

Abstract
Adaptive, seamless, multisponsor, multitherapy clinical trial designs executed as large scale platforms, could create superior evidence more efficiently than single‐sponsor, single‐drug trials. These trial PIPELINEs also could diminish barriers to trial participation, increase the representation of real‐world populations, and create systematic evidence development for learning throughout a therapeutic life cycle, to continually refine its use. Comparable evidence could arise from multiarm design, shared comparator arms, and standardized endpoints—aiding sponsors in demonstrating the distinct value of their innovative medicines; facilitating providers and patients in selecting the most appropriate treatments; assisting regulators in efficacy and safety determinations; helping payers make coverage and reimbursement decisions; and spurring scientists with translational insights. Reduced trial times and costs could enable more indications, reduced development cycle times, and improved system financial sustainability. Challenges to overcome range from statistical to operational to collaborative governance and data exchange.
Expanding and adaptively connecting basket, umbrella, and real‐world evidence trials that simultaneously test multiple therapies (as single agents and as concomitant or sequential combinations) through appropriately powered, multicenter, multiarm designs could improve patient access to therapies, increase evidence for all stakeholders to employ in their decisions, and do so more efficiently and faster than a portfolio of individual trials. Achieving this potential will require overcoming operational, statistical, and organizational challenges.
The challenge of individually crafted clinical trials
Current drug development emphasizes individually crafted clinical trials that construct each study design from first principles, recruit a dedicated clinical network, and tailor clinical operations to the therapeutic characteristics and clinical hypothesis. This approach has served to create an armamentarium of nearly 1,500 drugs approved by the US Food and Drug Administration (FDA),1 but turning insights into therapies remains long, costly, and uncertain.2, 3 Exclusivity, expiry, and late‐development failures make financial sustainability an urgent issue for sponsors.4 Increasingly, this “one drug, one population, one indication, one phase” approach faces strains as science fragments diseases into ever smaller therapeutic segments, with hundreds of new therapies in clinical trials.5, 6, 7, 8 Simultaneously, demands are increasing from patients, providers, trialists, regulators, policy makers, and payers. While patients lose patience for access to promising therapies, patient safety advocates express concerns, and payers increasingly require evidence of real‐world effectiveness and favorable benefit vs. cost.9, 10, 11, 12 These demands, in turn, mandate innovative drug development approaches.
PIPELINEs: an evolutionary response
Numerous enabling components based on the methods and infrastructures of platform trials have emerged over the past decade. Basket and umbrella trial programs demonstrate that multiproduct, multisite trials can be both informative and feasible. Adaptive clinical trial designs improve the probability of success and clinical trial efficiency.13, 14, 15, 16, 17, 18 Precision medicine using biomarker panels can increasingly screen all presenting patients to identify subpopulations for targeted treatment.19, 20, 21 Real‐world evidence is beginning to demonstrate the ability to assess effectiveness, safety, and perhaps new indication opportunities.22 Linking the best platform practices, extending their scope across clinical development, and expanding their scale could create a new approach to clinical development that we term PIPELINE (Portfolio of Innovative Platform Engines, Longitudinal Investigations and Novel Effectiveness).
PIPELINEs (Figure 1) assemble existing single‐phase trial platforms and their components into a larger design to generate more, faster, and lower‐cost knowledge than the current “one drug, one population, one indication, one phase” clinical trial approach. A PIPELINE may combine basket, confirmatory basket, and adaptive umbrella trial platforms with seamless graduation across the clinical evidence spectrum. We further propose extending PIPELINEs into real‐world evidence designs to study effectiveness and natural disease history to inform both ongoing therapeutic decisions and next‐generation medicines innovation. While much of the inspiration and early examples arise from oncology, we believe the approach could apply to many, if not most, therapeutic areas including neurodegenerative, psychiatric, inflammatory, infective, and metabolic disorders.
Figure 1.
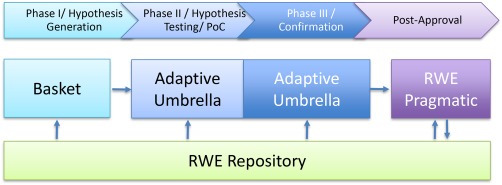
Illustrative simple PIPELINE design showing a hypothesis generation basket trial feeding candidates to seamlessly linked Phase II Proof of Concept (PoC)/Phase III umbrella platforms. A pragmatic clinical trial platform then continues studying effectiveness and regimen optimization. A real world patient repository collects observational and pragmatic trial information to prompt new hypothesis generation, aid propensity scoring and representativeness analysis of the inclusion/exclusion criteria, and natural history progression to refine endpoint impact estimates.
PIPELINEs enable Adaptive Biomedical Innovation
Adaptive Biomedical Innovation (ABI), as described in a companion article in this issue, transforms a siloed process separated by independent decisions involving few stakeholders at each point into a more holistic process, with greater stakeholder engagement throughout.23, 24, 25, 26 ABI emphasizes prospectively planned iterations of clinical evidence development throughout the therapeutic lifespan to improve patient access and safety through staged and contingent decision‐making. To succeed, ABI will require an inclusive approach that reliably produces credible, comparable, longitudinal clinical evidence for multiple candidate therapeutics, across many indications, to improve financial sustainability for all, and to increase appropriate patient access to therapies. PIPELINEs could provide a practical means to deliver that evidence.
COMPLEXITY SCIENCE OFFERS AN ANALYTIC FRAMEWORK
Unlike engineers optimizing mechanical flows on a factory floor, drug developers work in a complex adaptive system in which stakeholders with varying agendas perform independent actions with uncertain outcomes while also responding to the actions of others.27, 28, 29, 30, 31 Complex adaptive systems are not centrally commanded but rather emerge from stakeholder needs and actions to satisfy their individual objectives, ideally, while not violating those of the other stakeholders. Such systems may exhibit initially surprising behaviors driven by underlying, understandable features, including self‐assembling subgroups to accomplish local goals and feedback loops that can amplify (or dampen) resource use and system outputs.
We use this complexity science framework to examine PIPELINE prospects in the drug development ecosystem. We describe the:
Components of trial designs and operational innovations that could be assembled into PIPELINEs.
PIPELINE concept, which assembles the components and stakeholders to create a higher performing drug development ecosystem.
Feedback loops that may propel or inhibit PIPELINEs formation and growth.
Stakeholder impacts to explore whether each benefits, and so would willingly participate in a PIPELINE ecosystem.
PIPELINE governance models that might emerge to orchestrate, fund, operate, and divide the benefits (including intellectual property rights).
Finally, we close with a perspective on areas for future research, limitations, and suggested initial actions.
PIPELINE COMPONENTS
PIPELINEs assemble components from existing, demonstrated clinical trial designs and recent innovations in clinical trial operations, which we review below and summarize in Table 1.
Table 1.
PIPELINE components
| Clinical trial platforms | Operational innovations |
|---|---|
| Basket | Demonstrated |
| • Hypothesis generation (Signature, NCI‐MATCH) | • Master Protocols |
| • Confirmatory (proposed) | • Consistent Informed Consents |
| Umbrella (Lung‐MAP) | • Screening |
| Adaptive Platform (I‐SPY family) | • Quantitative decision making |
| SMART | Emerging |
| SMART | • Repositories |
| Real‐world evidence | • EHR/CRF convergence |
| • Biostatistics analysis pipelines |
Trial designs enabling PIPELINEs
PIPELINEs combine multiple clinical trial platform designs serially and in parallel to create a more efficient, patient‐centered, and informative clinical development ecosystem. Due to varied clinical trial design historical origins and evolution, overlapping term use complicates taxonomic descriptions. We broadly group them into basket, umbrella, adaptive platform, SMART, and real‐world evidence designs, described below. Similarly, the transitions between the clinical trial platforms may blend deterministic and adaptive approaches.
Basket trials test therapies across a portfolio of populations in which a therapy would traditionally receive a distinct regulatory indication (e.g., in oncology organ‐of‐origin populations).32 In oncology, basket trials often employ biomarker‐driven designs that associate a specific therapy with a unique and often rare biomarker, such as a genotypic or phenotypic aberration.33, 34 With this model, the biomarker must be a hypothesized response predictor with a clinically feasible assay. “Pooling” efficacy information across populations to create a single composite outcome endpoint increases power, if statistical methods can accommodate the “pruning” of failed cohorts and the variability among populations, especially varying progression, comparators, and endpoint criteria.35 Likewise, distinguishing traditional indications as separate inferential entities, and “borrowing” information across them via Bayesian statistical methods, can reduce required patient cohorts.36
NCI‐MATCH (http://www.cancer.gov/about‐cancer/treatment/clinical‐trials/nci‐supported/nci‐match#1)37, 38 and Signature (http://www.signaturetrial.com/en)39 trials are basket trials that use pooling and borrowing, respectively. NCI‐MATCH is also a trial platform testing multiple therapies simultaneously. The increased scale reduces screening failures, improving accrual rates and cost‐effectiveness. As seen in Figure 2, NCI‐MATCH is a master protocol that opens and closes substudies, each an independent arm studying a therapy‐biomarker association, whereas Signature operates separate studies for each therapeutic, united by a protocol template. Both trials feature the flexibility of adding or closing arms as new information becomes available, but neither has biomarker‐negative cohorts nor a randomization strategy. A companion article in this issue details a general design concept for a confirmatory basket trial capable of generating pivotal results for regulatory submission.35
Figure 2.
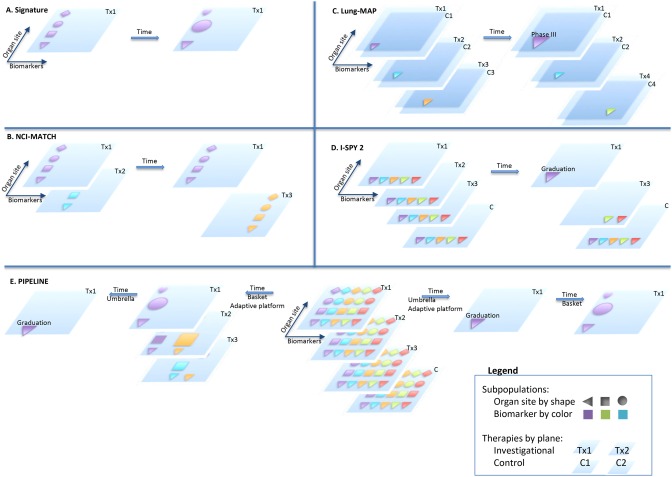
Schematic Clinical Trial Designs. (a) Signature adds and closes cohorts, using Bayesian methods. (b) NCI‐MATCH adds and closes treatment arms, within a master protocol. (c) With its master protocol Lung‐MAP adds and closes treatment arms, which are each randomized, and graduates winning arms to phase III seamlessly with inferential linking. (d) I‐SPY 2 adaptively randomizes to efficiently find the graduates for I‐SPY 3 or other phase III trials. (e) PIPELINEs will have the scale and diversification of options to allow information to flow in multiple directions. To the left, an adaptive basket approach could identify subpopulations for treatment, which are then compared with appropriate control in an umbrella approach. To the right, an umbrella type adaptive basket finds a graduate for phase III and supplemental indications are then explored in a basket trial.
Umbrella trials test a portfolio of therapies for a particular traditional indication. In a simple biomarker‐driven design, a hypothesized therapy‐biomarker association stratifies patients into separate treatment substudies. Randomization for each biomarker defined group, and adaptive features such as opening and closing cohorts or modifying the standard‐of‐care treatment arm based on new knowledge, are utilized by Lung‐MAP (Figure 2).20, 40, 41 Lung‐MAP also exemplifies seamless design, which integrates different response outcomes and clinical development phases into one clinical program to eliminate the “dead space” between phases.17, 42 In operationally seamless designs, algorithms graduate therapeutic candidates to the next phase immediately, whereas in inferentially seamless designs, consistent treatment regimens and endpoints allow including the data from the prior phase patients (e.g., phase II) in later phase (e.g., pivotal phase III) analysis.
Adaptive clinical trials, by definition, use prespecified algorithms to dynamically modify trial parameters in response to internal interim evidence.16, 17, 43, 44, 45 Parameters that may be subjects of adaptive designs include randomization schedule, treatment arms, dose options, and sample size.46 Many, but not all, adaptive designs use a Bayesian framework. These designs may better address a priori design element uncertainties and so may increase the probability of success, may improve trial efficiency by requiring fewer participants, or both. Response‐adaptive randomization can tune the randomization ratio between winning arms and the losing (toxic or ineffective) and control arms to reduce overall trial sizes and improve patient efficacy and safety.15, 18 A concern is that inadequately designed adaptive clinical trials risk increased false‐positive results. Premature interim analyses risk increased investigator bias if their results escape. Rigorous design, trial simulations, triple‐blind operations, computerized randomization, validated short‐term endpoints, and disciplined data monitoring committees can mitigate these risks. Implementing adaptive designs requires rapid data collection and facile communication across trial sites, and may require timely endpoints. Additional adaptive features could include dynamic modification based on external evidence, and are not explicitly included in the formal Center for Drug Evaluation and Research (CDER) guidance definition. Adhoc external adaptations (e.g., changing a standard‐of‐care comparator arm based on external treatment guidelines) risk increasing false‐positive rates but may improve the ultimate utility. Prospectively defined adaptations implemented by prespecified algorithms mitigate this risk (e.g., maturing phase II data prospectively governing an adaptation in the subsequent phase III study).
Adaptive clinical trial features can be inserted into basket or umbrella trial designs. Dose and other regimen titration studies can also benefit from adaptive designs.48
I‐SPY 2 is the prototypical adaptive platform trial, which infuses adaptive features into an umbrella‐like design. 13, 14, 18, 49, 50, 51, 52, 53, 54 It uses response‐adaptive randomization, borrowing by Bayesian methods, and other adaptive features to increase efficiency as it simultaneously explores the efficacy and safety of multiple therapies, alone and in combination, in multiple biomarker‐defined breast cancer subpopulations.55, 56, 57, 58, 59, 60 I‐SPY 2 also employs seamless design to I‐SPY 3, using a Bayesian 85% predictive probability of phase III success as its graduation criteria for any individual substudy arm.61, 62, 63 See Supplemental Materials (Section M) for a more complete description of the I‐SPY platform family, and Figure 2 for an illustration of I‐SPY 2, other trial designs, and some combinations of these different trial designs that would be enabled by PIPELINEs.
Sequential, Multiple Assignment, Randomized Trials (SMART) enable flexibility for patients and trialists by permitting multiple randomization steps within a single trial. Patients may be rerandomized to other treatment options at decision points within a trial, retaining the ability for comparisons among treatment options at individual points, while also enabling evaluation of entire treatment sequences.64, 65, 66 This approach addresses the problem of “drop‐outs”—participants with a clear lack of response or those suffering serious side effects who would switch to an alternate therapy in routine practice but must terminate participation in traditional trials, losing information and prohibiting formalized understanding regarding sequential application of treatments. I‐SPY 2 is integrating SMART design currently.
Real‐World Evidence (RWE) approaches use a variety of evidence‐gathering tools to produce results that can be generalized and applied in routine practice settings, and inform clinical trial design. RWE includes observational studies that prospectively monitor the natural history of diseases such as Alzheimer's to generate potential new clinical endpoints, prognostic factors, and biomarkers including deep phenotyping using cognitive, functional, and other novel assessments.67 For example, new prognostic factors (or a scalar function of multiple factors) could be dynamically incorporated into interventional studies with adaptive designs via covariate‐adaptive randomization in order to balance the prognostic factors among the treatment arms.68 RWE also includes interventional trials. Pragmatic trials, a type of randomized trial that evaluates the effectiveness of interventions in real‐world patient populations, recruit patients more representative of the community intended‐to‐treat population by relaxing randomized controlled trial (RCT) inclusion and exclusion criteria that seek homogeneous populations to limit heterogeneity of disease stage, comorbidity, adherence, and other potential confounding factors.69 Pragmatic trials may prove especially helpful in determining real‐world effectiveness, e.g., in “efficacy‐to‐effectiveness” (E2E) designs, the effectiveness trial commences seamlessly upon completion of the efficacy trial.70, 71 As standard‐of‐care screening moves oncology towards treating all patients with biomarkers similarly to the clinical trial selection criteria, nonrandomized RWE intervention trials have demonstrated their ability to generate hypotheses, evaluate side effect profiles, and test patient‐reported outcomes (PROs) as a first step towards their qualification as RCT endpoints. For example, ASCO's TAPUR (http://www.tapur.org/) uses approved treatments off‐label in a basket platform design in order to generate hypotheses regarding toxicity and supplemental indication. Payer restrictions on off‐label therapeutic use may limit future studies, but multiple other RWE applications exist including dose titration, regimen optimization, mechanistic studies, and prognostic vs. predictive biomarker studies where using very large, heterogeneous patient populations offers the advantage of generalizability. Ultimately, patients, providers, regulators, and payers are demanding comparative effectiveness evidence, so efforts to converge the internal validity of clinical trial information (reliability or accuracy of results) with the external validity of real‐world population data (generalizability of results to nontrial patient populations) will help close the gap between efficacy and effectiveness.
Operational components enabling PIPELINEs
Operational innovations are making clinical trial platforms practical. Some have now been demonstrated while others are just now emerging from research projects. While arcane to many, without these critical components PIPELINEs could not be created.
Demonstrated operational components
Master protocols that allow multidrug, multisubstudy, biomarker‐driven studies with dynamic arm additions and terminations, dynamic patient assignment, and rerandomization, and even treatment regimen changes, have now been demonstrated. These master protocols are not only breakthroughs for their adaptive trial designs, but also for their avoidance of de novo Institutional Review Board (IRB) reviews for each arm. Some individual site IRBs even delegate their decisions on future arms to a master protocol steering committee, which regularly reports graduation or closure of biomarker or therapeutic strategies to ensure site‐level transparency.72, 73, 74 A corollary advance is the Master IND structure, originally proposed by the FDA's Dr. Woodcock, which enables protocols to initiate new arms without the 30‐day FDA review period.55 The invention of master protocols with master INDs significantly enables PIPELINE construction.
Standardized informed consent forms can promote ethical consistency across sites, anticipate crossover or other arm‐switching designs, allow future testing of biological samples, and enable operational efficiency. By isolating each site's unique language into a separate informed consent template section, I‐SPY 2 has streamlined, if not eliminated, the process. The NIH NHGRI (National Human Genome Research Institute) has developed consent forms that anticipate indefinite biobanking, future testing, and reinterpretation of historic testing (https://www.genome.gov/27026588/).
The one million patient NIH Precision Medicine Initiative Cohort Program working group (PMI‐WG) recommendation that “…a standardized consent protocol to ensure consistency in the terms and conditions that all PMI cohort participants agree to”73 is providing further momentum for both standardized informed consent and master protocol acceptance (https://www.nih.gov/precision‐medicine‐initiative‐cohort‐program).
Screening that rapidly identifies and matches patients to trial arms is critical for both patients and trialists. For example, centralized molecular panels have been demonstrated by individual trials like NCI‐MATCH, I‐SPY 2, and others (above). This innovation is transforming screening from an individual physician process into a semiautomated process that prioritizes patient trial opportunities for providers to share with each patient. Fully collaborative molecular screening programs like the EORTC SPECTA coordinates several disease‐specific platforms on a pan‐European level with the aim of identifying, at an early stage, specific druggable aberrations and offer specific targeted treatment to patients within clinical trials (http://spectacolor.eortc.org/).76 While invented for cancer, the approach is applicable to any therapeutic area with diagnostic driven trial criteria.
Quantitative decision making using decision analysis and simulations can refine individual platform and PIPELINE trial designs overall and for individual therapeutics. Some approaches attempt to mathematically optimize a single “utility function” that captures the net benefit of the system, while other approaches show the statistical or deterministic implications of a scenario. Both approaches depend on the quality of their inputs, the interpretation of their results as those inputs vary, and being placed in context in the larger decision‐making process. Regardless of the approach, the models require an interdisciplinary team spanning research to commercial with supporting technical modelers. Applications range from single trial designs (classic, biomarker, and adaptive) to inform an individual development team,47, 77 to full clinical evidence development plans and company portfolio models,78, 79 to complete therapeutic lifespan models to support multistakeholder decisions.33, 34, 80, 81, 82
Emerging operational components
Emerging patient electronic health record (EHR) repositories useful for virtual patient recruitment can be built from provider electronic medical record systems, payer claims databases, disease‐specific patient registries (EPAD Register, to be discussed later, http://ep‐ad.org/), or social media patient communities (PatientsLikeMe https://www.patientslikeme.com/). EHR‐based clinical trial alerts and “infobuttons” can identify eligible patients in the clinical context.83, 84 At the population level, Gassull and team85 demonstrated that EHRs could identify sufficient patients at less than a dozen sites to potentially fill, at standard conversion rates, Alzheimer's, cardiovascular, and lung cancer trials. PIPELINEs with their larger scale can better leverage these innovations than individually crafted trials.
Beyond basic criteria screening, repositories could aid trials by facilitating deep phenotyping, ethnic and socioeconomic balancing, subpopulation identification, and comorbidities exploration. Including biobanking would further increase repository value. Repository utility is diminished, unfortunately, by incomplete, inaccurate, inconsistent, and delayed data that is technically, legally, or commercially difficult to access at the patient level.
Point‐of‐care EHR, case report form convergence would enable the elimination of dual data entry for clinical trial participants and extend Good Clinical Practice (GCP) data quality to real‐world populations for pragmatic and other RWE trials.
Point‐of‐care structured data capture for both medical care and clinical trial reporting could increase quality and streamline processes for both domains. The FDA is interested in using electronic source data in case report forms to simplify the onerous process of data cleaning and verification.86, 87 The UCSF project management office in collaboration with I‐SPY and Salesforce has developed TRANSCEND to integrate multiple data sources and enable real‐time adaptive randomization.88 The UCSF/I‐SPY OneSource effort (http://www.quantumleaphealth.org/onesource) has the further goal to integrate the EHR and GCP clinical trial reporting systems. When this migration occurs, patients at involved clinical trial sites could routinely participate in real‐world clinical studies, with commensurate learning acceleration and lower study costs.
Biostatistics analysis pipelines have been uncommon for clinical trials. The “omics” disciplines often rely on fully‐ and semiautomated data collection, assembly, annotation, curation, and signature creation pipelines. Similarly, each domain of clinical trial data from patient history, to diagnostics, to imaging, consists of raw data, processed information, and decision‐guiding “signatures.” An early example of such a clinical trial biostatistics tool was caINTEGRATOR.89, 90 Renewed efforts in this field could yield significant dividends in quality and efficiency.
THE PIPELINE CONCEPT
We propose assembling multiproduct, multiarm clinical designs such as umbrella, basket, and pragmatic trials into large‐scale adaptively connected clinical evidence development systems that span from phase Ib hypothesis generation trials or Proof‐of‐Concept phase II trials through postapproval monitoring and effectiveness studies. The I‐SPY family of trials represents a prototype, as their individually adaptive multiarm designs not only span from phase Ib through confirmation trials for regulatory submission, but also adaptively and relatively seamlessly continue successful arms from earlier phases to subsequent ones (see Supplemental Box S1).
The primary goal is to provide patients early, appropriate, and sustained access to new medicines, based on credible, comparable evidence for all decision makers from sponsors to regulators, payers, providers, and patients. Secondary goals include: producing such evidence more efficiently and thereby decreasing exposure to ineffective or toxic regimens while reducing cost and time investments; increasing the number of studied medicines and their indications; optimizing the clinical regimens of individual medicines including dosing; and understanding the best combinations and sequences of treatments.
Core characteristics
We suggest that multiple PIPELINE styles could emerge, building on existing platform trials. Some may emphasize a single condition, and others might feature exploration of combinations and supplemental indications across conditions.
All the varieties share at least these characteristics:
Multiple investigational therapies studied comparatively as monotherapies, and when possible, in combination;
Multidevelopment phase scope from early development through confirmatory trials and into postapproval real‐world evidence;
Adaptive trial designs with individual arms being added, evaluated, modified, and terminated based on interim analyses, other studies within the coherent PIPELINE entity, and external data with analysis of statistical issues;
Standardization of selection criteria (stage, comorbidity, prior treatment, and others), screening protocols, diagnostic assays, biomarker analysis, allocation algorithms, statistical designs, regimens, and endpoint measurement;
Formal inferential linking between linked studies when appropriate, guided by standardized operating procedures, especially regarding population definitions, regimens, endpoints, and graduation criteria;
Systematic regimen exploration of variables including endpoints, biomarkers, safety monitoring, dosing, or treatment;
Shared control populations among investigational arms when appropriate;
Continuous enrollment operationally enabled by master protocols and informed consent processes that anticipate arm modifications, multiple reuse biobanking, and data transparency;
Operations infrastructure to streamline patient recruitment, data collection, and trial logistics;
Data exchange mechanisms to enable data pooling and information borrowing among willing participants.
Expanding umbrella designs
An umbrella‐based PIPELINE adaptively connects multiple single phase, umbrella trial platforms (Figure 3). In the breast cancer‐focused I‐SPY family, the individual platforms each employ adaptive patient enrollment. I‐SPY 2 is introducing SMART multipoint adaptive randomization. All are enabled by master protocols, and master informed consent forms and routinized patient screening.
Figure 3.
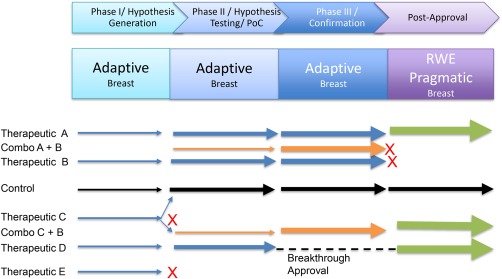
Adaptive umbrella‐based PIPELINEs test multiple therapeutics (blue) and combinations (orange) in parallel with a shared comparator arm (black), usually in a single broad indication with multiple sub‐populations. Adaptive randomization occurs within each phase with operational and inferential seamless graduation between phases. Real‐world evidence may continue the adaptive umbrella designs or use more classic designs. Not all indications succeed as indicated by “X”. Arrow thickness connotes patient numbers.
Predictable, rapid advancement between phases based on defined success criteria distinguish PIPELINEs from single stage platform trials. For example, in 2014 neratinib graduated from I‐SPY 2 to I‐SPY 3 when it met the Bayesian predictive probability threshold for success in a 300‐patient phase III trial in at least one of 10 predefined biomarker signatures.62, 63, 91
Extending into real‐world settings would enable PIPELINEs to provide increased evidence regarding patient quality of life, regimen titration, comparative effectiveness, and further subpopulation hypothesis generation.
EPAD (http://ep‐ad.org/) is an emerging Alzheimer's disease PIPELINE that links RWE with applied adaptive umbrella designs.92 Starting with a 24,000 pre‐Alzheimer's patient register, 6,000 will be invited into a research cohort for rich characterization including biomarker, cognitive, clinical, risk factor, and genetic data. Based on the hypotheses generated in that cohort, 1,500 will be invited into an early‐stage adaptive clinical trial to test Alzheimer's preventative agents. Adaptive platforms have also been developed in other therapeutic areas.93
These emerging, partial PIPELINE demonstrations illustrate feasibility and suggest potential for more complete adaptive, umbrella‐based PIPELINEs.
Expanding basket designs
Basket trials test multiple indications for one or more therapeutics such as Signature and MATCH, described above. A basket‐based PIPELINE (Figure 4) would couple a confirmatory, pivotal trial basket platform (as described in a companion article in this issue) with earlier stage baskets. TAPUR or similar real‐world approaches could generate hypotheses for baskets and continue to monitor approved indication baskets for effectiveness. Extensive off‐label use may not continue, given valid payer concerns, but real‐world evidence on biomarkers and comorbidities may likely provide many concepts worth testing. The coupling may be discrete, or more continuous, as implied in Figure 4.
Figure 4.
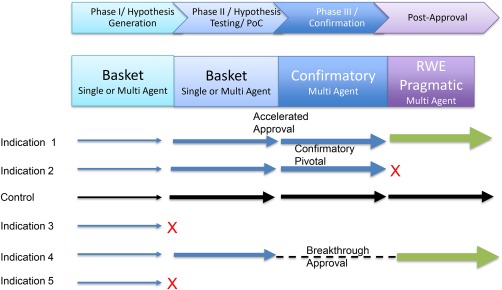
Basket‐based PIPELINE employs hypothesis generation designs early in development followed by confirmatory basket designs capable of creating pivotal data for regulatory submission. Real‐world evidence pragmatic designs could employ either basket or more classic approaches. Each row indicates a therapeutic being tested in an indication. Not all indications succeed as indicated by “X”. Arrow thickness connotes patient numbers. For simplicity, simultaneous multiple therapeutics are not shown.
As with all PIPELINEs, these would benefit from the operational innovations and allow compounds to pursue regulatory approval from individual indication arms whenever appropriate while continuing in others. Compared with umbrella‐based PIPELINEs, basket‐based leverage adds scope across indications or even therapeutic areas, providing more supplemental indication opportunities.
Establishing basket‐based platforms beyond cancer might prove attractive for any therapeutic areas with consistent underlying mechanisms, such as autoimmunity or infectious disease, e.g., multidrug‐resistant organism‐specific biomarkers could be leveraged across multiple infection sites to speed development of novel antibiotics.94 Extending beyond oncology to areas such as metabolic diseases will require richer phenotyping, including using insights from RWE hypothesis‐generating pilots.
Precision medicine presents unique challenges that PIPELINE scale and design mitigate.19 Preclinical models often do not recapitulate condition nuances, biomarker behavior, or both. For example, immuno‐oncology biomarkers can be confounded by variable immune response and cellular and cytokine interactions in the tumor microenvironment. Multiple candidate biomarker profiles, each rare, may require exploration. Biomarkers may need to include more than a single genetic aberration, protein, or other analyte. No biomarkers perform perfectly. The resulting false‐positives and false‐negatives present clinical, ethical, and economic challenges as well as scientific ones.34, 95, 96, 97 False‐positives expose nonresponders to side effects and may delay them seeking alternative, better treatment. False‐negatives may deny care to those who would benefit. If based on continuous biomarkers, such as protein or RNA expression levels, determining biomarker cutoffs can be difficult. Success can also be affected by unforeseen signaling pathway interaction and feedback. Intrapatient disease heterogeneity98 and evolutionary dynamics may affect progression and drug resistance, creating complex relationships between short‐term endpoints and ultimate clinical benefit.99 For these reasons, most biomarker development must occur in clinical trials simultaneously with therapeutic development.100 PIPELINEs address these issues with their multiarm, adaptive population assignment, multiagent designs, operational screening efficiencies for studying low prevalence subpopulations, and RWE trials that may allow the exploration of complex therapeutic strategies.
The full PIPELINE
The most ambitious PIPELINE variety (Figure 5) combines the prior varieties to creatively connect the individual platform advantages and to further increase scale. This ecosystem possesses larger trial infrastructure efficiency, data exchange, and knowledge creation potential. It integrates the seamless, efficient comparative evidence generation of adaptive umbrella designs with the multiple indication discovery and confirmation efficiency of the basket designs, and the combination and therapeutic lifespan regimen refinement evidence of big data‐enabled pragmatic and real‐world evidence monitoring studies. Combining these platforms should also increase the therapeutics studied and individual arms conducted. Portfolio decision analysis47, 101 and simulations would further suggest optimal trial sizes to avoid over accruing in any single deterministic or adaptive trial arm.
Figure 5.
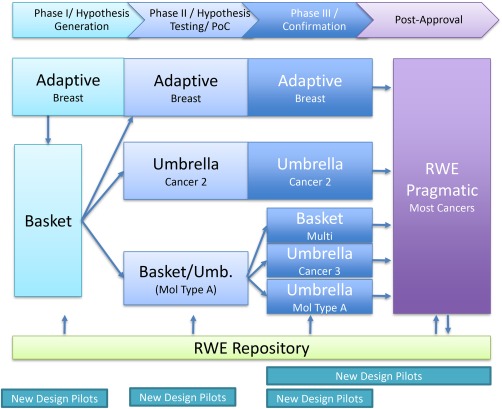
PIPELINE variety 3: integrated designs link basket, umbrella, adaptive and pragmatic platforms and pilot new designs that may be integrated such as the SMART design into I‐SPY2 adaptive platform. Phases are indicated by color. Abutting platforms use inferential and operational seamless graduation. RWE repositories aid all platforms in patient identification, population representativeness assessment, propensity scoring, historical control definitions, natural history understanding and hypothesis generation. New design pilots systematically test approaches and standard operating procedures prior to their incorporation into the PIPELINE.
The scope, scale, and standardization of PIPELINEs combined with their inherent design flexibility would facilitate examining exploratory biomarkers, regimens, and endpoints. Adaptive trials are designed to accelerate knowledge turns to provide more efficient learning with more efficient resource use. The integrated PIPELINE approach provides natural mechanisms to facilitate both forward and reverse adaptive feedback. As trial infrastructure and operational efficiencies grow, upgrading patient care systems to also provide GCP compliant data not only might avoid double data entry and reduce auditing costs, but also might enable large‐scale real‐world evidence collection, so that every patient has the potential to add to the evidence base, according to their consent. PIPELINE scale, standardization, and infrastructure could further multiply these effects. Nonetheless, pilots always will be needed to drive further innovations that if proven successful would be included in, or replace, existing PIPELINE components.
PIPELINEs must overcome multiple challenges to achieve these potential benefits. They require changes to traditional trial operations. Leadership activities from overall governance and trial design to standards setting and clinical site operations all expand under PIPELINEs. Statistical design, modeling, data handling rules, and analysis issues must be solved and implemented. Operating infrastructure from staffing to computer systems must efficiently support that expansion and add the needed capabilities such as continuous recruitment, screening, and enrollment.
We next discuss the feedback loops that PIPELINEs could initiate to propel their growth, examine the impacts on each stakeholder group to assess their motives to participate, and explore the governance, funding, and intellectual property issues PIPELINEs must navigate.
POSITIVE FEEDBACK LOOPS
Successful complex adaptive systems possess positive feedback loops that accelerate their adoption and sustain their stability. PIPELINEs benefit from three feedback loops that also reinforce each other.
Increased learning via standardization
Maintaining numerous standards are essential for PIPELINEs to generate data that are comparable, credible, and able to be pooled to provide key information for regulators, payers, providers, patients, scientists, and policy makers for their decision making. Without standards, PIPELINEs will replicate the unproductive variability of traditional, individual clinical trials.
Core standards include: endpoints and their measurement; master trial protocols and associated IRB processes; patient informed consent policies; patient population definitions, including condition staging, molecular typing, and other inclusion and exclusion criteria; and to the extent possible, the treatment regimens themselves. Standardization enables: sharing comparator arms among intervention arms; comparing intervention effects; pooling and information borrowing techniques to increase effect detection power; and switching comparator arms for analysis—even to previous intervention arms. Disciplined design and enforcement of standard operating procedures (SOPs) at the point of data collection through multisite aggregation and biostatistical analysis for the core data elements are critical for PIPELINE success.
PIPELINE standardization could advance companion diagnostics development, e.g., the unknown concordance of distinct PD1/PDL1 overexpression tests required a post‐hoc consortium to conduct the Blueprint concordance study.102, 103 A PIPELINE ecosystem might have catalyzed harmonization earlier due to its requirements for population definitions, and enabled comparisons more seamlessly through its ability to add alternative assays and exploratory biomarkers consistently across arms.
Efficacy endpoint variety exemplifies the potential benefits and challenges for PIPELINE standards. Stakeholder opinions vary when defining clinically meaningful improvements. Surrogate endpoints are particularly controversial, utilized for their timeliness and accessibility despite the risk of erroneous extrapolation. Today, without a required standardized endpoint set, development teams independently select endpoints and their measurement methods. The resulting diversity among trials diminishes meta‐analyses ability to inform efficacy, safety, and new hypothesis development.
PIPELINEs must employ certain core designs, protocols, and metrics to ensure comparability. Simultaneously, they must allow for additional monitoring, biomarkers, and regimen flexibility to accommodate the specific properties and clinical context of each candidate medicine. Standards evolution must also be anticipated (e.g., endpoints, biomarkers, regimens, or success metrics). PIPELINEs could provide broad evidence to inform those discussions by systematically introducing and comparing alternatives, often perhaps by initially adding elements to adapt to the specific needs of individual therapeutics. By considering flexibility to study new standards as an inherent design feature through pilots, PIPELINEs may include explicit evolution mechanisms, preventing the ossification that standardization could otherwise incur. Balancing innovative evolution with the benefits of historic standards for consistency and efficiency will be an ongoing PIPELINE challenge.
This interaction of standards and learning can create a self‐reinforcing feedback loop. Standards‐enabled credible, comparable, and fit‐for‐purpose evidence can motivate treatment guideline creators, regulators, and payers preferentially to seek and use such evidence and speed regulator and payer decision‐making. Sponsor participation incentives multiply for those with superior products who benefit from the more comparable, credible evidence to drive faster adoption, greater market share, and perhaps higher reimbursement.
Product sponsors, who lack early evidence of superiority, may decline to participate and might hamper adoption of PIPELINEs, but ultimately payers likely will demand comparative effectiveness evidence. Paradoxically, lower initial participation by these sponsors with marginal therapeutics may ultimately increase PIPELINE credibility. These positive and negative feedback loops should combine over time to impel increasing fractions of product sponsors to join PIPELINEs.
Increased learning via economies of scope
PIPELINEs span multiple therapeutics, combinations, regimens, and development phases to provide more learning opportunities for individual therapies and therapeutic areas than classic single therapeutic clinical trial approaches. Economists refer to cost improvements due to producing more than one product type as economies of scope.104, 105, 106 PIPELINE hypothesis‐generating baskets, cross‐arm meta‐analyses, and big data exploration of their RWE clinical repositories could generate more putative combinations, supplemental indications, and regimen refinements. PIPELINE standing confirmatory platforms and larger patient flows can then test those hypotheses faster. PIPELINE scope could enable testing the Chen‐Beckman counterintuitive simulation result that more, lower‐powered Proof‐of‐Concept trials generate more successes than fewer, better‐powered trials.47, 77, 78, 107
Multiple stakeholders will drive the feedback loops. Faster and more indications, combinations, and refined treatment regimens done with smaller arm sizes provide sponsor incentives.108 Academic participation will be rewarded with greater publication and funding opportunities. Regulators and payers will gain from more granular understanding of subpopulation, dosing, and combination effects. Patient accrual may increase from more personalized approaches. Stakeholder feedback loops from PIPELINE scope create a better ecosystem for patients seeking experimental therapies and for other stakeholders seeking to maximize learning from each participating patient.
Increased speed and efficiency via economies of scale and scope
In addition to the economies of scope from studying more therapeutics across more phases, indications, combinations, and regimens, PIPELINEs also generate economies of scale from simply increased volume in activities such as screening. Both types combine to drive operating efficiencies.
Today, each independent trial must receive IRB approval, contract sites, train those sites, institute patient recruitment processes, create infrastructure and implement procedures, among other activities. After design, it can require 2 years to open a trial for accrual, with North American median benchmarks ranging from 6–9 months, with Europe and Asia being even longer.109, 110, 111 Typically, insufficient staffing and inefficient procedures elongate patient recruitment times, with IMS Health median oncology phase II and III recruitment times in North America being 22.5 and 32 months, respectively. The treatment time (time from first dose to endpoint measurement) is then added. Figure 6 illustrates how operational efficiencies from efficient design of adaptive seamless platforms, with master protocols and continuous enrollment in a network, have reduced oncology development time in ISPY2/3 by about 4 years, from 117 months to 71 months, assuming 12‐month phase II and 24‐month phase III treatment periods. The time from opening to first and last patient recruitment for individual arms is 50–67% faster than the median benchmarks. Beyond these savings, biostatistics automation can reduce study result analysis time. Platforms reduce the planning time significantly but this time is not quantified in this analysis.
Figure 6.
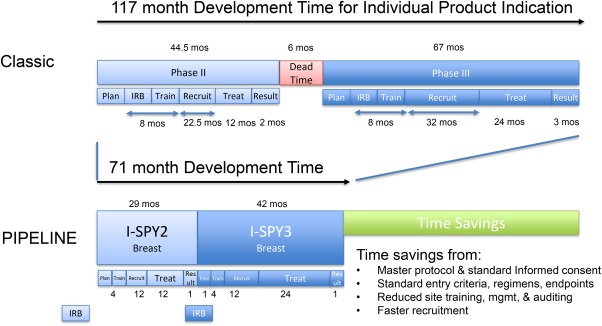
Operational efficiency leads to multiyear time‐saving potential. Classic case benchmark data from published IMS Health, Medidata, Tufts Center for Drug Development analysis and academic center studies. PIPELINE benchmarks taken from current I‐SPY platform performance levels.
These efficiencies create another positive feedback loop. PIPELINE therapeutic sponsors reach market sooner at lower cost than nonparticipants. Patients and clinical trialists both benefit from the improved care from more efficient operations, which may garner higher patient satisfaction and improved center reputation, which increase patient flows and center desire to affiliate.
Positive spiral from three feedback loops
Multiple positive feedback loops can create a positive spiral (Figure 7). Economies of scale that lower costs reinforce the economies of scope by enabling more indications, combinations, and refinements. Standardization, in turn, supports economies of scale and scope while benefiting from increased willingness to standardize due to those achievements.
Figure 7.
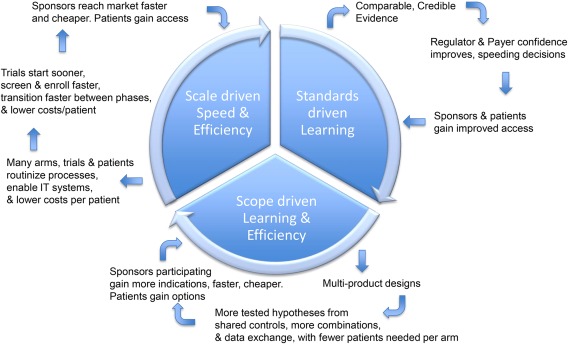
PIPELINE feedback loops create a virtuous cycle for their growth and sustainability by providing stakeholder benefits.
To begin this positive spiral, PIPELINEs must attain a minimal initial scale and demonstrated success. Thanks to initial federal and charitable funding, some component trial platforms such as Lung‐MAP, MATCH, and the I‐SPY platform family have begun building operating scale within their scope, but none have yet fully activated the positive spiral.
STAKEHOLDER AND SYSTEM NET IMPACTS
According to complex adaptive systems theory, PIPELINEs will develop and succeed only if they satisfy the needs of all stakeholders—in this case patients, providers, trialists, therapeutic developers, regulators, and payers. We speculate on impacts based on experience with PIPELINE prototypes and their components.
Patients would gain faster and more geographically distributed access to experimental therapies overall. Adaptive designs reduce patients exposed to standard‐of‐care, ineffective, or toxic agents by strategies that optimize tailoring available individual and combination therapies to more personalized subpopulations. Adaptive switching reassures patients that they can pursue alternative therapies if the initial arm proves unsuccessful. Comparative longitudinal evidence will provide better information for treatment decisions, informed by faster discovery of unanticipated side effects or benefits, including PROs from RWE approaches. PIPELINE cost and speed efficiencies will enable testing more therapies, combinations, and indications, ultimately resulting in more therapeutic options, especially for small population conditions. Efforts to improve the informed consent process and to include patient advocacy groups (disease‐based foundations, philanthropies, and public health advocates) in governance would enable patient influence, on the individual and collective level.
Provider and patient interests are largely aligned on the issues above, in particular comparative longitudinal evidence will engender more accurate and tailored treatment guidelines. As community centers join trial efforts, opportunities increase for research, publication, and leadership. Providers under shared risk contracts may also benefit from PIPELINE efficacy comparability, effectiveness tracking, and clinical utility information.
Clinical trialists benefit from the operational efficiencies that allow a larger number and greater variety of therapies and biomarkers, which could increase translational research opportunities. Increased competition from community trialists, decreased funding for investigator‐initiated trials, or both, could create tension. However, PIPELINEs provide new career paths in platform trial design, standards creation, operations management, and therapy development leadership. New models of academic credit may need to evolve, as in other fields where the value of team science is increasingly recognized.
Therapeutic developers, from industry, philanthropic, or publicly funded organizations, will benefit from PIPELINE speed and cost efficiencies. Commercial entities, the majority of sponsors, will gain revenue increases from earlier market entry, early mover market share increases, and more monotherapy and combination indications for smaller investment of money, time, and other resources. PIPELINE comparative evidence that includes cost‐effectiveness and PROs will also aid coverage and reimbursement negotiations with payers, at least for those developers with superior products. PIPELINE efficiencies do, however, require developers to relinquish some control over development because of the standardized features.
PIPELINE efficiencies could empower midsized biotechnology companies to develop their candidate therapies more independently, increasing innovation and competition with possible changes in commercial partnering patterns.
Regulators will benefit from the credible, comparable, and consistent generation of high‐quality evidence throughout the therapeutic lifespan, including postapproval. Industry funds most clinical trials, sometimes causing concerns of bias. Moving more clinical development to PIPELINEs that are standardized and semi‐independent may mitigate those concerns. Dossiers from umbrella and basket trials may allow clinical utility comparisons of therapeutics, subpopulations, and predictive biomarkers across indications.
Payers, health technology assessment agencies, and public health advocates benefit like regulators from the improved preapproval efficacy and safety evidence quality. In addition, the comparable, rigorous effectiveness evidence that PIPELINEs can generate as they extend into real‐world settings could help inform payers' coverage and reimbursement decisions. Credible PIPELINE evidence could also enhance patient and provider acceptance of those decisions. Expanding PIPELINE funding sources to include payers, health technology assessment programs, and others who would benefit from improved comparative evidence development might further reduce the developer bias concerns raised above.
Establishing endpoints and other aspects of these designs will require early regulatory and payer input, initially increasing their workloads, and more compounds and combinations will result in more applications. Once established, PIPELINEs should reduce individual application review workloads through familiarity and comparability, so the net impact on total health authority staffing needs is unclear.
Stakeholders generally share many goals. PIPELINEs can facilitate consensus regarding implementation of these goals through stakeholder collaboration on design strategy, standards, and other features with good governance and management.
OVERCOMING GOVERNANCE AND ORGANIZATIONAL CHALLENGES
Complex adaptive systems theory suggests that multiple PIPELINE structures will emerge, each tuned to characteristics of therapeutic areas, synergies with existing organizations, and individual leadership. Each PIPELINE needs a trusted managing entity to design and operate the components of its multisponsor, multiagent, multiphase system as well as fulfill the requirements of any clinical trial operator.
Balancing intellectual property rights
For straightforward monotherapy trial designs with arms that essentially parallel individual trials but with a shared comparator, intellectual property and financial issues need not be much more complex than independent proprietary trials.
For PIPELINE designs that combine information among the arms, or explore combination treatment in a single arm, the managing entity responsibilities concerning therapeutic selection, funding, and intellectual property expand. Clarity regarding which organizations have what rights to which data uses and any resulting intellectual property, as well as financial obligations, will require negotiation. Secure systems for data sharing patterned after FDA Sentinel,112 MIT Enigma projects,113, 114 or others, may help resolve some confidentiality concerns. Such negotiations have historically been artfully individualized processes. The scale and number of parties involved in a PIPELINE will require reasonably standard terms to provide fairness to all while maintaining situational flexibility and appropriate confidentiality for each therapeutic sponsor. No clear best model has yet emerged among the platform pilots. These negotiations remain idiosyncratic, difficult, and time‐consuming processes that require champions, creativity, openness, and trust to resolve for the PIPELINE overall and for each specific therapeutic.
Organizational structures
A PIPELINE managing entity may select among many organizational models and leaderships styles. Ideally, its processes should incorporate multiple stakeholder perspectives including sponsors, regulators, payers, providers, patient, public health officials, and academics, since each has interests that the PIPELINE must directly or indirectly satisfy.
PIPELINE managing entities may likely emerge from the existing platform trials, which often use an independent nonprofit structure (EORTC, I‐SPY, Lung‐MAP, NCI‐MATCH). Large philanthropic development organizations such as MMV (Medicines for Malaria Venture, http://www.mmv.org/) and the Gates Foundation (http://www.gatesfoundation.org) are in many ways creating PIPELINEs to serve developing country needs.115, 116, 117 Academic centers might orchestrate PIPELINEs, especially those mainly funded via NIH or philanthropy. Existing commercial contract research organizations, which have large networks of clinical trial sites, may attempt to evolve those networks into the more coordinated and standardized platforms that PIPELINEs require. While it appears unlikely to the authors, one could also imagine a single company with a large portfolio in a therapeutic area establishing a PIPELINE solely for its own use and that of its collaboration partners. Additionally, one might imagine an independent government operated PIPELINE similar in spirit to the NIH‐operated genomics national centers or NASA.
Each framework has advantages and disadvantages. For‐profit structures may raise the needed capital and talent more rapidly, but risk higher costs to sponsors and perhaps suspicion from other stakeholders, lowering their incentives to participate. Academic structures may generate greater scientific knowledge, but risk underemphasizing speed and overemphasizing academic rewards at the risk of not satisfying sponsors' needs. Nonprofit structures may combine either the best or worst features of the other models, but inherently face capital constraints and may have diminished initial access to academic clinical sites if they have no prior reputation. Government‐led PIPELINEs could quickly generate scale but government capacity to form, fund, and operate them currently appears limited.
The degree of centralization within each framework also poses trade‐offs. Those that emphasize collaboration could likely build greater credibility, but risk losing nimbleness, decisiveness, and operational excellence. Those that emphasize centralized control may risk the opposite.
Complex adaptive systems theory asserts that a vibrant ecosystem will experiment with multiple approaches in parallel and over time. Multiple PIPELINE structures should similarly be expected. Those ultimately successful may prove to have some inherent, fit‐for‐purpose structural advantage, but they just as likely may leverage success from stronger leadership, superior execution, higher initial funding, and first‐mover advantage starting the feedback loops sooner.
CONCLUSION
Concept summary
Benefits
PIPELINEs could improve patient access to therapies, increase the useful evidence for all stakeholders to employ in their decisions, and do so more efficiently and faster than a portfolio of individually crafted clinical trials. These shared goals would enable all stakeholders to engage in PIPELINE development in a collaborative Adaptive Biomedical Innovation framework.
Concept and proposal
The proposed PIPELINE approach employs a connected clinical knowledge engine from efficacy hypothesis testing (phase Ib/IIa) through real‐world effectiveness by expanding and adaptively linking basket, umbrella, adaptive, and real‐world evidence trial platforms. This would leverage clinical operations advances in master protocols, generalized informed consents, patient screening, patient record repositories, and the convergence of EHRs with clinical trial reporting systems and biostatistics analysis pipelines to simultaneously develop multiple therapies (as single agents and as concomitant or sequential combinations).
Feedback loops
Once initiated, PIPELINEs will likely generate three mutually reinforcing feedback loops that should create incentives for all stakeholders to participate and propel their growth. First, increased learning and efficiencies occur from standardization of master protocols, population definitions, screening, endpoints, data collection, and regimens. Second economies of scope arise from the variety of therapeutics, number of indications, and linked lifecycle phases. Finally, economies of scale ensue from patient volumes, shared control arms, and infrastructure.
Governance
PIPELINEs possess broad management responsibilities. Their governance structures must determine designs, select and enforce standards, set phase graduation requirements, and construct site networks. They must also implement infrastructure, from information systems to legal support. Pipeline managers must also build capabilities to solicit therapeutic developers, manage intellectual property rights, establish data transparency and publication protocols, and negotiate the financial structures to appropriately share costs and distribute any resulting gains. Multiple legal and management structures can be envisioned from the highly collaborative to command and control, from distributed to centralized, and from government to nonprofit to commercially led. It is too early to predict which forms will prove most effective. Complex adaptive systems theory suggests that multiple forms are likely feasible.
We also expect that multiple PIPELINEs may emerge and compete in any single, or across multiple, therapeutic areas. We view this competition as generally beneficial to the system, as long as PIPELINE minimum efficiency scale is maintained, although competition can prove uncomfortable at times for the participants. A balance must be maintained between operational efficiency and scope for individual creativity.
Extensions
While initially inspired by US oncology adaptive clinical trial platforms, PIPELINEs could extend globally and arise in many therapeutic areas including antiinfective, inflammatory, autoimmune, neurodegenerative, and perhaps psychiatric disorders.
Limitations and areas for future work and monitoring
To succeed, PIPELINEs must overcome multiple challenges and continue to advance operational, statistical, and organizational innovations. This article has only been able to address the major concepts and has drawn upon only a few of the largest emergent examples. Continued biostatistical advances are required, particularly in multiphase adaptive designs with therapeutic graduations and “information borrowing.” Operational scale efficiencies are not guaranteed. Further work will be required on patient screening and recruitment, rapid site start‐up, and information infrastructures. Operational efficiencies also will require disciplined PIPELINE governance in areas such as: setting appropriate standards based on well‐understood data handling rules; limiting core trial element complexity; focusing individual study customization on the most critical elements; and inducing high site performance through both rewards and penalties. PIPELINE organizational structures will require further elucidation to ensure adequate control while maintaining appropriate community participation and diversity of ideas, to fairly balance and adjudicate stakeholder interests, and to confirm sustainable financial models.
We believe PIPELINE organizational structures that engage patients as full research partners and formally include payer interests and concerns will prove critical to success. Drug development is a global activity and PIPELINEs too will need to include international sites and perspectives, which will add scale and scope, and will create challenges from the operational to the legal, cultural, and ethical. Allying with existing adaptive platforms such as I‐SPY and EPAD could prove productive first steps.
Developments in allied fields continue advancing rapidly. PIPELINEs must monitor and incorporate the best innovations while not becoming distracted. Areas of particular interest include: big data designs, methods and tools; patient reported outcomes; social media networks; remote patient monitoring; and precision medicine breakthroughs.
Call to action and suggested initial actions
The individually crafted “one drug, one population, one indication, one phase at a time” clinical trial system has diminishing returns in oncology and other therapeutic areas. It will not help reduce the time it takes to reach market nor enable more efficient evaluation of the promising funnel of agents. Adaptive clinical trial platforms, such as ISPY2, NCI‐MATCH, and Lung‐MAP in the US and EPAD in Europe point the way to the future. They exemplify a new response to the pressures of rapid scientific discoveries and stakeholder demands for more, and more credible, knowledge regarding benefits, harms, value, and uncertainties.
The growth of I‐SPY into a family of platforms and the rapid emergence of CoNNCT (Collaborative Novel‐Novel Combination Therapies) in response to NCI‐MATCH illustrate that evolution continues.118 To achieve the potential benefits for all stakeholders we suggest the following actions:
Continued support by and consultation with regulators.
Engagement of private and public payers directly or through funding bodies such as PCORI to help define PIPELINE study objectives and participate in developing patient screening and recruitment.
Public agency, foundation, and private support for research and pilots that address PIPELINE challenges related to biostatistical designs, operations, organizational structures, and information technology platforms.
Expanded professional organizational attention to PIPELINEs at meetings, working groups, and journals.
Policy changes to allow and then encourage platforms.
Investments to establish standard operating procedures for point‐of‐care information capture and data exchange for adaptive trials as a more precision medicine approach.
Catalytic funding to help existing platforms and new PIPELINEs reach sufficient scale to initiate the sustaining feedback loops.
Establishing PIPELINEs present significant challenges and will require adaptation by all stakeholders. On balance, we suggest that the PIPELINE potential for increased comparable and credible clinical knowledge for use by all stakeholders in their decision making to improve patient health outcomes is worth the investment.
CONFLICT OF INTEREST
MT is President of Co‐Bio Consulting, which provides management consulting services to biomedical firms. ZA is a full‐time employee of and stockholder in Amgen, Inc., and owns stock options in Cytel, Inc. RAB is a stockholder in Johnson & Johnson, Inc. He is the founder and Chief Scientific Officer of Onco‐Mind, LLC, founded to foster study of the effect of intratumoral heterogeneity and evolutionary dynamics on optimal personalized treatment strategies for cancer. CC is a full‐time employee of Merck & Co., Inc.
Supporting information
Supporting Information S1
Supporting Information S2
Supporting Information S3
Supporting Information S4
ACKNOWLEDGMENTS
The first two authors contributed equally to this work. We thank the following for their assistance and comments: Weili He and Jane Perlmutter.
References
- 1. Kinch, M.S. , Haynesworth, A. , Kinch, S.L. & Hoyer, D. An overview of FDA‐approved new molecular entities: 1827‐2013. Drug Discov. Today 19, 1033–1039 (2014). [DOI] [PubMed] [Google Scholar]
- 2. DiMasi, J.A. , Grabowski, H.G. & Hansen, R.W. Innovation in the pharmaceutical industry: new estimates of R&D costs. J. Health Econ. 47, 20–33 (2016). [DOI] [PubMed] [Google Scholar]
- 3. BIO . Clinical development success rates 2006–2015. Report 1–12 (2016). [Google Scholar]
- 4. Terry, C. & Lesser, N. Measuring the return from pharmaceutical innovation 2015 Transforming R & D returns in uncertain times. Deloitte Centre for Health Solutions. <https://www.bio.org/sites/default/files/Clinical Development Success Rates 2006‐2015 — BIO, Biomedtracker, Amplion 2016.pdf> (2015). [Google Scholar]
- 5. Lloyd, I. Pharma R & D Annual Review 2012 . <https://citeline.com/pharmaprojects‐pharma‐rd‐annual‐review‐2016/> (2016).
- 6. FDA . Novel Drugs 2015 Summary . www.Fda.Gov/Drugs. <http://www.fda.gov/downloads/Drugs/DevelopmentApprovalProcess/DrugInnovation/UCM481709.pdf> (2016).
- 7. Jenkins, J.K. 2015: Another Strong Year for Patients in Need of New Drug Therapies . <http://blogs.fda.gov/fdavoice/index.php/2016/01/2015‐another‐strong‐year‐for‐patients‐in‐need‐of‐new‐drug‐therapies> (2016).
- 8. Trusheim, M.R. & Berndt, E.R. The segmentation of therapeutic populations in oncology. Heal. Manag. Policy Innov. 1, 19–34 (2012). [Google Scholar]
- 9. Turkewitz, J. Patients seek ‘right to try’ new drugs. New York Times A16 (2015). [Google Scholar]
- 10. Relation between pharmaceuticals regulatory framework and timely access of medicines to patients? Reflection on difficulties and opportunities Summary of comments from member states. European Commission 1–10 (2014). <http://ec.europa.eu/health/files/committee/73meeting/pharm672_regulatory_framework_and_early_access_annex.pdf>. [Google Scholar]
- 11. Safra, E.J. & Rodwin, M.A. Independent drug testing to ensure drug safety and efficacy. J. Health Care Law Policy 18, 43 (2015). [Google Scholar]
- 12. Patients demand access to investigational therapies. Managed Care Executive. <http://managedhealthcareexecutive.modernmedicine.com/managed‐healthcare‐executive/news/patients‐demand‐access‐investigational‐therapies>. [Google Scholar]
- 13. Saville, B.R. & Berry, S.M. Efficiencies of platform clinical trials: A vision of the future. Clin. Trials 13, 358–366 (2016). [DOI] [PubMed] [Google Scholar]
- 14. Berry, S.M. , Connor, J.T. & Lewis, R.J. The platform trial: an efficient strategy for evaluating multiple treatments. JAMA 313, 1619–1620 (2015). [DOI] [PubMed] [Google Scholar]
- 15. Berry, D.A. Emerging innovations in clinical trial design. Clin. Pharmacol. Ther. 99, 82–91 (2016). [DOI] [PubMed] [Google Scholar]
- 16. Gallo, P. et al Adaptive designs in clinical drug development—an Executive Summary of the PhRMA Working Group. J. Biopharm. Stat. 16, 275–283; discussion 285–291, 293–298, 311–312 (2006). [DOI] [PubMed] [Google Scholar]
- 17. Yong Zang, J.J.L. Adaptive clinical trial designs in oncology. Chinese Clin. Oncol. 3, (2014). [DOI] [PubMed] [Google Scholar]
- 18. Legocki, L.J. et al Clinical trialist perspectives on the ethics of adaptive clinical trials: a mixed‐methods analysis. BMC Med. Ethics 16, 27 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Trusheim, M.R. , Berndt, E.R. & Douglas, F.L. Stratified medicine: strategic and economic implications of combining drugs and clinical biomarkers. Nat. Rev. Drug Discov. 6, 287–293 (2007). [DOI] [PubMed] [Google Scholar]
- 20. Suh, J.H. et al Comprehensive genomic profiling facilitates implementation of the national comprehensive cancer network guidelines for lung cancer biomarker testing and identifies patients who may benefit from enrollment in mechanism‐driven clinical trials. Oncologist 21, 684–691 (2016). doi:10.1634/theoncologist.2016-0030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Erdmann, J. All aboard: Will molecular tumor boards help cancer patients? Nat. Med. 21, 655–656 (2015). [DOI] [PubMed] [Google Scholar]
- 22. FDA . Sentinel program interim assessment.
- 23. Eichler, H.‐G. et al Adaptive licensing: taking the next step in the evolution of drug approval. Clin. Pharmacol. Ther. 91, 426–437 (2012). [DOI] [PubMed] [Google Scholar]
- 24. Eichler, H.‐G. et al From adaptive licensing to adaptive pathways: delivering a flexible life‐span approach to bring new drugs to patients. Clin. Pharmacol. Ther. 97, 234–246 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hirsch, G. & Trusheim, M.R. Referencing companion article on ABI.pdf. Clin. Pharmacol. Ther. (2016); e‐pub ahead of print. [Google Scholar]
- 26. Baird, L.G. , Trusheim, M.R. , Eichler, H.‐G. , Berndt, E.R. & Hirsch, G. Comparison of stakeholder metrics for traditional and adaptive development and licensing approaches to drug development. Ther. Innov. Regul. Sci. 47, 474–483 (2013). [DOI] [PubMed] [Google Scholar]
- 27. Gupta, A. & Anish, S. Insights from complexity theory: understanding organizations better. <http://tejas.iimb.ac.in/articles/12.php>.
- 28. Mitleton‐Kelly, E.T. (Ed.). Ten Principles of Complexity and Enabling Infrastructures Pergamon, Amsterdam (2003).
- 29. McClennan, B. Evolutionary Psychology: Complex Systems and Social Theory . <http://web.eecs.utk.edu/∼mclennan/papers/EPCSST.pdf>.
- 30. Rouse, W. Health care as a complex adaptive system: implications for design and management. In The Bridge (National Academy of Engineering ) 38, 17–25. pp. 7–25 (2008). [Google Scholar]
- 31. Rickles, D. , Hawe, P. & Shiell, A. A simple guide to chaos and complexity. J. Epidemiol. Community Health 61, 933–937 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bedard, P.L. , Hansen, A.R. , Ratain, M.J. & Siu, L.L. Tumour heterogeneity in the clinic. Nature 501, 355–364 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Trusheim, M.R. et al Quantifying factors for the success of stratified medicine. Nat. Rev. Drug Discov. 10, 817–833 (2011). [DOI] [PubMed] [Google Scholar]
- 34. Trusheim, M.R. & Berndt, E.R. The clinical benefits, ethics, and economics of stratified medicine and companion diagnostics. Drug Discov. Today 20, 1439–1450 (2015). [DOI] [PubMed] [Google Scholar]
- 35. Beckman, R.A. , Antonijevic, Z. , & Kalamegham, R. Adaptive design for a confirmatory Phase 3 basket trial in multiple tumor types based on a putatitive predictive biomarker. Clin. Pharmacol. Ther. (2016). [DOI] [PubMed] [Google Scholar]
- 36. Berry, S.M. , Broglio, K.R. , Groshen, S. & Berry, D.A. Bayesian hierarchical modeling of patient subpopulations: efficient designs of phase II oncology clinical trials. Clin. Trials 10, 720–734 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. McNeil, C. NCI‐MATCH launch highlights new trial design in precision‐medicine era. J. Natl. Cancer Inst. 107, (2015). [DOI] [PubMed] [Google Scholar]
- 38. Mullard, A. NCI‐MATCH trial pushes cancer umbrella trial paradigm. Nat. Rev. Drug Discov. 14, 513–515 (2015). [DOI] [PubMed] [Google Scholar]
- 39. Kang, B.P. et al The signature program: bringing the protocol to the patient. Clin. Pharmacol. Ther. 98, 124–126 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Herbst, R.S. et al Lung Master Protocol (Lung‐MAP)‐a biomarker‐driven protocol for accelerating development of therapies for squamous cell lung cancer: SWOG S1400. Clin. Cancer Res. 21, 1514–1524 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Redman, M.W. & Allegra, C.J. The master protocol concept. Semin. Oncol. 42, 724–730 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bretz, F. , Schmidli, H. , König, F. , Racine, A. & Maurer, W. Confirmatory seamless phase II/III clinical trials with hypotheses selection at interim: general concepts. Biomec. J. 48, 623–634 (2006). [DOI] [PubMed] [Google Scholar]
- 43. Berry, D.A. Bayesian clinical trials. Nat. Rev. Drug Discov. 5, 27–36 (2006). [DOI] [PubMed] [Google Scholar]
- 44. Kux, L. Draft Guidance for Industry and Food and Drug Administration Staff; eCopy Program for Medical Device Submissions; Availability. Fed. Regist. 77, 63837–63839 (2012). [Google Scholar]
- 45. FDA . Adaptive design clinical trials for drugs and biologics. Draft Guid. 50 <http://www.fda.gov/downloads/Drugs/.../Guidances/ucm201790.pdf> (2010). [DOI] [PubMed] [Google Scholar]
- 46. Menis, J. , Hasan, B. & Besse, B. New clinical research strategies in thoracic oncology: clinical trial design, adaptive, basket and umbrella trials, new end‐points and new evaluations of response. Eur. Respir. Rev. 23, 367–378 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Beckman, R.A. , Clark, J. & Chen, C. Integrating predictive biomarkers and classifiers into oncology clinical development programmes. Nat. Rev. Drug Discov. 10, 735–748 (2011). [DOI] [PubMed] [Google Scholar]
- 48. Yuan, J. et al Seamless phase IIa/IIb and enhanced dose‐finding adaptive design. J. Biopharm. Stat. 1–12 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bates, S.E. et al Advancing clinical trials to streamline drug development. Clin. Cancer Res. 21, 4527–4535 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Berry, D.A. Emerging innovations in clinical trial design. Clin. Pharmacol. Ther. 99, 82–91 (2016). [DOI] [PubMed] [Google Scholar]
- 51. McClellan, M. et al Pioneering Statistical Approaches to Accelerate Drug Development through Adaptive Trial Designs . <https://custom.cvent.com/00BF8C9066844371A697530EA9BB54B7/files/875adda52b2440afa33004ed4689f664.pdf> (2016).
- 52. Alexander, B.M. et al Biomarker‐based adaptive trials for patients with glioblastoma—lessons from I‐SPY 2. Neuro. Oncol. 15, 972–978 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Berry, D.A. How to take clinical research to the next level. Fortune Insider. <http://fortune.com/author/donald‐berry/> (2015). [Google Scholar]
- 54. Berry, D.A. The Brave New World of clinical cancer research: adaptive biomarker‐driven trials integrating clinical practice with clinical research. Mol. Oncol. 9, 951–959 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Esserman, L.J. A model for accelerating identification and regulatory approval of effective investigational agents. Cureus 4, (2012). [Google Scholar]
- 56. Esserman, L.J. et al Chemotherapy response and recurrence‐free survival in neoadjuvant breast cancer depends on biomarker profiles: results from the I‐SPY 1 TRIAL (CALGB 150007/150012; ACRIN 6657). Breast Cancer Res. Treat. 132, 1049–1062 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Esserman, L. The I‐SPY Master Trials: a model for accelerating the pace of getting the right drugs to the right patients. <http://vitaltransformation.com/wp-content/uploads/2014/10/Laura-Esserman-Innovation-in-Cliical-Trials-London-0ct-2014.pdf>.
- 58. Barker, A.D. et al I‐SPY 2: an adaptive breast cancer trial design in the setting of neoadjuvant chemotherapy. Clin. Pharmacol. Ther. 86, 97–100 (2009). [DOI] [PubMed] [Google Scholar]
- 59. Esserman, L.J. & Woodcock, J. Accelerating identification and regulatory approval of investigational cancer drugs. JAMA 306, 2608–2609 (2011). [DOI] [PubMed] [Google Scholar]
- 60. Yee, D. et al Adaptive trials in the neoadjuvant setting: a model to safely tailor care while accelerating drug development. J. Clin. Oncol. 30, 4584–4586 (2012). [DOI] [PubMed] [Google Scholar]
- 61. Esserman, L. I‐SPY 2 & 3 Trials . <http://www.nationalacademies.org/hmd/∼/media/Files/Activity Files/Quality/VSRT/IC Meeting Docs/DLC 5‐30‐14/Davis.pdf>.
- 62. Al, H.S.R. et al I‐SPY 2 TRIAL adaptive randomization of veliparib‐carboplatin treatment in breast cancer. N. Engl. J. Med. 375, 23–24. (July 7, 2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Al, P. et al I‐SPY 2 TRIAL adaptive randomization of neratinib in breast cancer. N. Engl. J. Med. 375, 11–12 (July 7, (2016). [Google Scholar]
- 64. Almirall, D. , Nahum‐Shani, I. , Sherwood, N.E. & Murphy, S.A. Introduction to SMART designs for the development of adaptive interventions: with application to weight loss research. Transl. Behav. Med. 4, 260–274 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Almirall, D. & Blalock, J.A. Summary of the SRNT treatment network Webinar: getting SMART about developing individualized sequences of health interventions. Nicotine Tob. Res. 16, 252 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Lei, H. , Nahum‐Shani, I. , Lynch, K. , Oslin, D. & Murphy, S.A. A ‘SMART’ design for building individualized treatment sequences. Annu. Rev. Clin. Psychol. 8, 21–48 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Ausiello, D. & Lipnick, S. Real‐time assessment of wellness and disease in daily life. Big Data 3, 203–208 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Zang, Y. & Lee, J.J. Adaptive clinical trial designs in oncology. Chinese Clin. Oncol. 3, 49 (2014). [DOI] [PubMed] [Google Scholar]
- 69. Goettsch, W. & Makady, A. IMI GetReal‐Project No. 115546 WP1: Deliverable D1.3: Glossary of Definitions of Common Terms . <https://www.imi-getreal.eu/LinkClick.aspx?fileticket=PfkLm8v32KM%3d&portalid=1>. (2015).
- 70. Eichler, H.‐G. et al Bridging the efficacy‐effectiveness gap: a regulator's perspective on addressing variability of drug response. Nat. Rev. Drug Discov. 10, 495–506 (2011). [DOI] [PubMed] [Google Scholar]
- 71. Selker, H.P. et al A proposal for integrated efficacy‐to‐effectiveness (E2E) clinical trials. Clin. Pharmacol. Ther. 95, 147–153 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Herbst, R. et al Design of a Disease‐Specific Master Protocol. Issue Br. Conf. Clin. Cancer Res. 1–11 (2012). [Google Scholar]
- 73. Ledford, H. ‘Master protocol’ aims to revamp cancer trials. Nature 498, 146–147 (2013). [DOI] [PubMed] [Google Scholar]
- 74. Herbst, R. et al Design of a disease‐specific master protocol. Issue brief. Conference on clinical cancer research. November 2012. Friends of Cancer Research (2012). [Google Scholar]
- 75. Precision Medicine Initiative (PMI) Working Group . The precision medicine initiative cohort program — building a research foundation for 21st century medicine. Precision Medicine Initiative (PMI) Working Group Report to the Advisory Committee to the Director, NIH Sept 17, (2015).
- 76. Lacombe, D. et al European perspective for effective cancer drug development. Nat. Rev. Clin. Oncol. 11, 1–7 (2014). [DOI] [PubMed] [Google Scholar]
- 77. Chen, C. & Beckman, R.A. Maximizing return on socioeconomic investment in phase II proof‐of‐concept trials. Clin. Cancer Res. 20, 1730–1734 (2014). [DOI] [PubMed] [Google Scholar]
- 78. Chen, C. & Beckman, R.A. Optimal cost‐effective designs of phase II proof of concept trials and associated go‐no go decisions. J. Biopharm. Stat. 19, 424–436 (2009). [DOI] [PubMed] [Google Scholar]
- 79. Chen, C. & Beckman, R.A. Optimal cost‐effective go‐no go decisions in late‐stage oncology drug development. Stat. Biopharm. Res. 1, 159–169 (2009). [Google Scholar]
- 80. Graf, A. , Posch, M. & König, F. Adaptive designs for subpopulation analysis optimizing utility functions. Biometr. J. 57, 76–89 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Ondra, T. et al Optimizing trial designs for targeted therapies. oral presentation at 51st annual Drug Information Association (DIA) meeting, June 14‐18, 2015, Washington, DC (2015).
- 82. Ondra, T. et al Optimizing trial designs for targeted therapies. ArXiv ID 1606.03987 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Embi, P.J. et al Effect of a clinical trial alert system on physician participation in trial recruitment. Arch. Intern. Med. 165, 2272–2277 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Long, J. , Hulse, N.C. & Tao, C. Infobutton usage in Patient Portal MyHealth. AMIA Jt. Summits Transl. Sci. Proc. AMIA Summit Transl. Sci. 112–116 (2015). [PMC free article] [PubMed] [Google Scholar]
- 85. Kermani, F. Database exploitation could slash clinical trial durations, cut costs. The Pink Sheet Daily. Article 14141029001. (2014). [Google Scholar]
- 86. FDA pressuring sponsors to go digital in case report forms. Clin. Trials Advis. 18, 1–4 (2013). [Google Scholar]
- 87. FDA . Guidance for Industry: electronic source data in clinical investigations. <http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm328691.pdf> (2014).
- 88. Warner, J.L. et al Development, implementation, and initial evaluation of a foundational open interoperability standard for oncology treatment planning and summarization. J. Am. Med. Inform. Assoc. 22, 577–586 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Wiley, A. NCI Wiki: caIntegrator . <https://wiki.nci.nih.gov/display/caIntegrator/caIntegrator;WIKISESSIONID=D3560EA9100ACA3A8E4A7E7B54B8B24D> (2015).
- 90. Jackel, L. NCI Wiki. caIntegrator Retirement Announcement . <https://wiki.nci.nih.gov/display/caIntegrator/caIntegrator+Retirement+Announcement;WIKISESSIONID=B5DFF09AEC12A1F6302D9A850324EF9F> (2016).
- 91. Neratinib Graduates to I‐SPY 3. Cancer Discov. 4, 624 (2014). [DOI] [PubMed] [Google Scholar]
- 92. Stephenson, D. et al Charting a path toward combination therapy for Alzheimer's disease. Expert Rev. Neurother. 15, 107–113 (2015). [DOI] [PubMed] [Google Scholar]
- 93. Berry, S.M. et al A response adaptive randomization platform trial for efficient evaluation of Ebola virus treatments: a model for pandemic response. Clin. Trials 13, 22–30 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Infectious Diseases Society of America . White paper: recommendations on the conduct of superiority and organism‐specific clinical trials of antibacterial agents for the treatment of infections caused by drug‐resistant bacterial pathogens. Clin. Infect. Dis. 55, 1031–1046 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Dabbs, D.J. et al High false‐negative rate of HER2 quantitative reverse transcription polymerase chain reaction of the oncotype DX test: An independent quality assurance study. J. Clin. Oncol. 29, 4279–4285 (2011). [DOI] [PubMed] [Google Scholar]
- 96. Eberhard, D.A. et al Mutations in the epidermal growth factor receptor and in KRAS are predictive and prognostic indicators in patients with non‐small‐cell lung cancer treated with chemotherapy alone and in combination with erlotinib. J. Clin. Oncol. 23, 5900–5909 (2005). [DOI] [PubMed] [Google Scholar]
- 97. Perez, E.A. et al HER2 testing in patients with breast cancer: poor correlation between weak positivity by immunohistochemistry and gene amplification by fluorescence in situ hybridization. Mayo Clin. Proc. 77, 148–154 (2002). [DOI] [PubMed] [Google Scholar]
- 98. Yates, L.R. et al Subclonal diversification of primary breast cancer revealed by multiregion sequencing. Nat. Med. 21, 751–759 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Beckman, R.A. , Schemmann, G.S. & Yeang, C.‐H. Impact of genetic dynamics and single‐cell heterogeneity on development of nonstandard personalized medicine strategies for cancer. Proc. Natl. Acad. Sci. U. S. A. 109, 14586–14591 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Trusheim, M.R. , Austin, M.J.F. , Rausch, C. & Berndt, E.R. Uncertain prognosis for high‐quality diagnostics: clinical challenges, economic barriers and needed reforms. Pharmacogenomics 14, 325–334 (2013). [DOI] [PubMed] [Google Scholar]
- 101. Antonijevic, Z. The impact of adaptive design on portfolio optimization. Ther. Innov. Regul. Sci. 50, 615–619. (2016). doi:10.1177/2168479016640020. [DOI] [PubMed] [Google Scholar]
- 102. AACR . A blueprint proposal for companion diagnostic comparability. <http://www.fda.gov/downloads/MedicalDevices/NewsEvents/WorkshopsConferences/UCM439440.pdf>.
- 103. Schalper, K.A. , Kaftan, E. & Herbst, R.S. Predictive biomarkers for PD‐1 axis therapies: the hidden treasure or a call for research. Clin. Cancer Res. 22, 2102–2104 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Willig, J.C.P.R.D. Economies of scale in multi‐output production. Q. J. Econ. 91, 481–493 (1977). [Google Scholar]
- 105. Willig, J.C.P.R.D. Economies of scope. Am. Econ. Rev. 71, 262–272 (1981). [Google Scholar]
- 106. Chandler, A.D. & Takashi, H. Scale and scope: The dynamics of industrial capitalism Harvard University Press, Cambridge, MA, (2009). [DOI] [PubMed]
- 107. Beckman, R.A. & Chen, C. Translating predictive biomarkers within oncology clinical development programs. Biomark. Med. 9, 851–862 (2015). [DOI] [PubMed] [Google Scholar]
- 108. Trusheim, M.R. & Berndt, E.R. Economic challenges and possible policy actions to advance stratified medicine. Per. Med. 9, 413–427 (2012). [DOI] [PubMed] [Google Scholar]
- 109. Getz, K. , Lamberti, M.J. & Tufts Center for the Study of Drug Development . In Parexel Biopharmaceutical R&D Statistical Sourcebook 2015/2016 p. 337. PAREXEL International Corporation, Waltham, MA, USA.
- 110. Various enrollment benchmarks from StudyOptimizer. In Parexel Biopharmaceutical R&D Statistical Sourcebook 292–296. PAREXEL International Corporation, Waltham, MA, USA.
- 111. Winhusen, T. Not getting lost in translational science: A tool for navigating the pre‐implementation phase of multi‐site pharmacological clinical trials. Appl. Clin. Trials 23, 36–39 (2014). [PMC free article] [PubMed] [Google Scholar]
- 112. Curtis, L.H. et al Design considerations, architecture, and use of the Mini‐Sentinel distributed data system. Pharmacoepidemiol Drug Saf. 21, 23–31 (2012). [DOI] [PubMed] [Google Scholar]
- 113. Zyskind, G. , Nathan, O. & Pentland, A. Enigma: decentralized computation platform with guaranteed privacy. <http://enigma.media.mit.edu>. (2015).
- 114. MIT Internet Trust Consortium . Project Enigma . <http://www.mit‐trust.org/projects/>.
- 115. Wells, T.N.C. , Alonso, P.L. & Gutteridge, W.E. New medicines to improve control and contribute to the eradication of malaria. Nat. Rev. Drug Discov. 8, 879–891 (2009). [DOI] [PubMed] [Google Scholar]
- 116. Bathurst, I. & Hentschel, C. Medicines for Malaria Venture: sustaining antimalarial drug development. Trends Parasitol. 22, 301–307 (2006). [DOI] [PubMed] [Google Scholar]
- 117. Gates Foundation . Discovery & Translational Sciences Strategy Overview. <http://www.gatesfoundation.org/What-We-Do/Global-Health/Discovery-and-Translational-Sciences>.
- 118. Chang, D. , Scarlett, U. , Murtagh, T. & Flaherty, K. The CoNNCT Initiative: accelerating novel combinations for cancer. In Vivo (Brooklyn). 34, (2016). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information S1
Supporting Information S2
Supporting Information S3
Supporting Information S4