (1.1%) | 10 | 180 (0.7%) | 10 | Tyrosine protein kinase |
| IPR001841 | 358 (1.1%) | 11 | 180 (0.7%) | 34 | Zn-finger, RING |
| IPR003599 | 347 (1.0%) | 12 | 174 (0.7%) | 8 | Immunoglobulin subtype |
| IPR000504 | 346 (1.0%) | 13 | 156 (0.6%) | 21 | RNA-binding region RNP-1 (RNP recognition motif) |
| IPR003600 | 345 (1.0%) | 14 | 170 (0.7%) | 6 | Immunoglobulin-like |
| IPR001849 | 326 (1.0%) | 15 | 128 (0.5%) | 33 | Pleckstrin-like |
| IPR002965 | 299 (0.9%) | 16 | 102 (0.4%) | 11 | Proline-rich extensin |
| IPR001452 | 296 (0.9%) | 17 | 138 (0.6%) | 32 | SH3 domain |
Figure 1.
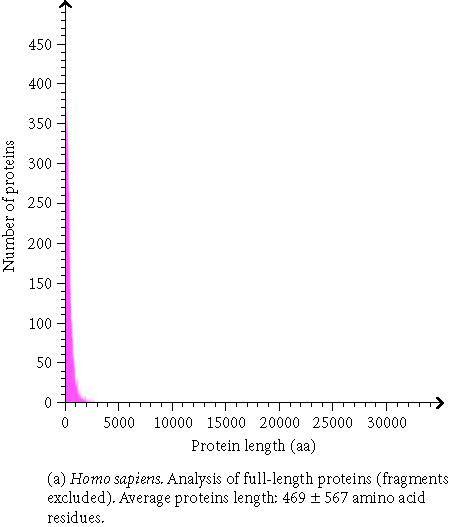
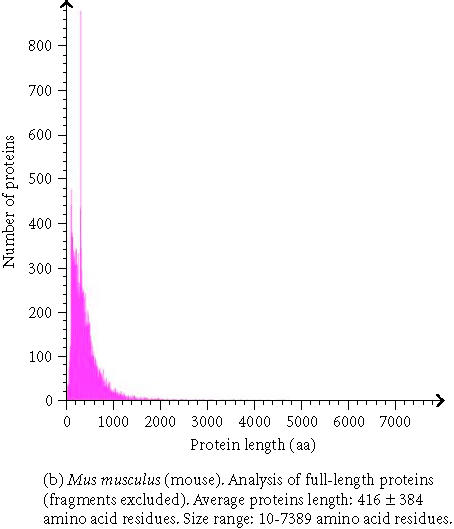
Protein length distribution of Homo sapiens and Mus musculus.
Figure 2.
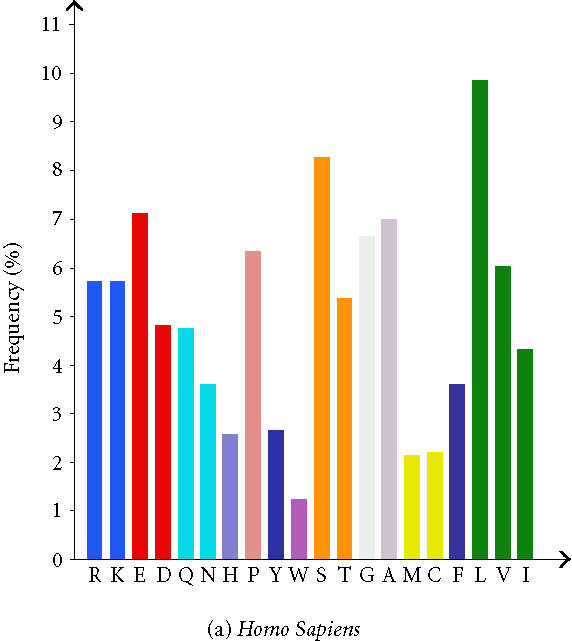
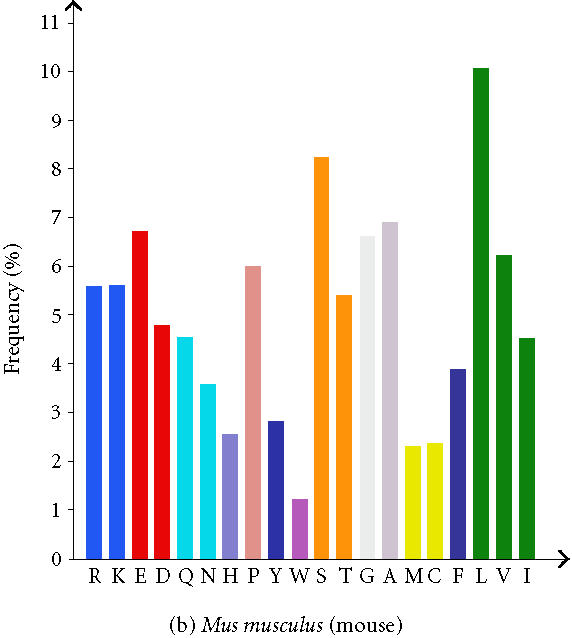
Amino acid composition of Homo sapiens and Mus musculus. (The total number of each amino acid in each proteome is given additionally as well as the frequency in (%).)
Other tools important especially for laboratory scientists are image analysis tools, laboratory information management systems, and software for the characterisation from mass spectrometric data.
Acknowledgments
DISCUSSION
In the last years there has been a tremendous increase in the amount of data available concerning the human genome and more particularly the molecular basis of genetic diseases. Every week, new discoveries are made that link one or more genetic diseases to defects in specific genes. To take into account these developments, the SWISS-PROT protein sequence database for example is gradually enhanced by the addition of a number of features that are specifically intended for researchers working on the basis of human genetic diseases as well as the extent of polymorphisms. The latter are very important too, since they may represent the basis for differences between individuals, which are particularly interesting for some aspects of medicine and drug research. Such comprehensive sequence databases are mandatory for the use of proteome analysis tools like the proteome analysis database, which combines the different protein sequences of a given organism to a complete proteome. This proteome can be regarded as a whole new unit, analysable according to different points of view (like distribution of domains and protein families, and secondary and tertiary structures of proteins), and can be made comparable to other proteomes. In general, for using the proteomics data for healthcare and drug development, first the characteristics of proteomes of entire species—mainly the human—have to be understood before secondly differentiation between individuals can be surveyed.
But although the number of proteome analysis tools and databases is increasing and most of them are providing a very good quality of computational efforts and/or annotation of information, the user should not forget that automated analysis always can hold some mistakes. Data material in databases is reliable, but only to a certain point. Automatic tools which use data derived from databases can thus be error-prone, rules built on their basis can be wrong, and sequence similarities can occur due to chance and not due to relationship. Users of bioinformatics tools should in no way feel discouraged in their using, they only should keep in mind the potential pitfalls of automated systems and even of humans—and be encouraged to check all data as far as possible and not blindly rely on them.
References
- Stoesser G, Baker W, van den Broek A, et al. The EMBL nucleotide sequence database. Nucleic Acids Res. 2001;29(1):17–21. doi: 10.1093/nar/29.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C H, Huang H, Arminski L, et al. The Protein Information Resource: an integrated public resource of functional annotation of proteins. Nucleic Acids Res. 2002;30(1):35–37. doi: 10.1093/nar/30.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bairoch A, Apweiler R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 2000;28(1):45–48. doi: 10.1093/nar/28.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat T N, Bourne P, Feng Z, et al. The PDB data uniformity project. Nucleic Acids Res. 2001;29(1):214–218. doi: 10.1093/nar/29.1.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson P L, Francomano C, Foster P, Bocchini C, Li P, McKusick V A. The status of online Mendelian inheritance in man (OMIM) medio 1994. Nucleic Acids Res. 1994;22(17):3470–3473. doi: 10.1093/nar/22.17.3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Donovan C, Apweiler R, Bairoch A. The human proteomics initiative (HPI) Trends Biotechnol. 2001;19(5):178–181. doi: 10.1016/s0167-7799(01)01598-0. [DOI] [PubMed] [Google Scholar]
- Bairoch A, Apweiler R. The SWISS-PROT protein sequence database: its relevance to human molecular medical research. J Mol Med. 1997;75(5):312–316. [PubMed] [Google Scholar]
- Fleischmann W, Moller S, Gateau A, Apweiler R. A novel method for automatic functional annotation of proteins. Bioinformatics. 1999;15(3):228–233. doi: 10.1093/bioinformatics/15.3.228. [DOI] [PubMed] [Google Scholar]
- Kretschmann E, Fleischmann W, Apweiler R. Automatic rule generation for protein annotation with the C4.5 data mining algorithm applied on SWISS-PROT. Bioinformatics. 2001;17(10):920–926. doi: 10.1093/bioinformatics/17.10.920. [DOI] [PubMed] [Google Scholar]
- Kriventseva E V, Fleischmann W, Zdobnov E M, Apweiler R. CluSTr: a database of clusters of SWISS-PROT+TrEMBL proteins. Nucleic Acids Res. 2001;29(1):33–36. doi: 10.1093/nar/29.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W, Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22(12):2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- Dodge C, Schneider R, Sander C. The HSSP database of protein structure-sequence alignments and family profiles. Nucleic Acids Res. 1998;26(1):313–315. doi: 10.1093/nar/26.1.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm L, Sander C. The FSSP database: fold classification based on structure-structure alignment of proteins. Nucleic Acids Res. 1996;24(1):206–209. doi: 10.1093/nar/24.1.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Conte L, Brenner S E, Hubbard T J.P, Chothia C, Murzin A G. SCOP database in 2002: refinements accommodate structural genomics. Nucleic Acids Res. 2002;30(1):264–267. doi: 10.1093/nar/30.1.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearl F M.G, Martin N, Bray J E, et al. A rapid classification protocol for the CATH Domain Database to support structural genomics. Nucleic Acids Res. 2001;29(1):223–227. doi: 10.1093/nar/29.1.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoogland C, Sanchez J C, Tonella L, et al. The 1999 SWISS-2DPAGE database update. Nucleic Acids Res. 2000;28(1):286–288. doi: 10.1093/nar/28.1.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apweiler R, Biswas M, Fleischmann W, et al. Proteome Analysis Database: online application of InterPro and CluSTr for the functional classification of proteins in whole genomes. Nucleic Acids Res. 2001;29(1):44–48. doi: 10.1093/nar/29.1.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apweiler R, Attwood T K, Bairoch A, et al. The InterPro database, an integrated documentation resource for protein families, domains and functional sites. Nucleic Acids Res. 2001;29(1):37–40. doi: 10.1093/nar/29.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball C A, Blake J A, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25(1):25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruitt K D, Maglott D R. RefSeq and LocusLink: NCBI gene-centered resources. Nucleic Acids Res. 2001;29(1):137–140. doi: 10.1093/nar/29.1.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard T, Barker D, Birney E, et al. The Ensemble genome database project. Nucleic Acids Res. 2002;30(1):38–41. doi: 10.1093/nar/30.1.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnhammer E L, von Heijne G, Krogh A. A hidden Markov model for predicting transmembrane helices in protein sequences. In: Glasgow J, Little-John T, Major F, Lathrop R, Sankoff D, Sensen C, editors. AAAI Press; Menlo Park, Calif: 1998. pp. 175–182. Proceedings of the 6th International Conference on Intelligent Systems for Molecular Biology. [PubMed] [Google Scholar]
- Nielsen H, Brunak S, von Heijne G. Machine learning approaches for the prediction of signal peptides and other protein sorting signals. Protein Eng. 1999;12(1):3–9. doi: 10.1093/protein/12.1.3. [DOI] [PubMed] [Google Scholar]