

Structured Abstract
Purpose
High-throughput sequencing (HTS) of immunoglobulin heavy chain genes (IGH) in unselected clinical samples for minimal residual disease (MRD) in B lymphoblastic leukemia (B-ALL) has not been tested. As current methods for MRD detection, such as flow cytometry or patient-specific qPCR are complex or difficult to standardize in the clinical laboratory, sequencing may enhance clinical prognostication.
Experimental design
We sequenced IGH in paired pre- and day 29 post-treatment samples using residual material from consecutive, unselected samples from Children's Oncology Group AALL0932 trial to measure MRD as compared to flow cytometry. We assessed the impact of on-going recombination at IGH on MRD detection in post-treatment samples. Lastly, we evaluated a subset of cases with discordant MRD results between flow cytometry and sequencing.
Results
We find clonal IGH rearrangements in 92 of 98 pre-treatment patient samples. Further, while on-going recombination of IGH was evident, index clones typically prevailed in MRD-positive post-treatment samples, suggesting that clonal evolution at IGH does not contribute substantively to tumor fitness. MRD was detected by sequencing in all flow cytometry positive cases with no false negative results. Additionally, in a subset of patients, MRD was detected by sequencing, but not by flow cytometry, including a fraction with MRD levels within the sensitivity of flow cytometry. We provide data that suggest that this discordance in some patients may be due to the phenotypic maturation of the transformed cell.
Conclusion
Our results provide strong support for high-throughput sequencing of IGH to enhance clinical prognostication in B-ALL.
Keywords: Leukemias and lymphomas, Minimal residual disease, high-throughput sequencing, IGH
Introduction
B lymphoblastic leukemia/lymphoma (B-ALL) is an aggressive, immature B-cell neoplasm in which substantial progress in pediatric and adult outcomes has been achieved (1). In addition to conventional prognostic factors, such as cytogenetics and karyotype, post-treatment detection of minimal residual disease (MRD) in B-ALL is important for identifying patients who may require treatment intensification (2-7). MRD measurement is accepted for prognostication in pediatric B-ALL, and may also surpass other prognostic markers in adult B-ALL (2-4, 7-9). However, while MRD assessment by flow cytometry or qPCR has been incorporated widely in clinical studies, broader implementation has been limited by the challenges of ensuring accurate test performance in the clinical laboratory. For flow cytometry, MRD assessment is confounded by observer variation due to difficulty of consistently interpreting complex multi-dimensional data in the context of therapy-induced immunophenotypic drift and marrow regeneration. For qPCR, in which patient-specific clonal rearrangements of the immunoglobulin heavy chain (IGH) or T-cell receptors (TRG, TRB, TRD, and TRA) are tracked, MRD assessment requires significant laboratory and institutional commitment to maintain staffing to ensure continuity and quality of testing.
We recently reported the first application of high-throughput sequencing for the assessment of MRD in acute T-lineage lymphoblastic leukemia, demonstrating the potential of sequencing to improve clinical prognostication (10). Subsequently, Faham et al. demonstrated the feasibility of sequencing for MRD detection in B-ALL (11). However, their study did not appear to evaluate the potential, widespread applicability of sequencing IGH for MRD detection in B-ALL, because they required all samples to have a known clonal IGH gene rearrangement for inclusion. Gawad et al. recently reported evidence for massive clonal evolution (on-going recombination) in IGH as detected by deep sequencing (12). However, they focused primarily on diagnostic B-ALL samples and did not systematically evaluate the impact of this on-going recombination of IGH on MRD detection.
Here, we report on high-throughput sequencing of IGH for MRD in B-ALL using residual material from unselected samples derived from patients enrolled in Children's Oncology Group clinical trial AALL0932. We show that high-throughput sequencing of IGH is broadly applicable in most patients with B-ALL (91 of 98 patients considered), and find that sequencing detected MRD in all cases that were positive by flow cytometry, with no false negative results. We further demonstrate the use of a synthetic immune system to minimize bias and ensure accurate results in multiplexed PCR. Notably, given the enhanced precision and specificity of high-throughput sequencing, we provide the first comprehensive assessment of clonal evolution of IGH during induction therapy. Lastly, we show that in rare cases, index clonal IGH sequences may be identified within an immunophenotypically mature B-cell population in post-treatment samples, suggesting that in some patients, tumor cells may undergo therapy-induced immunophenotypic maturation. Taken together, our data suggest that high-throughput sequencing of IGH could significantly improve clinical prognostication in B-ALL.
Materials and Methods
Clinical samples
Clinical samples (pre-treatment: peripheral blood or bone marrow aspirate; and day 29 post-treatment: bone marrow aspirates) were collected from 99 individuals with informed consent by local clinicians, as part of enrollment into Children's Oncology Group Study AALL0932 (registered as #NCT01190930 (13)). All patients had a diagnosis of B-ALL established locally, and then COG-coded pre- and post-treatment samples were submitted to the University of Washington Hematopathology Laboratory for routine flow cytometry analysis for MRD. Samples were not otherwise pre-selected, such as on the basis of having prior evidence of a clonal IGH gene rearrangement (11).
Flow cytometry
Multi-parametric flow cytometry was performed at the University of Washington as part of the routine evaluation for MRD (14). Samples were labeled with the antibody combinations: CD20 FITC, CD10 PE, CD38 PerCP-Cy5.5, CD19 PE-Cy7, CD58 APC, and CD45 APC-H7 and 2) CD9 FITC, CD13/CD33 PE, CD34 PerCP-Cy5.5, CD19 PE-Cy7, CD10 APC and CD45 APC-H7, and processed using NH4Cl+0.25% formaldehyde for red cell lysis with >750,000 events acquired on a Becton-Dickinson LSRII. Clusters of events differing from normal B-cell maturation were defined as MRD by B.W., and quantified relative to total mononuclear cells and total nucleated cells. Data were analyzed using Woodlist software version 2.7 (in-house software written by B. Wood, equivalent software is commercially-available) (14).
High-throughput sequencing
Sequencing CDR3 regions
IGH CDR3 regions were amplified and sequenced from 400 ng of pre-treatment and 6 μg of Day 29, post-treatment DNA samples, or in a subset of cases using all available extracted DNA. Amplification and sequencing of IGH CDR3 regions was carried out using the ClonoSEQ™ platform (Adaptive Biotechnologies, Seattle, WA) in which a set of multiplexed forward primers matching V and D gene segment sequences are combined with a set of reverse primers matching J gene segment sequences to amplify both mature VDJ and immature DJ IGH rearrangements. Sequencing was performed starting from the J gene segment and extending 87 base pairs into the CDR3 region. Sequences for IGH CDR3 regions were delineated according to criteria established by the International ImMunoGeneTics collaboration with a standard algorithm to identify V, D, and J gene segments (15, 16). Rearranged CDR3 sequences were classified as non-productive if insertions or deletions were identified that resulted in frame-shifts or premature stop codons.
Identifying CDR3 sequences and defining clonality
DNA from bone marrow isolated from 9 healthy individuals was used to amplify IGH sequences using the same protocol as the day 29, post-treatment B-ALL samples. The frequency of the most common IGH gene rearrangement from the marrow aspirate of these individuals averaged 0.08±0.04% of total nucleated cells (mean±SD) (Supplementary Figure 1). For this study, a very conservative definition of a neoplastic clone was chosen as representing a minimum of 10% of nucleated cells. For samples in which the two most common IGH sequences were of comparable frequency, bi-allelic rearrangement was assumed with both sequences considered to represent the lymphoblast clone.
In day 29 post-treatment samples, MRD was identified by searching for CDR3 sequences that identically matched the clonal sequence defined in pre-treatment samples, requiring a complete (87 base pair) match. Both the presence and the frequency of the MRD clone relative to the total IGH repertoire and total nucleated cells were determined. To determine if the clonal CDR3 sequences identified were specific, all post-treatment samples were screened for the presence and frequency of all identified pre-treatment IGH CDR3 clones (17).
Use of a synthetic template to minimize bias in multiplexed PCR
Since accurate quantification of lymphoblast clones for MRD detection is critical, we developed an approach to ensure minimal bias in multi-plex PCR (17). Briefly, each potential VDJ rearrangement of the IGH locus contains one of nine J segments, one of 27 D segments and one of 124 V segments, many of which have disparate nucleotide sequences. In order to amplify all possible VDJ combinations, we used a single tube, multiplex PCR assay with 84 V and 15 D forward and 9 J reverse primers. To remove potential PCR bias, every possible V-J and D-J pair was chemically synthesized as a template with specific barcodes (17). These templates were engineered so as to be recognizable as non-biologic and have universal 3′ and 5′ ends to permit amplification with universal primers and subsequent quantification by HTS. This synthetic immune system can then be used to calibrate the multiplex PCR assay. Iteratively, the multiplex pool of templates is amplified and sequenced with our IGH V/D- and J-specific primers, and the primer concentrations are adjusted to re-balance PCR amplification (Supplementary Figure 2). Once the multiplex primer mixture amplifies each V and J template nearly equivalently, residual bias is removed computationally.
Quantifying the fraction of B cells in bone marrow or blood
The ClonoSEQ™ assay amplifies and sequences rearranged IGH molecules. We quantify the amount of DNA input into the assay and convert this to the equivalent total number of nucleated cells, assuming ∼6.4 pg genomic DNA per diploid cell. ClonoSEQ™ then amplifies and sequences the molecules with rearranged IGH. Since the ClonoSEQ™ assay includes analysis of DJ rearrangements, most B cells produce two sequences, one of which is a complete VDJ rearrangement and the other either a VDJ or an incomplete DJ rearrangement. To estimate the number of starting templates with IGH rearrangements that were in the sample, the average number of sequence reads for each starting template is measured. Synthetic control templates are also spiked at limiting dilution into each sample such that each template may be present at most as a single copy. One then can compute the average number of reads for each sequenced spiked synthetic template. The total number of B cells sequenced is then derived as the total number of sequencing reads divided by this computed average reads per template (fold-coverage), further divided by two, because there are two alleles per B cell. The total fraction of B cells in the mixture is this number of B cells divided by the total number of input cells as determined by DNA input quantity. This method is precise across a large range of initial B cell fractions (Supplementary Figure 3).
Identifying evolved IGH clonotypes
To detect on-going recombination at the IGH locus (so-called clonal evolution), we applied the algorithm proposed by Gawad et al. (12). Briefly, this approach defines evolved clonotypes of IGH as those that: (i) share an identical JH sequence with the index clone, (ii) share at least six bases of identical NDN sequence with the index clone (starting from the J-segment boundary, with D-segment matches collapsed to two effective NDN bases), and (iii) are annotated with a different VH gene than the index clone (12). We verified the specificity of this approach using cross-patient comparisons and find that among clones that share a JH sequence, only 0.005% were considered “evolved” in other patients, as compared to 60.9% of JH-matching clones within the same patient, confirming that this algorithm has a low false positive rate (12).
Results
Using unselected, consecutive residual material derived from 99 paired pre- and day 29 post-treatment B-ALL samples from patients enrolled in the Children's Oncology Group AALL0932, we sequenced the complementarity-determining region 3 (CDR3) of IGH using the equivalent of approximately 300,000 genomes for pre-treatment samples and 1,000,000 genomes for day 29, post-treatment samples. One case unexpectedly failed at the outset with inadequate DNA from the extraction of the pre-treatment sample and was excluded from further consideration. The pre-treatment sequencing analysis permitted us to define for 91 of 98 patients the unique, recombined IGH gene sequences (V-DJ complete rearrangements > 10%), representing the patient's clonal, neoplastic B lymphoblasts (Table 1, Figure 1A). The remaining seven patients without a B lymphoblast clone with a complete IGH rearrangement of 10% or higher of total cells in the bone marrow were studied individually. One patient had a large B lymphoblast clone with two D-J rearrangements, but no complete V-DJ IGH rearrangements. A second patient had a clone comprising ∼4% of nucleated cells in the bone marrow, which still represents a statistically significant expansion compared to healthy donors (Supplementary Figure 1), and this value agreed with the corresponding flow cytometry result. The remaining 5 patient samples did not have an identifiable clonal IGH sequence at diagnosis, defined as that comprising more than 10% among total nucleated cells of a complete (V-J) or incomplete D-J IGH sequence, and were not considered further herein. By flow cytometry, these 5 cases did not have a distinguishing or common pre-treatment immunophenotype (Supplementary Figure 4, Supplementary Table 1). Thus, of 98 unselected samples, 92 cases at diagnosis had a clonal IGH complete or incomplete DJ-rearrangement meeting our definition of a clonal B-cell population comprising more than 10% of total cells, with one additional case having a clonal IGH rearrangement at 4%, matching flow cytometry.
Table 1. Summary of 99 consecutive patient cases.
| Failed initial DNA extraction | 1 |
| Adequate DNA | 98 |
| Pre-treatment sequencing | |
| Pre-treatment clonal IGH (V-J) | 91 |
| Pre-treatment clonal IGH (D-J) only | 1 |
| Pre-treatment bi-allelic IGH | 26* |
| Post-treatment MRD | |
| HTS+/mpFC+ post-treatment MRD | 23 |
| HTS+/mpFC− post-treatment MRD | 28 |
| HTS−/mpFC− post-treatment MRD | 40 |
| 8 additional mpFC–negative cases, triple-flow cytometry sorted for HTS | |
| Evidence of clonal, pre-treatment IGH in sorted mature B-cell fraction | 1 |
| No evidence of clonal, pre-treatment IGH in sorted mature B-cell fraction in samples with adequate DNA | 7 |
Adequate DNA was only available to test for IGH D-J rearrangements (and therefore bi-allelic samples) in 84 of the 98 cases. Samples with three or more highly expanded IGH sequences were also not considered bi-allelic.
Figure 1. Pre- and day 29 post-treatment B lymphoblast frequencies by high-throughput sequencing (HTS) versus multi-parametric flow cytometry (mpFC).
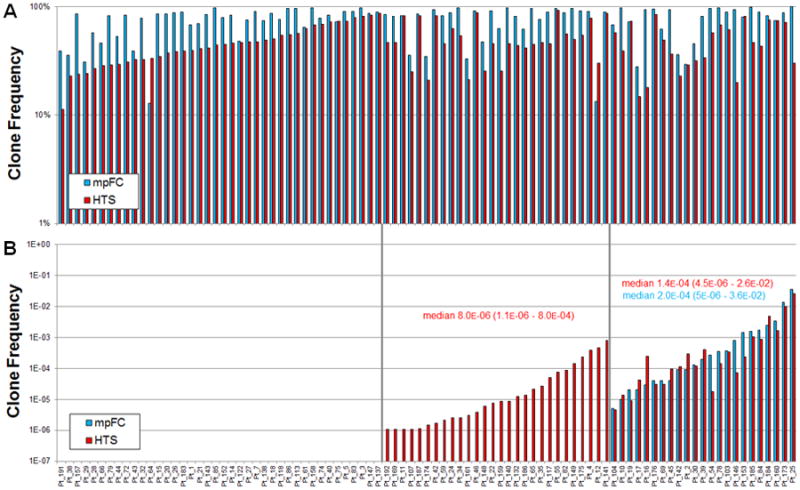
Pre-treatment and Day 29, post-treatment, clonal B lymphoblasts were identified by HTS (red) or by mpFC (blue) (N=91). HTS data are reported as the frequency of the clonal sequence of total rearranged IGH sequences; mpFC data are reported as the percentage of total nucleated cells by mpFC. Cases without a complete IGH gene rearrangement in pre-treatment samples (n=7) are not shown. (A) High-throughput sequencing identifies pre-treatment, clonal IGH sequences in 91 of 98 patients. (B) High-throughput sequencing of IGH identifies post-treatment MRD detected by flow cytometry and in an additional subset of patients for whom flow cytometry analysis was negative. Day 29, post-treatment samples (n=91) includes only those cases with complete V-J rearrangement and excludes 1 case with only a D-J rearrangement and 5 cases for which no clonal IGH rearrangement was seen. Three subsets of cases are identified in post-treatment samples (left to right): 1) MRD not detected by either HTS or mpFC, 2) MRD detected by HTS, but not by mpFC, and 3) MRD detected by both HTS and mpFC.
84 samples were examined with an assay designed to amplify V-DJ and D-J rearrangements.58 of these samples had at least two IGH sequences with frequencies higher than 10%: 26 had two clonal IGH above 10% and the third most frequent clone at least 10-fold smaller than the second most frequent, providing the potential to improve specificity for this assay by post-treatment tracking both alleles. The use of a synthetic IGH immune system in which templates of all VH and JH primer pairs are used to adjust primer concentrations minimizes multiplexed PCR bias. Among these 84 patients, incomplete D-J rearrangements with a frequency of at least 10% were observed in 22 samples, one of which showed no evidence of any V(D) J clonal expansion (thus making the 92 samples in which we find a clonal IGH rearrangement (Table 1).
With knowledge of the index IGH CDR3 sequences of the patient's clonal population in the pre-treatment samples (Figure 1A), we examined the potential for HTS to identify the same sequence at day 29 post-treatment and compared these findings to results obtained by multi-parametric flow cytometry (mpFC) performed as part of the Children's Oncology Group trial (Figure 1B). In those patients with bi-allelic IGH rearrangements, quantified MRD for the two alleles was highly correlated (Figure 2). High-throughput sequencing to detect the patient's original clonal IGH sequences in day 29 post-treatment samples revealed three sub-groups of patients in this cohort: 1) those for whom MRD was not detected by either HTS or mpFC, 40 cases; 2) those for whom MRD was detected only by HTS but not mpFC, 28 cases; and 3) those in which MRD was detected by both HTS and mpFC, 23 cases (Figure 1B). Importantly, there were no cases (false-negative results) for which MRD was detected only by mpFC, but not by HTS of IGH. For the 28 cases for which HTS did and mpFC did not detect MRD, the MRD was, on average, 10-to-100-fold lower than for the 23 cases for which both mpFC and HTS detected MRD (median 8.0 ×10-6 vs. 1.4×10-4, p = 7.0 ×10-5 by two-tailed U test; Figure 1B). As a conservative estimate, there was approximately a 10-fold increase in the lower detection limit for MRD detection by HTS as compared to mpFC. Tracking of IGH clones could be improved by following both rearranged alleles in tumors that had evidence of bi-allelic rearrangement (Figure 2). However, for the purposes of subsequent comparisons between flow cytometry and sequencing, tracking of only a single dominant sequence was performed.
Figure 2. Comparison of clonal frequency of MRD in cases with presumed bi-allelic rearrangement of IGH versus mpFC.
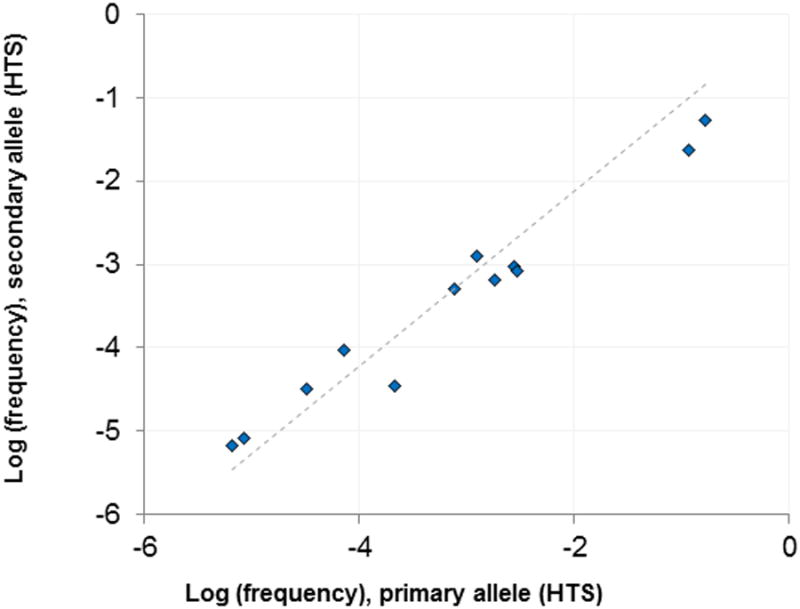
In samples with bi-allelic rearrangement, tracking of both alleles provides enhanced specificity for quantification of MRD. 26 diagnostic samples generated clear evidence for bi-allelic rearrangements (exactly 2 dominant IGH sequences): in these samples, the frequency of each allele was tracked in post-treatment samples. In 10 samples, neither allele was observed post-treatment. In 4 samples, only one allele was observed post-treatment (at an average frequency of 1.2 × 10-5). In the remaining 12 samples, both alleles were observed post-treatment. The frequencies of the dominant and secondary alleles in post-treatment samples are charted. Post-treatment allele frequencies in bi-allelic samples are highly correlated (r2 = 0.97).
As previously reported (11, 12), we also find widespread evidence of on-going clonal evolution of IGH in these B-ALL samples, both in the pre- and MRD-positive post-treatment (Table 2). In post-treatment samples, the predominant clone in pre-treatment samples was typically the most frequent clone. Clones consistent with VH replacement were found in 19 patients. In one patient, the only MRD detected was a single clone consistent with VH replacement at a level of ∼1 in 1,000,000. The clinical significance of this low level MRD is unclear. In the other 18 post-treatment MRD positive cases with evidence of VH replacement, the dominant clone identified pre-treatment was also dominant post-treatment. On average, 3.5% of total IGH rearrangements matched the dominant clone post-treatment, while only 0.036% of IGH rearrangements were consistent with VH replacement of the major clone. Among pre-treatment samples in which VH replaced clones were detected, all VH replaced clones together were 0.4% as large as the dominant clone on average (median). Among post-treatment samples, VH replaced clones were on average 0.6% as large as the dominant clone. These trends are illustrated for a particular patient with MRD and a large number of VH-replaced clonotypes in Supplementary Figure 5. Taken together, these data that generally there is little change in the relative proportions of the dominant clone and VH replaced sub-clones through induction chemotherapy.
Table 2. Summary evolved clonotypes in pre-treatment clonal IGH CDR3 sequences.
Clonal IGH CDR3 sequences were identified in nearly all patients as either complete V-DJ or incomplete D-J rearrangements. Among the 91 patients in which a complete V-DJ rearrangement was observed, the number of clones due to ongoing IGH evolution by VH replacement at diagnosis is indicated, as well as the proportion of total clone size (major clone plus all evolved clonotypes) accounted for by these VH-replaced clones. We used the definition of an evolved IGH clonotype initially proposed by Gawad et al. (12).
| N (percentage) | Mean, median (range) | |
|---|---|---|
| No VDJ clone | 7/98 (7.1%) | - |
|
| ||
| Pre-treatment VDJ clone identified | 91/98 (92.9%) | - |
|
| ||
| No evidence of evolved clonotypes | 6/91 (6.6%) | - |
|
| ||
| Evidence of evolved clonotypes | 85/91 (93.4%) | - |
|
| ||
| Number of evolved clonotypes | Mean 195, median 22, (range 1 to 1,953) | |
|
| ||
| Proportion of evolved clonotypes of total clone | Mean 3.4%, median 0.4%, (range 0.0010% to 59.8%) | |
To evaluate the specificity of HTS for assessment of MRD, we evaluated the frequency of the specific clonal IGH sequence from each patient being present in the other 97 patient post-treatment samples at day 29. This estimate provides an assessment of the relative likelihood post-treatment that B cells may by chance have the exact same clonal index IGH sequence. Prior analysis has reported that non-specific amplification of IGH rearrangements in B-ALL may be dependent on the time point post-therapy and also the gene segment and therefore target rearrangement being considered (18). In our cohort, at the level of 1 cell in 100,000, 9-shared sequences of 8,918 IGH comparisons (0.10%) in cross-patient comparisons were detected. This rate is slightly higher (0.72%) when clones at a frequency of 1 cell in 1,000,000 are considered. Of note, the level of these coincidental clones was lower by approximately one order of magnitude on average as compared to true MRD positives (i.e. index sequences found in the corresponding post-treatment samples, and not in cross-patient samples).
Of note then, 52 of 91 unselected patients had MRD clones at the level of 1 cell in a million or higher that was detectable by HTS of IGH alone, compared to only 23 cases of MRD positivity by flow cytometry (Figure 1B). As many of the samples that were MRD negative by flow cytometry and MRD positive by HTS had levels of MRD within the expected range of detection of flow cytometry (∼1 in 10,000 to 100,000), it is possible that flow cytometry may be missing relatively high level MRD in a subset of patients. This could be due to post-treatment changes in the antigen expression resulting in immunophenotypic normalization (19). We therefore sought to determine if the detection of clonal sequences matching the original diagnostic samples could be derived from the mature B-cell fraction of samples. In order to test this hypothesis, we analyzed 10 additional, paired consecutive pre- and post-treatment B-ALL MRD samples for which residual material was available, and for which there was no evidence of minimal residual disease as determined by flow cytometry. This sample size was chosen because approximately 15 to 20% of post-treatment samples were MRD negative by flow cytometry, yet had MRD detected by high-throughput sequencing within the sensitivity of flow cytometry (Figure 1B). We triple flow cytometry-sorted the mature B-cell fraction from the post-treatment marrow aspirate samples to isolate “mature” B cells and then sequenced these fractions to define the B-cell repertoire in these post-treatment samples, while subsequently sequencing the paired, triple-sorted pre-treatment samples to minimize the possibility of contamination of the post-treatment samples by the pre-treatment ones. Eight samples had sufficient DNA after flow cytometry sorting for subsequent DNA sequencing. While 7 of 8 post-treatment samples showed no evidence of clonal IGH sequence overlap between the post-treatment, flow cytometry-sorted mature B-cell fractions and the pre-treatment diagnostic samples, there was one case in which the pre-treatment IGH V-J sequence was found within the mature B-cell compartment, consistent with the concept of immunophenotypic normalization. The level of this sequence was 0.01% of total cells. By flow cytometry, this leukemia clearly had an abnormal antigenic phenotype at diagnosis (Supplementary Figure 6A), with aberrant immunophenotypic expression of CD10 (increased), CD38 (decreased to absent), CD45 (decreased), and CD58 (slightly increased). This aberrant immunophenotype was not evident in the post-treatment, day 29 sample (Supplementary Figure 6B) from which the mature B-cell fraction was sorted.
Discussion
Accurate risk stratification is important for the personalized treatment of patients with B-lineage acute lymphoblastic leukemia (2-5). Recent clinical studies have confirmed the importance of MRD in guiding patient care in both pediatric and adult B-ALL (9, 20). However, widespread implementation of diagnostic assays to accurately stratify patient risk in the clinical laboratory has been challenging. Each existing method of analyzing MRD, such as flow cytometry or qPCR, has its own drawbacks and technical challenges. Here, using a cohort of patient samples submitted to our institution for flow cytometric analysis of MRD, we confirm the broad potential of sequencing of IGH to permit highly specific molecular detection of clonality in nearly all-comers (91 of 98, ∼93%) without the laborious requirement of individualized PCR or allele-specific oligonucleotide design, implicit in current molecular-based approaches for monitoring MRD (8, 21).
While conventional practice has focused on identifying and following at least 2 clonal molecular markers for MRD detection (22), recent data from the AIEOP-BFM ALL 2000 study of pediatric and adolescent B-ALL suggests patients with only one marker can be similarly stratified as they have outcome results quite similar to those patients followed by two markers (20). Specifically, of 3,184 patients, 454 had only one marker and event free survival results in this subset of patients was 5-year EFS of 94.1% for standard-risk (versus 92.3% EFS for 2 markers) or 83.3% for intermediate-risk (77.6% for 2 markers) (20). Thus, the ability to detect clonal IGH rearrangements in a large proportion of patients in our unselected cohort of B-ALL patients suggests that high-throughput sequencing of IGH should be, at the minimum, a viable strategy for MRD detection in many patients with B-ALL. Moreover, our data further support the analysis for both complete (VDJ) and incomplete (DJ) IGH rearrangements in B-ALL for MRD assessment, and highlight the potential utility of this approach to improve accuracy of quantification of MRD (Figure 2). Additionally, in patients with bi-allelic IGH rearrangements, tracking of both alleles can help to confirm the detection of minimal residual disease.
Prior work by Gawad et al. using next-generation sequencing has identified remarkable diversity in pre-treatment diagnostic samples due to on-going clonal evolution of IGH, with some tumors being composed of thousands of apparent sub-clones with respect to IGH and which appear to be derived from an ancestral clone due to a VH replacement model (11, 12) or alternately, on-going V(D) J recombination of a clone with incomplete DJ recombination. We similarly identify herein evidence of on-going recombination of IGH in pre-treatment samples. However, in contrast to analysis by Gawad et al. who did not evaluate the impact of this on MRD detection (12), we show here that clonal diversity as defined by evolved IGH clonotypes does not substantively change in day 29 post-treatment samples. Further, in MRD-positive, post-treatment sample, the dominant clone found at relapse by sequencing was identical to the dominant clone seen at diagnosis, and the relative proportions of the dominant clone versus the sub-clones was similar in pre- and post-treatment samples. These findings together suggest that within this early time frame, on-going rearrangement at the IGH locus does not substantially alter the fitness of lymphoblast clones during induction chemotherapy. Additional studies at subsequent time points will need to be performed to further document how IGH repertoire may change over time, as several studies have shown that late relapsed clones can often be retrospectively identified in pre-treatment samples (23). Nevertheless, our data provide the first picture of clonal IGH evolution through induction chemotherapy.
Similar to our findings in T-ALL (10), high-throughput sequencing can identify MRD in a subset of cases for which flow cytometry evaluation is negative. Indeed, in this cohort, 5 cases had MRD detected by HTS at a level of >1 cell in 10,000 that should have been detected by flow cytometry (Figure 1B). To consider the possibility that these detected IGH sequences may be derived from cells with an altered immunophenotype, we examined an additional subset of eight cases that had detectable MRD by next-generation sequencing, but were negative by flow cytometry. Of these, one of the eight patients had a clonal MRD sequence identified in the post-treatment sample that matched the patient's index clone and was found within a triple flow cytometry-sorted B-cell fraction that appears to be immunophenotypically mature. Our finding of rare index IGH sequences within the mature B-cell fraction raises the possibility that in some patients, the clonal lymphoblast population detected by sequencing may undergo substantial antigenic maturation during therapy. We do not believe this finding is artifact due to contamination or detection of cell-free IGH sequences in plasma as shown for some mature aggressive B-cell lymphomas (24) or residual DNA associated with dead B lymphoblasts as shown in rare instances in which PCR detects MRD but not flow cytometry (25), as we minimized these possibilities by performing triple flow cytometry sorting and sequencing of the post-treatment sample before analysis of the pre-treatment sample in all cases. Further, we do not believe this to be coincidental identification of a clonally unrelated, reactive B-cell population with identical VDJ-recombination of IGH, because in our analysis of post-treatment samples in our cohort, we did not detect coincidental clonal IGH sequences at the level that was seen in this patient at ∼ 0.01%, even among the very rare examples of shared sequences. Our finding of antigenic-shift is similar to that reported by Gawad et al. who showed in their diagnostic pediatric B-ALL samples that a portion of evolved clones may have substantial antigenic differences as compared to the index clone (12). Given that the malignant potential of a clone such as this with antigenic maturation is not known, its clinical import is undetermined despite its relatively high frequency in this patient.
The patient samples considered in this study were derived from the COG AALL0932 trial, from patients who were regarded as being generally low to standard risk for relapse. However, as shown in our findings, (Figure 1B), many of these cases (51 of 92) have clonal sequences detectable by high-throughput sequencing but are flow cytometry negative. While the clinical significance of these MRD levels below current diagnostic threshold of <0.01% is outside the scope of this study, the significance of these low level sequences requires further study. It is likely that MRD positivity by sequencing will be clinically important for a subset of patients. Several studies support this claim. First, Paganin et al. show that detectable, but not quantifiable MRD below the level of <10-4 had an intermediate event-free-survival (EFS) of 45% at 3 years versus patients who had MRD results that were either outright negative or outright positive at a level ≥ 10-4(26). The EFS for these patients were 73% and 19% for negative versus positive-MRD, respectively. Second, Stow et al. showed in a study involving 455 clinical samples and using PCR analysis of MRD at one time point (day 46, at end of remission-induction) that levels below the conventional clinical threshold of 0.01% was important for predicting relapse (27). In this work, patients in whom PCR was able to detect very low levels of MRD (0.001% to <0.01%) had a 12.7% (+/- 5.1%; SE) cumulative risk of relapse at 5 years compared to 5% (+/-1.5%) for those below the MRD level of 0.01% (p <0.047). Stow et al. conclude that MRD positivity below the standard threshold of 0.01% has prognostic significance for some patients, and that patients with low-level positivity by PCR should be closely monitored (27). Third, Pulsipher et al. reported data showing that patients with pre-transplant detectable MRD by deep sequencing of IGH had a significantly increased cumulative incidence of relapse when compared with those patients with no evidence of disease by sequencing (57% versus 4.4%; p=0.008) in a cohort of 64 COG patients who underwent TBI-based myeloablative transplant for high-risk ALL in CR1 or CR2 (28). Furthermore, while patients from this same cohort who were MRD negative based on flow cytometry had a cumulative risk of relapse of nearly 25%, by comparison, patients who were MRD-negative by deep sequencing of IGH using the identical platform/assay described herein had a cumulative risk of relapse of less than 5%. Lastly, recent work comparing quantitative PCR-based methods versus flow cytometry for MRD detection in a cohort of 1,547 children with acute lymphoblastic leukemia enrolled into the AIEOP-BFM ALL 2000 trial has suggested that the outcome of patients with discordant MRD results between PCR-based methods and flow cytometry is likely to be different than for those cases for which MRD results are concordant between the two approaches (25). Therefore, low-level minimal residual disease detected by sequencing is likely to be meaningful for a subset of patients and suggests that that these patients, at the very least, should be closely monitored.
Indeed, the increased sensitivity of MRD determination by HTS offers the opportunity to clinically assess for trends in MRD over time at time-points when the level of disease may be beneath the detection limit of flow cytometry. Prior work has suggested that in patients with B-ALL, there is an inverse correlation between the level of MRD detected versus time to late relapse (29). Data from the AIEOP-BFM ALL 2000 study further support these findings, as patients with high MRD levels at day 33, but no MRD detected by molecular analysis at day 78 had a decreased 5-year cumulative relapse incidence of 20.7% versus 40.7% in patients who were positive for MRD at day 78, but had persistent MRD at a level less than 10-3. Additional work suggests that oftentimes, the clone that represents late relapse may be identified retrospectively in the index sample (20, 23, 29). Our studies demonstrate robust analytic ability to quantify IGH rearrangements (Supplementary Figure 3).
In summary, we demonstrate the utility of HTS of IGH genes for the detection of MRD in B-ALL. Despite on-going recombination of IGH, the analysis of IGH gene rearrangement permits robust identification of the neoplastic lymphoblast population in a high proportion of unselected cases. For cases in which no clonal IGH gene rearrangement was identified, evaluation of other loci, such as IGK, TRB, TRG, and TRD genes may be helpful, as previously suggested by others and Faham et al. (11). Since HTS could greatly reduce the complexity associated with patient-specific molecular analysis of MRD and improve on observer variation in flow cytometry, it offers the potential to improve clinical prognostication in B-ALL. Further evaluation of sequencing-based MRD detection, however, must be confirmed in subsequent studies involving independent laboratories. However, the synthetic IGH templates that we use herein to ensure accurate and unbiased, multiplex PCR could be one approach to ensure standardized assay performance. Clinical laboratories adopting sequencing for MRD could use this synthetic immune system to confirm consistent test performance, permitting broad implementation of this technology.
Additionally, it will be important to understand the clinical implications of cases in which relatively higher-level of MRD was missed by flow cytometry but detected by sequencing. Although a limited observation, we show that clonal IGH index sequences in some patients can be identified within the immunophenotypically apparent mature B-cell subset, suggesting that some B lymphoblast clones may undergo antigenic drift and/or maturation. The clinical significance of this finding merits further study and is underway.
Supplementary Material
Statement of Translational relevance.
This study evaluates the potential for high-throughput sequencing (HTS) of the immunoglobulin heavy chain gene, IGH, to detect minimal residual disease (MRD) in patients with acute precursor B lymphoblastic leukemia (B-ALL). We show using unselected, consecutive paired pre- and day 29 post-treatment samples from patients enrolled in Children's Oncology Group ALL00932 trial that MRD can be readily detected by sequencing with no false negative results as compared to flow cytometry. As prior work exploring next-generation sequencing in B-ALL included only patients with prior confirmed evidence of a clonal IGH rearrangement, our results show that sequencing of IGH alone is broadly applicable for unselected patients with B-ALL. Further, we measure IGH repertoire during therapy and show that on-going recombination at IGH does not contribute to enhanced tumor fitness that represents day 29 relapse. We contend that the enhanced specificity of high-throughput sequencing of IGH for MRD detection should greatly improve patient-prognostication.
Acknowledgments
We thank the staff of the UW Hematopathology Laboratory for their excellent support of the flow cytometry analysis and sorting experiments and DNA preparation.
Grant Support: This work was supported by the University of Washington, Department of Laboratory Medicine (to D.W. and B.W.), and by Adaptive Biotechnologies, Seattle, WA. In addition, this work was also supported by NIH (award number CA98543-09 via the Children's Oncology Group to B.W.). Lastly, this work was supported by a Chair's Grant U10 CA98543 and Human Specimen Banking Grant U24 CA114766 of the Children's Oncology Group from the National Cancer Institute, National Institutes of Health, Bethesda, MS, USA (to M.L.).
Footnotes
Conflicts of Interest: H.R. and C.C. have consultancy, equity ownership, patents & royalties with Adaptive Biotechnologies; R.E, A.S., B.H., J.V., M.R., I.K., C.C. and D.W.W., have employment and equity ownership with Adaptive Biotechnologies; B.W. has research funding from Becton, Dickinson and Company, NJ; D. Wu, B.W., with H.R. and Adaptive Biotechnologies are collaborating on a related research project examining next-generation sequencing for mature B- and T-cell lymphomas. M.L.L. and A.A. have no relevant conflicts of interest to disclose.
Authors' Contributions: Conception and design: D.Wu, B.W., H.R.
Development of methodology: H.R., R.E., A.S., B.H., J.V., M.R., I.K., C.C., D.Williamson
Acquisition of data: D.Wu, B.W., H.R., R.E., M.L.L., A.A., A.S., B.H., J.V., M.R., I.K., D.Williamson, and C.C.
Analysis and interpretation of data: All
Writing, review and/or revision of manuscript: All
Administrative, technical or material support: D.Wu, H.R., B.W., R.E., B.H.
Study supervision: D.Wu, B.W., H.R.
References
- 1.Pui CH, Evans WE. Treatment of acute lymphoblastic leukemia. N Engl J Med. 2006;354:166–78. doi: 10.1056/NEJMra052603. [DOI] [PubMed] [Google Scholar]
- 2.Borowitz MJ, Devidas M, Hunger SP, Bowman WP, Carroll AJ, Carroll WL, et al. Clinical significance of minimal residual disease in childhood acute lymphoblastic leukemia and its relationship to other prognostic factors: a Children's Oncology Group study. Blood. 2008;111:5477–85. doi: 10.1182/blood-2008-01-132837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bruggemann M, Raff T, Kneba M. Has MRD monitoring superseded other prognostic factors in adult ALL? Blood. 2012;120:4470–81. doi: 10.1182/blood-2012-06-379040. [DOI] [PubMed] [Google Scholar]
- 4.Campana D. Progress of minimal residual disease studies in childhood acute leukemia. Curr Hematol Malig Rep. 2010;5:169–76. doi: 10.1007/s11899-010-0056-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Campana D. Minimal residual disease in acute lymphoblastic leukemia. Hematology Am Soc Hematol Educ Program. 2010;2010:7–12. doi: 10.1182/asheducation-2010.1.7. [DOI] [PubMed] [Google Scholar]
- 6.Cazzaniga G, Biondi A. Molecular monitoring of childhood acute lymphoblastic leukemia using antigen receptor gene rearrangements and quantitative polymerase chain reaction technology. Haematologica. 2005;90:382–90. [PubMed] [Google Scholar]
- 7.van Dongen JJ, Seriu T, Panzer-Grumayer ER, Biondi A, Pongers-Willemse MJ, Corral L, et al. Prognostic value of minimal residual disease in acute lymphoblastic leukaemia in childhood. Lancet. 1998;352:1731–8. doi: 10.1016/S0140-6736(98)04058-6. [DOI] [PubMed] [Google Scholar]
- 8.van der Velden VH, Cazzaniga G, Schrauder A, Hancock J, Bader P, Panzer-Grumayer ER, et al. Analysis of minimal residual disease by Ig/TCR gene rearrangements: guidelines for interpretation of real-time quantitative PCR data. Leukemia. 2007;21:604–11. doi: 10.1038/sj.leu.2404586. [DOI] [PubMed] [Google Scholar]
- 9.Bruggemann M, Raff T, Flohr T, Gokbuget N, Nakao M, Droese J, et al. Clinical significance of minimal residual disease quantification in adult patients with standard-risk acute lymphoblastic leukemia. Blood. 2006;107:1116–23. doi: 10.1182/blood-2005-07-2708. [DOI] [PubMed] [Google Scholar]
- 10.Wu D, Sherwood A, Fromm JR, Winter SS, Dunsmore KP, Loh ML, et al. High-throughput sequencing detects minimal residual disease in acute T lymphoblastic leukemia. Sci Transl Med. 2012;4:134ra63. doi: 10.1126/scitranslmed.3003656. [DOI] [PubMed] [Google Scholar]
- 11.Faham M, Zheng J, Moorhead M, Carlton VE, Stow P, Coustan-Smith E, et al. Deep-sequencing approach for minimal residual disease detection in acute lymphoblastic leukemia. Blood. 2012;120:5173–80. doi: 10.1182/blood-2012-07-444042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gawad C, Pepin F, Carlton VE, Klinger M, Logan AC, Miklos DB, et al. Massive evolution of the immunoglobulin heavy chain locus in children with B precursor acute lymphoblastic leukemia. Blood. 2012;120:4407–17. doi: 10.1182/blood-2012-05-429811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Children's Oncology Group. ClinicalTrialsgov [Internet] Bethesda (MD): National Library of Medicine (US); 2000. [cited 2014 Feb 12]. Risk-Adapted Chemotherapy in Younger Patients With Newly Diagnosed Standard-Risk Acute Lymphoblastic Leukemia. Available from: http://www.clinicaltrials.gov/ct2/show/NCT01190930 NLM Identifer: NCT01190930. [Google Scholar]
- 14.Wood B. 9-color and 10-color flow cytometry in the clinical laboratory. Arch Pathol Lab Med. 2006;130:680–90. doi: 10.5858/2006-130-680-CACFCI. [DOI] [PubMed] [Google Scholar]
- 15.Lefranc MP, Giudicelli V, Ginestoux C, Bosc N, Folch G, Guiraudou D, et al. IMGT-ONTOLOGY for immunogenetics and immunoinformatics. In Silico Biol. 2004;4:17–29. [PubMed] [Google Scholar]
- 16.Yousfi Monod M, Giudicelli V, Chaume D, Lefranc MP. IMGT/JunctionAnalysis: the first tool for the analysis of the immunoglobulin and T cell receptor complex V-J and V-D-J JUNCTIONs. Bioinformatics. 2004;20(Suppl 1):i379–85. doi: 10.1093/bioinformatics/bth945. [DOI] [PubMed] [Google Scholar]
- 17.Carlson CS, Emerson RO, Sherwood AM, Desmarais C, Chung MW, Parsons JM, et al. Using synthetic templates to design an unbiased multiplex PCR assay. Nature Communications. 2013;4:2680. doi: 10.1038/ncomms3680. [DOI] [PubMed] [Google Scholar]
- 18.van der Velden VH, Wijkhuijs JM, van Dongen JJ. Non-specific amplification of patient-specific Ig/TCR gene rearrangements depends on the time point during therapy: implications for minimal residual disease monitoring. Leukemia. 2008;22:641–4. doi: 10.1038/sj.leu.2404925. [DOI] [PubMed] [Google Scholar]
- 19.Chen W, Karandikar NJ, McKenna RW, Kroft SH. Stability of leukemia-associated immunophenotypes in precursor B-lymphoblastic leukemia/lymphoma: a single institution experience. Am J Clin Pathol. 2007;127:39–46. doi: 10.1309/7R6MU7R9YWJBY5V4. [DOI] [PubMed] [Google Scholar]
- 20.Conter V, Bartram CR, Valsecchi MG, Schrauder A, Panzer-Grumayer R, Moricke A, et al. Molecular response to treatment redefines all prognostic factors in children and adolescents with B-cell precursor acute lymphoblastic leukemia: results in 3184 patients of the AIEOP-BFM ALL 2000 study. Blood. 2010;115:3206–14. doi: 10.1182/blood-2009-10-248146. [DOI] [PubMed] [Google Scholar]
- 21.van der Velden VH, Panzer-Grumayer ER, Cazzaniga G, Flohr T, Sutton R, Schrauder A, et al. Optimization of PCR-based minimal residual disease diagnostics for childhood acute lymphoblastic leukemia in a multi-center setting. Leukemia. 2007;21:706–13. doi: 10.1038/sj.leu.2404535. [DOI] [PubMed] [Google Scholar]
- 22.van der Velden VH, van Dongen JJ. MRD detection in acute lymphoblastic leukemia patients using Ig/TCR gene rearrangements as targets for real-time quantitative PCR. Methods Mol Biol. 2009;538:115–50. doi: 10.1007/978-1-59745-418-6_7. [DOI] [PubMed] [Google Scholar]
- 23.Li A, Zhou J, Zuckerman D, Rue M, Dalton V, Lyons C, et al. Sequence analysis of clonal immunoglobulin and T-cell receptor gene rearrangements in children with acute lymphoblastic leukemia at diagnosis and at relapse: implications for pathogenesis and for the clinical utility of PCR-based methods of minimal residual disease detection. Blood. 2003;102:4520–6. doi: 10.1182/blood-2003-05-1455. [DOI] [PubMed] [Google Scholar]
- 24.Armand P, Oki Y, Neuberg DS, Faham M, Cummings C, Klinger M, et al. Detection of circulating tumour DNA in patients with aggressive B-cell non-Hodgkin lymphoma. Br J Haematol. 163:123–6. doi: 10.1111/bjh.12439. [DOI] [PubMed] [Google Scholar]
- 25.Gaipa G, Cazzaniga G, Valsecchi MG, Panzer-Grumayer R, Buldini B, Silvestri D, et al. Time point-dependent concordance of flow cytometry and real-time quantitative polymerase chain reaction for minimal residual disease detection in childhood acute lymphoblastic leukemia. Haematologica. 97:1582–93. doi: 10.3324/haematol.2011.060426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Paganin M, Zecca M, Fabbri G, Polato K, Biondi A, Rizzari C, et al. Minimal residual disease is an important predictive factor of outcome in children with relapsed ‘high-risk’ acute lymphoblastic leukemia. Leukemia. 2008;22:2193–200. doi: 10.1038/leu.2008.227. [DOI] [PubMed] [Google Scholar]
- 27.Stow P, Key L, Chen X, Pan Q, Neale GA, Coustan-Smith E, et al. Clinical significance of low levels of minimal residual disease at the end of remission induction therapy in childhood acute lymphoblastic leukemia. Blood. 2010;115:4657–63. doi: 10.1182/blood-2009-11-253435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pulsipher MA, Carlson CS, Mark K, Wall DA, Schultz KR, Bunin N, et al. Striking predicitve power for relapse and decreased survival associated with detectable minimal residual disease by IGH VDJ deep seqencing of bone marrow pre- and post-allogeneic transplant in children with B-Lineage ALL: a sub-analysis of the COG ASCT0431/PBMTC ONCO51 Study. Blood. 2013;122:919. [Google Scholar]
- 29.Choi S, Henderson MJ, Kwan E, Beesley AH, Sutton R, Bahar AY, et al. Relapse in children with acute lymphoblastic leukemia involving selection of a preexisting drug-resistant subclone. Blood. 2007;110:632–9. doi: 10.1182/blood-2007-01-067785. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.