

Summary
The type-1 ryanodine receptor (RyR1) is an intracellular calcium (Ca2+) release channel required for skeletal muscle contraction. Here we present cryo-EM reconstructions of RyR1 in multiple functional states revealing the structural basis of channel gating and ligand-dependent activation. Binding sites for the channel activators Ca2+, ATP and caffeine were identified at interdomain interfaces of the C-terminal domain. Either ATP or Ca2+ alone induce conformational changes in the cytoplasmic assembly (‘priming’), without pore dilation. In contrast, in the presence of all three activating ligands, high-resolution reconstructions of open and closed states of RyR1 were obtained from the same sample, enabling analyses of conformational changes associated with gating. Gating involves global conformational changes in the cytosolic assembly accompanied by local changes in the transmembrane domain, which include bending of the S6 transmembrane segment and consequent pore dilation, displacement and deformation of the S4-S5 linker, and conformational changes in the pseudo-voltage-sensor domain.
Introduction
Ryanodine receptors (RyRs) are homotetrameric intracellular calcium (Ca2+) release channels of extraordinary size and complexity located in the sarco/endoplasmic reticula (SR/ER) of most metazoan cell types. RyR1 is the predominant isoform in mammalian skeletal muscle, where SR Ca2+ release via RyR1 is required for excitation-contraction (EC) coupling. Paracrystalline RyR1 arrays on the terminal cisternae interact directly with, and are regulated by, CaV1.1 voltage-gated Ca2+ channels on the transverse (T)-tubule membrane (Franzini-Armstrong et al., 1999). Every-other RyR1 channel associates with a cluster of four CaV1.1 channels (also known as dihydropyridine receptors, or DHPRs) whereas the intervening RyR1 channels are not associated with DHPRs. RyR1 channels in these arrays exhibit coupled gating (Marx et al., 1998).
In planar lipid bilayers, the open probability (Po) of RyR1 channels has a biphasic dependence on cytosolic Ca2+ concentration, with a maximal Po in the 10-100 μM range and decreased activity at both higher and lower levels of Ca2+ (Bezprozvanny et al., 1991). RyR2, the predominant isoform in cardiac muscle, is activated primarily by Ca2+ entry through CaV1.2 via Ca2+-induced Ca2+-release (CICR) (Fabiato, 1983). The plant alkaloid ryanodine is a specific RyR channel modifier. It locks the channel in a reduced conductance state at low concentration and blocks it at higher concentrations (Nagasaki and Fleischer, 1988). Ryanodine receptors are also potently activated by ATP, which binds at a relatively low-affinity, non-hydrolyzing site. Given the affinity of this site and the high cytosolic concentration of ATP in muscle (Kushmerick et al., 1992), it is likely that RyR1 in skeletal muscle is constitutively bound to ATP or one of its analogues.
A number of proteins bind and regulate RyR activity, including calmodulin (Hamilton and Reid, 2000) and the immunophilins FKBP12/FKBP12.6, also called calstabin1/calstabin2, which are subunits of the RyR1/RyR2 channel complexes, respectively. FKBP12 and FKBP12.6 regulate the coupled gating of adjacent receptors (Marx et al., 1998) and also modulate the open probability of both RyR1 (Brillantes et al., 1994) and RyR2 (Kaftan et al., 1996) in planar lipid bilayer experiments, but do so without altering gating properties with respect to Ca2+, ATP or ryanodine. The high affinity and specificity of the RyR-FKBP interaction has been exploited here for purification of the receptor (Zalk et al., 2015).
Recently, three single-particle cryo-electron microscopy (cryo-EM) studies have described the architecture of the closed state of RyR1 (Efremov et al., 2015; Yan et al., 2015; Zalk et al., 2015). In this study, we have sought to understand Ca2+-dependent gating and activation of RyR1, and its modulation by key ligands by analyzing structures in multiple functional states. Earlier structural studies of RyR1 showed that gating involves dilation of the transmembrane pore accompanied by rearrangement of the cytosolic assembly with displacement of the peripheral regions of the latter towards the SR membrane (Samsó et al., 2009; Serysheva et al., 1999). In this report, we define ‘activation’ as the increase in open probability induced by the addition of a channel agonist, while ‘gating’ refers to the structural transition between the closed (non-conducting) and open (conducting) states of the channel which may occur under equilibrium conditions. We present structures of RyR1 in the presence and absence of channel modulators (such that open probability is expected to range from near zero to near-unitary), providing detailed insights into the molecular mechanisms of RyR1 activation and gating, and we identify the binding sites for Ca2+, ATP, caffeine and ryanodine. In the presence of Ca2+, ATP and caffeine we obtained structures of both open and closed states of the channel from the same sample using three-dimensional classification, allowing detailed characterization of the local and global structural changes associated with RyR1 gating.
Results
We have determined the structure of the RyR1:Calstabin2 (RyR1:Cs2) complex by cryo-EM under the following five conditions: 1) nominally Ca2+-free state (denoted ‘EGTA-only’); 2) activating [Ca2+] (denoted ‘Ca2+-only’); 3) activating Ca2+ plus ATP and caffeine (denoted ‘Ca2+/ATP/Caffeine’); 4) nominally Ca2+-free plus ATP and caffeine (denoted ‘EGTA/ATP/Caffeine’); and 5) activating Ca2+ plus ryanodine (denoted ‘ryanodine’). Conditions for each dataset are summarized in Table S1. Conditions 3 and 5 were intended to maximize open probability during data collection. Condition 5 was intended to lock the channel in a stable state characteristic of ryanodine-modified purified channels (Imagawa et al., 1987). Although we know that in single-channel experiments a mixture of micromolar Ca2+ and millimolar ATP is sufficient to achieve near-unitary open probability, it was unclear whether the same would hold true for rapidly vitrified receptors imaged under cryo-conditions. To maximize a highly populated open state we included the RyR channel activator caffeine in condition 3.
Single-channel recordings of purified RyR1/Cs2 reconstituted in planar lipid bilayers reveal that the purified protein is functional (Figure S1), with properties similar to those recorded from SR microsomes. No open channels could be detected in the EGTA-only condition. Under the Ca2+-only and EGTA/ATP/Caffeine conditions, the open probabilities of representative channels were 0.22 and 0.13, respectively, while under the Ca2+/ATP/Caffeine condition, the open probability was 0.91 (for the purified RyR1) or 0.92 (for RyR1 in SR microsomes). The addition of 10 μM ryanodine locked RyR1 in the characteristic sub-conductance state.
After initial 2D and 3D classification to eliminate poor-quality particles, all cryo-EM datasets were subjected to 3D classification into four classes with identical classification parameters to assess conformational heterogeneity (see Methods & Resources). Tables S1 and S3 summarize the numbers of particles and domain-specific resolutions for each dataset, Table S4 gives particulars for each class.
Improved resolution compared to published structures has allowed us to revise our previous model (Zalk et al., 2015) and extend sequence registration significantly. The overall resolution (using ‘gold-standard’ protocol and Fourier shell correlation=0.143) range from 3.8 Å for the Ca2+-only state to 4.5 Å for the EGTA-only state. In all cases the highest resolutions were obtained in the core of the channel (C-terminal 1400 residues). Thus, for the Ca2+/ATP/caffeine state (Classes 3&4), the overall resolution is 4.5 Å with a resolution of 4.1 Å in the core, while for the best reconstruction (Ca2+-only, all classes combined) the respective values are 3.8 and 3.6 Å. We show portions of the density maps at the narrowest part of the pore (I4937) and from the CTD in Figure 1A and in Figure 1B, respectively, for each condition after refinement of all particles together for each dataset, giving the ‘gold-standard’ core resolution for each reconstruction. Two maps are represented from the Ca2+/ATP/Caffeine dataset since both open and closed pore conformations are highly populated.
Figure 1. Structures of RyR1 in multiple functional states reveal conformational changes and ligand binding sites.
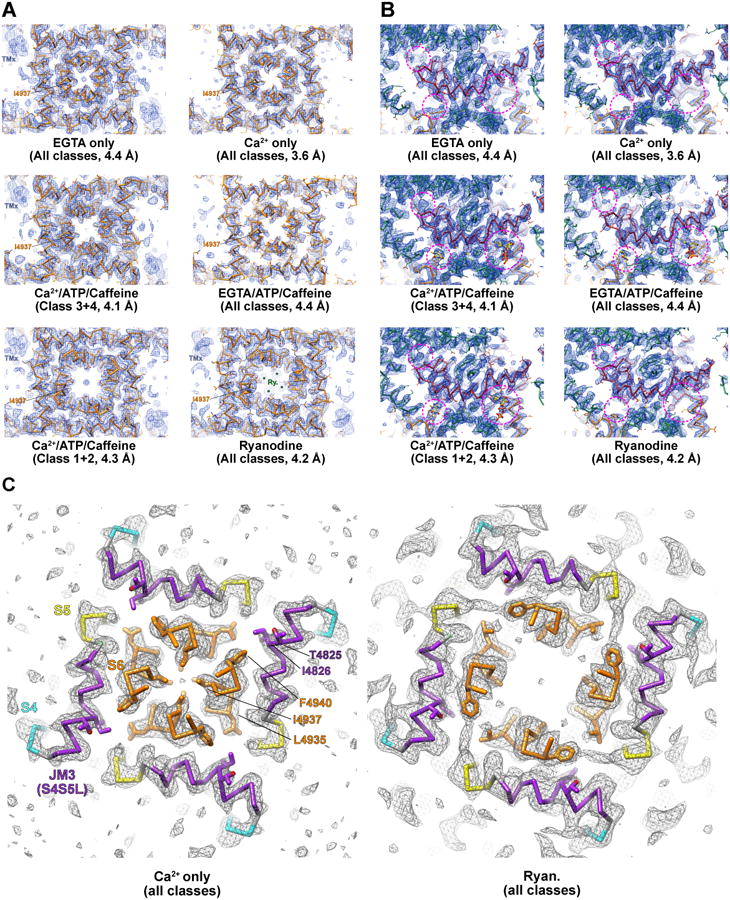
(A-B) For six representative refined reconstructions (identity as labeled), the atomic model, shown as a Cα trace with side-chains in stick representation, is depicted along with the density map, which is contoured at 4 σ and represented as a dark blue mesh. In (A), a 7 Å-thick slab is shown which is orthogonal to, and centered upon, the channel axis at the narrowest part of the pore, at the height of residue I4937 (labeled). In (B), an orthogonal view is represented, showing the cytoplasmic extension of S6 and the C-terminal domain, as well as the surrounding regions of the core solenoid and TaF domain. In all panels, the transmembrane region is colored in orange, the C-terminal domain in dark red, and the core solenoid and TaF domains in sea green. The three ligand binding sites around the C-terminal domain are highlighted with dashed magenta circles. The position of a transmembrane helix of uncertain provenance (TMx), is labeled in light blue when present. In the ryanodine map, putative ryanodine densities in the transmembrane pore are asterisked and labeled in dark green. (C) Identical views parallel to the channel axis are shown for the Ca2+-bound closed state (Ca2+-only, all classes) and the ryanodine-bound open state (all classes), with specific residues at the S6/JM3 (S4S5L) interface labeled. Notably, F4940 reorients during opening, coincident with straightening and displacement of S4S5L and expansion of the pore.
We fitted a largely complete RyR1 atomic model (4168 of 5037 residues) to the best map and then adapted it to the other states. With the resolution improvements we identified previously undescribed features, including a polyproline segment in S2S3, localization of calmodulin-binding peptides, and two additional putative transmembrane helices. Moreover, conformational changes between the different states led us to divide RyR1 into a cytosolic shell that comprises N-terminal domains through the end of the bridging solenoid from each protomer, and a channel and activation core from the C-terminal remainders. This core includes the core solenoid and its intimate associations with cytosolic extensions from the tetrameric pore. Because of hinge flexing now evident between BrA (residues 1657-2144) and BrB of the bridging solenoid, but relative rigidity at the BrB-BrC junction, we renamed BrA as the junctional solenoid (JSol) and BrB-BrC as the bridging solenoid (BSol). JSol is named for its joining of the N-terminal solenoid (NSol) with BSol and Csol (Figure S2). RyR1 domain definitions are in Table 1 and Figure S2.
Table 1.
RyR1 Domain Definitions.
| Domain Symbol | Identification | Residue Span |
|---|---|---|
| Cytosolic Shell | (1 – 3613) | |
| NTD-A | N-Terminal Domain A | (1-208) |
| NTD-B | N-Terminal Domain B | (209-392) |
| Nsol | N-Terminal Solenoid | (393-627) |
| SPRY1 | SP1a / Ryanodine receptor domain 1 | (628-849) |
| RY1&2 | RYR Repeats 1 and 2 | (850-1054) |
| SPRY2 | SP1a / Ryanodine receptor domain 2 | (1055-1241) |
| SPRY3 | SP1a / Ryanodine receptor domain 3 | (1242-1656) |
| JSol | Junctional Solenoid | (1657-2144) |
| BSol | Bridging Solenoid | (2145-3613) |
| (RY3&4) | RYR Repeats 3 and 4 | (2735-2938) |
| Channel and Activation Core | (3614-5037) | |
| SCLP | Shell-Core Linker Peptide, CaM & JSol Binding Sites | (3614-3666) |
| CSol | Core Solenoid | (3667-4174) |
| (EF1&2) | EF-hand pair | (4060-4134) |
| TaF | Thumb and Forefingers domain | (4175-4253) |
| TMx | Auxiliary transmembrane helices | (4322-4370) |
| pVSD | Pseudo voltage sensor domain | (4541-4819) |
| (S2S3) | Helical-bundle domain between S2 and S3 | (4666-4786) |
| Pore | Channel pore domain | (4820-4956) |
| (S6c) | Cytoplasmic extension of S6 | (4938-4956) |
| CTD | C-terminal domain | (4957-5037) |
| Cs2 | Calstabin-2 / FKBP12.6 |
Architectural Organization
The transmembrane pore shares the overall architecture of the six-transmembrane (6TM) cation channel family. A pseudo-voltage-sensor domain (pVSD) spanning helices S1-S4 is linked via a short amphipathic helix to the pore domain, composed of helices S5 and S6. RyR1 pore domain possesses unique structural features including the S2S3 helical bundle (S2S3 domain) inserted in the cytosolic loop between S2 and S3, a 30 Å cytosolic extension of S6 (S6c) terminating in a zinc-finger containing C-terminal domain (CTD), and an intrinsically disordered (glycine-rich) and highly acidic luminal loop between S1 and S2. The S2S3 domain is unique to the RyR family, as it is absent even from the closely related inositol 1,4,5-trisphosphate receptor (IP3R) channels (Fan et al., 2015). The majority of the cytosolic assembly forms an extensively interwoven scaffold of α-solenoid repeats, interspersed with compact domains of other folds, including two N-terminal β-trefoil domains (NTD-A and NTD-B) (Tung et al., 2010), three SPRY domains (SPRY1-SPRY3) (Lau and Van Petegem, 2014), two pairs of RYR repeats (RY1&2 and RY3&4) (Sharma et al., 2012; Yuchi et al., 2015) and a pair of EF-hands (EF1&2) (Xiong et al., 2006) inserted in CSol (Zalk et al., 2015).
Interprotomer Interfaces in the Cytosolic Shell
The inter-protomer contacts in the homotetrameric cytoplasmic shell can be divided into three main interaction sites. We denote interactions as X:X′ or X:X″, where X′ and X″ refer to domain X in the clockwise and anti-clockwise adjacent protomer as viewed from the cytosol, respectively. The three major sets of contacts coupling adjacent protomers are thus NTD-A:NTD-B′, NTD-A:BSol′ and SPRY2:BSol′. All have previously been implicated in RyR channel gating.
Contact between the NTDs and a region in the bridging solenoid including the DP4 peptide (2242-2477) is suggested as important in gating, and disruption by addition of synthetic DP4 sensitizes channels to activation by Ca2+ and caffeine (Yamamoto et al., 2000). In our model this region forms an extensive interface with NTD-A, making contacts with the 121-132 and 174-178 loops, and the 75-84 α-helix. Interestingly, the latter also contacts the CSol.
Auxiliary Transmembrane Helices (TMx)
We previously noted unassigned helical density in the TM region of RyR1 (Zalk et al., 2015). Now, with an improved map for the nominally Ca2+-free state, we can identify this density as TMx for a single transmembrane helix, a short luminal extension lacking definable secondary structure, and an associated density that could be from a partially unfolded second helix. This segment, located between the pVSD and pore domains of adjacent protomers, is not contiguous with other densities in the TM region. Such density was not present in other RyR1 reconstructions (Efremov et al., 2015; Yan et al., 2015), suggesting that TMx presence or stability depends on differences in protein purification procedures. Indeed, although it is clearly present in our Ca2+/ATP/caffeine, Ca2+-free and ryanodine-bound datasets, it is absent in the 30 μM Ca2+ and EGTA/ATP/caffeine datasets (Fig. 1A). TMx could be an unidentified RyR1-binding protein; however, we could not detect any additional proteins in our RyR1 preparations. TMx is more likely part of RyR1, presumably corresponding to the predicted TM hairpin in the region 4322-4370 (Du et al., 2002). Assuming that TMx is intrinsic to RyR1, we propose a revised TM-domain topology that has two TMx helices separated from the pVSD and pore domains by a long disordered cytoplasmic segment.
An RyR1-specific Polyproline Motif in the S2S3 Domain
Improved resolution in the RyR1 core allows complete sequence assignment of the S2S3 domain in pVSD, a domain unique to RyRs consisting of a bundle of five α-helices, the last of which is an amphipathic juxtamembrane helix (JM2). JM2 connects to S2S3 through a short, proline-rich linear motif, 4756-RKPDPPPGLL-4765 that is divergent among RyR isoforms, but conserved in RyR1 from different species. Having a conserved, proline-rich linear motif proximal to the core-gating machinery suggests a regulatory role, possibly as an SH3-domain binding site.
Binding Sites for Activating Ligands: Ca2+, ATP and Caffeine
It was apparent from inspection of density maps in the core of RyR1 that additional features, correlated with the presence and absence of Ca2+, ATP and caffeine, were located at CTD interdomain interfaces (Figure 1B). By comparing RyR1 structures in different states we find that the binding sites for activating ligands are located at interfaces between the CTD and, depending on the ligand, specific interacting domains (CSol, TaF, S2S3, and/or S6c) from the same protomer. Locally aligned difference density maps calculated between the reconstructions obtained for the Ca2+-free closed state and that in the presence of Ca2+, ATP and caffeine reveal three extra densities, clustered around the CTD (Figure 2A-B). Comparison of difference maps calculated from reconstructions that have Ca2+, but lack ATP/caffeine, and those that have ATP/caffeine, but lack Ca2+, allow us to uniquely assign a peak corresponding to Ca2+; density fit considerations allow us to infer which of the two remaining peaks is ATP and which is caffeine (Figure 2C & Supplemental Methods). Ca2+ binds at the interface of the CTD and CSol (Data File S1-1), ATP binds at the mutual junction of S6c, the CTD and TaF domains (Data File S1-2), and caffeine binds between the CTD and the S2S3 domain (Data File S1-3 & Figure 2D-F).
Figure 2. Ca2+, ATP and caffeine bind to the C-terminal domain of RyR1.
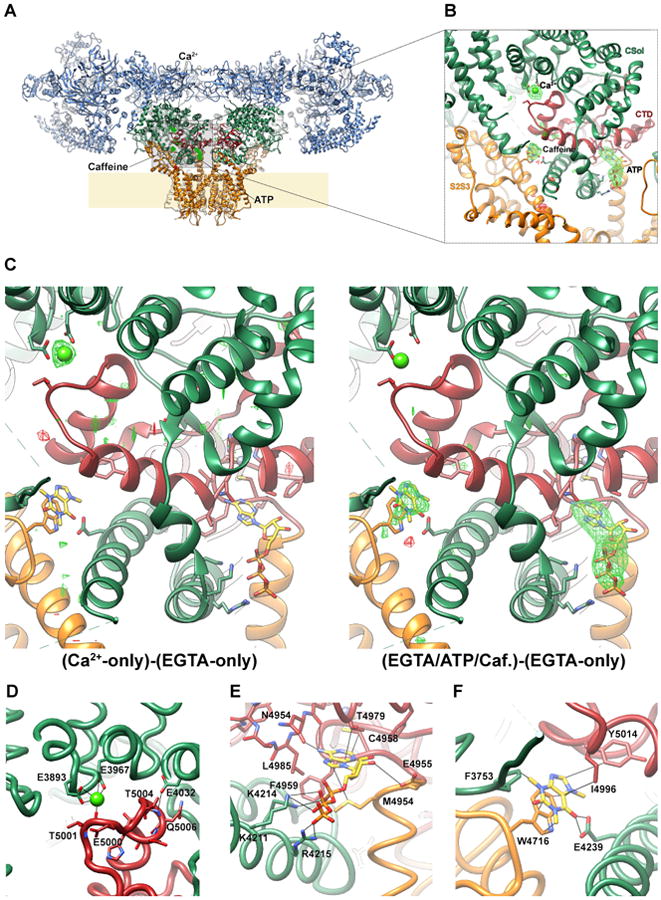
(A) The architecture of RyR1:Cs2 is depicted in ribbon representation, with the shell (residues 1-3666) colored light blue, the core solenoid (residues 3667-4253) dark green, the transmembrane region and S6c (residues 4540-4956) orange, and the CTD (residues 4957-5037) dark red. In the boxed area, a locally aligned difference density map calculated between the EGTA-only reconstruction (all classes combined) and the reconstruction obtained in the presence of Ca2+, ATP and caffeine (Classes 3&4) is depicted in green/red mesh, contoured at 5 σ Putative binding sites for Ca2+, ATP and caffeine are labeled. (B) Enlarged view of the inset in (A), with ligands and interacting residues on RyR1 depicted in stick representation. (C) Locally aligned difference maps, all contoured at 5σ, between the following respective pairs of structures: (Ca2+ only) - (EGTA only) (left) and (ATP/Caffeine/EGTA) - (EGTA only (right). (D-F) The binding sites of Ca2+, ATP, and caffeine, respectively, with likely interacting residues labeled and depicted in stick representation, and putative RyR-ligand interactions indicated as thin black sticks. The RyR1 backbone is shown in licorice/worm representation. One plausible orientation of caffeine is depicted, as it cannot be determined with certainty from the density alone.
The putative Ca2+-binding site is between the carboxylate side chains of E3893 and E3967 of the core solenoid, and the backbone carbonyl of T5001 from the CTD (Figure 2D, Figure S3A & Data File S1-1). In addition, Q3970 and H3895 contribute to the second coordination sphere of Ca2+ (Figure S3A & Data File S1-1). These five seeming Ca2+–interacting residues are conserved in both the RyR and IP3R families (Figure S3B), consistent with a possible role for this site in Ca2+-dependent activation of RyR and IP3R channels, both of which display a similar biphasic response to Ca2+ (Bezprozvanny et al., 1991). Mutation of residue E4032 severely impairs Ca2+-activation in both RyR1 and RyR2 (Fessenden et al., 2001; Li and Chen, 2001). E4032 appears to stabilize the CTD-CSol interface by directly H-bonding to the amide nitrogens at the end of one of the CTD helices, but it does not directly contribute to the Ca2+-binding site (Figure 2D, Figure S3 & Data File S1-1) and E4032 is not conserved in IP3R channels (Figure S3B).
The ATP binding site of RyR1 is located at the interface formed by the S6c-CTD junction and the C-terminal half of the TaF motif, with the entrance of the pocket facing away from the channel axis, not directly accessible from the permeation pathway. The adenine base is buried in a hydrophobic cleft lined by M4954, F4959, T4979 and L4985, while the ribose ring and triphosphate tail interact with S6c and the ‘forefingers’ of the TaF domain (Figure 2E & Data File S1-2). Three positively charged residues (K4211, K4214 and R4215) from the first TaF finger are positioned to interact with the triphosphate tail of the nucleotide, while E4955 appears likely to interact with the ribose ring. The adenine binding site is located directly adjacent to the recently identified Zn-binding site in the CTD (Yan et al., 2015).
The caffeine-binding site is situated between the S2S3 domain and the CTD. The orientation is ambiguous, given the resolution of the data, but it appears to be stacking parallel to the aromatic side-chain of W4716 from S2S3, with I4996 forming hydrophobic interactions with the other face of the molecule, and possible hydrogen bonds between one of the two carbonyls and the carboxylate moiety of E4239 of the TaF domain (Figure 2F & Data File S1-3).
Concerted Cytosolic Shell Movements in All States of Activation
RyR1 exhibits substantial conformational variability in the EGTA-only dataset (Figure 3A; Data File S2-1), characterized primarily by the presence of multiple states of the cytosolic shellup (away from) or down (towards) with respect to the plane of the SR membrane, in addition to a small degree of variability of the displacement of the Csol and TaF domains away from the channel verticalaxis. The population of particles can be classified into an arbitrary number of classes with distinct conformations, suggesting a continuum of conformational states due to oscillation of the shell in solution. These movement scan be modeled by rotations of multidomain units (comprising NTDABC, SPRY123-RY1/2, JSol, Cs2, and BSol′-RY3&&4′), each being rigid to a first approximation, about axes parallel to the membrane (Figure S4). In the absence of activating ligands the observed motion is confined exclusively to the cytosolic region (Figure 1A, Figure 1C, Figure 3A & Figure 3C). Similar cytosolic shell conformations persist upon additions of activating ligands (Figure 3B), but the variation in shell conformation are then over different and more limited ranges (Figure S4; Data File S2: 2-5).
Figure 3. Ligand-induced conformational changes in the RyR1 closed state.
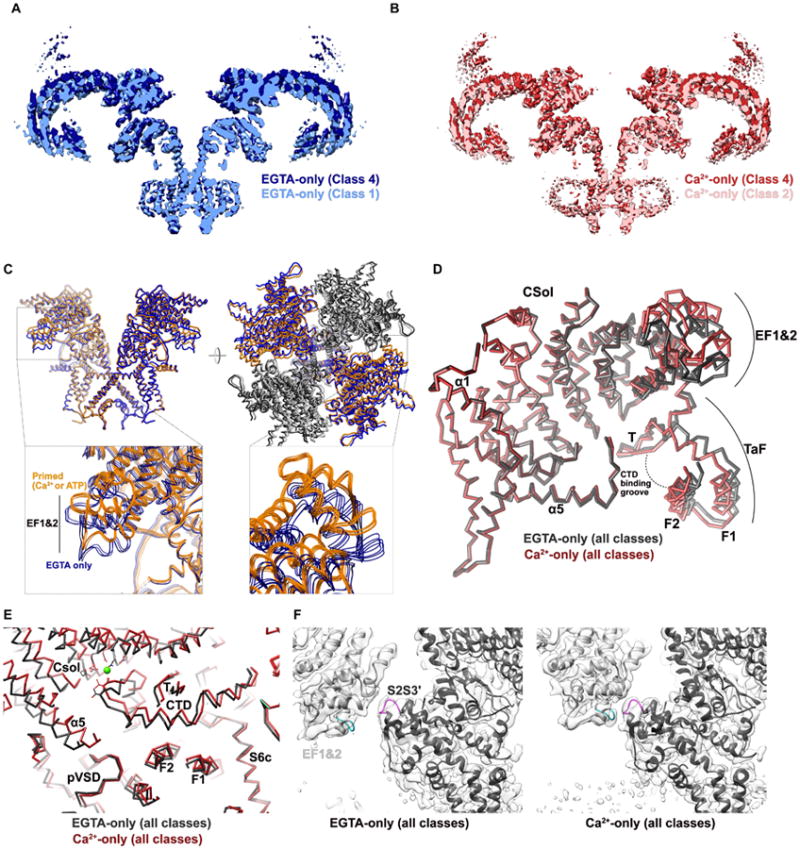
(A,B) Conformational variability of maps derived from three-dimensional classes in, respectively, the EGTA-only dataset (blue/light blue density, contoured at 3×r.m.s), and the Ca2+-only dataset (orange/red density, contoured at 3σ). In both cases only the two extremes are represented, as labeled; a comparison of all classes is provided for both datasets by, respectively, Data File S2: 1-2. (C) Conformational changes of the RyR1 core in the presence of Ca2+ alone or ATP/caffeine alone. The EGTA-only (all classes combined) structure (residues 3667-5037 of two opposing protomers; in the top view, the two alternating protomers are depicted in gray/charcoal to show the change in interprotomer interactions) is depicted in thin blue ribbons, while the Ca2+-only (all classes combined) structure is depicted in orange ribbons. In the inset, an overlay of all four EGTA-only classes (thin blue ribbons; EGTA-only classes 1-4) and all 8 “primed” structures (thin orange ribbons; EGTA/ATP/caffeine classes 1-4 and Ca2+-only classes 1-4) in the EF1&2 region are shown to illustrate the consistent conformational change observed between the apo- and primed states of the activation core. (D) The activation core undergoes flexible changes during priming. Here, residues 3639-4253 of the EGTA-only (charcoal) and Ca2+-only (dark red) structures are shown aligned on the core solenoid. The TaF and EF1&2 domains are displaced in opposite directions, changing their disposition with respect to the body of the core solenoid. (E) Change in orientation of the CTD with ligand binding. Here, a Cα trace of the atomic model for the EGTA-only reconstruction (all classes combined) and the Ca2+-only reconstruction (all classes combined) are shown in charcoal and dark red, respectively. The bound Ca2+ ion is represented as a green sphere, and the sidechains of coordinating residues are shown in stick representation. The view is the same as represented in Figure 1B. (F) The EF1&2/S2S3′ interface, viewed orthogonal to the channel axis, for the EGTA-only (left) and Ca2+-only (right) reconstructions, with density maps shown as transparent surfaces, contoured at 3σ. A loop in the EF1&2 domain is colored cyan, and a loop in the S2S3 domain is colored magenta, to improve visibility of the conformational change.
Activation Module Changes that Prime the Channel for Opening
In contrast to the Ca2+-free state, where the core is largely fixed despite shell movements, the addition of either Ca2+ or ATP/caffeine results in characteristic changes in the core (Figure 3C-F, Data File S1: 4-5, Data File S2: 6-10). Associated with binding of these activating ligands, cytoplasmic portions of the core adopt a new conformation while leaving the transmembrane portion of the channel closed, not significantly changed from the Ca2+-free state (Figure 1A & 1C). We call this cytoplasmic portion of the core the activation module, which comprises cytoplasmic core domains (CSol-EF1&2, TaF and CTD) and sub-domains that extend into the cytoplasm from the 6TM channel pore itself (S2S3 and S6c). Changes in the activation module include a plastic deformation of the core solenoid, presumably due to contacts with the CTD mediated by Ca2+ or ATP (Figure 2B-E). The CSol deformations lead to re-orientation of the TaF/CTD unit relative to CSol (Figure 3C, 3D & Data File S2-7) and also a change in the disposition of the EF1 & 2 pair (Figure 3C, 3E & Data File S2-10), which is displaced in the opposite direction from the TaF/CTD unit. Movement of the EF1 & 2 pair is particularly noticeable in the tetramer where it then directly contacts S2S3 of a neighboring protomer, whereas these features are separated in the absence of Ca2+ or ATP/caffeine (Figure 3F, Data File S2: 10-15).
Our observation that the channels remain closed under the conditions of Ca2+ alone or ATP/caffeine alone is qualitatively consistent with the single-channel properties (Figure S1A). The pore remains constricted at I4937 to an aperture insufficient to allow passage of a hydrated monovalent or divalent cation. We consider the common conformational state obtained with 30 μM Ca2+ or ATP/caffeine alone to be ‘primed’ for activation but held from opening by a barrier that can be overcome by the partner ligand.
Channel Opening upon Synergistic Activation of RyR1 by Ca2+ and ATP/caffeine
Binding of Ca2+, ATP and caffeine results in four distinct conformations (Data File S2-3); two of these have a constricted pore with an aperture similar to that observed in the closed states (Classes 3&4; Figure 1A, 4B & 4D) and two have adilated pore consistent with passage of hydrated ions (open state) (Classes 1&2; Figure 1A, 4C & 4D, Data File S1: 6-7) each comprising approximately 50% of the particles compatible with the high (but not unitary) open probability of RyR1/Cs2 in the presence of Ca2+, ATP and caffeine (Figure S1A).
Figure 4. Ryanodine receptor gating in the presence of Ca2+, ATP and caffeine.
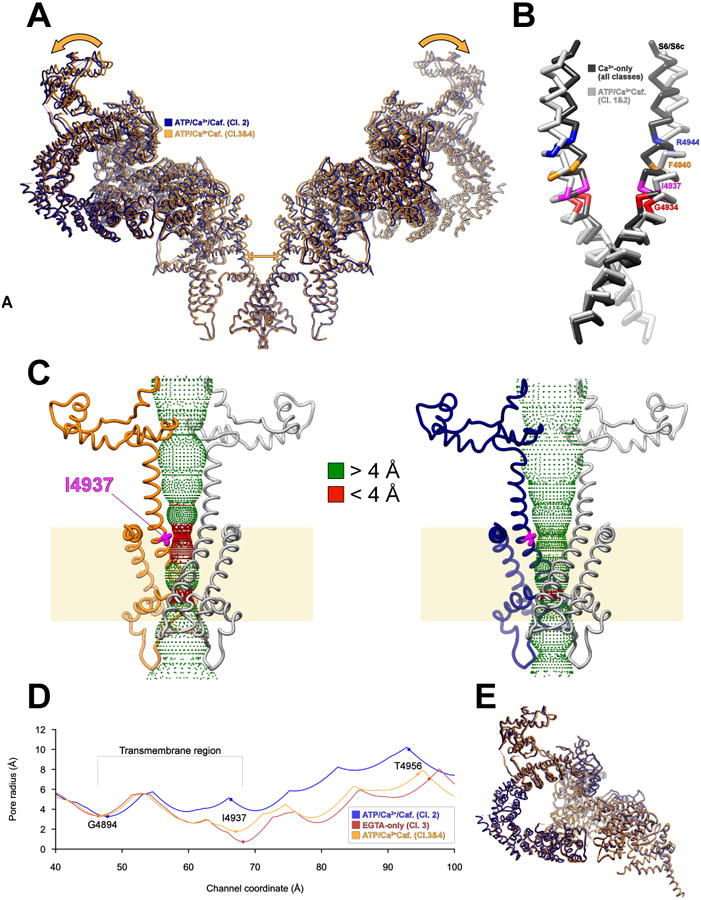
(A) Global conformational changes between the closed (orange, Ca2+/ATP/Caf. Class 3&4) and the open (blue, Ca2+/ATP/Caf. Class 1&2) states identified in the presence of Ca2+, ATP and caffeine. Arrows show the direction of movement of a semi-rigid entity encompassing S6c, the CTD, the core solenoid and the junctional and bridging solenoids. (B) Helix S6 bends during gating. Here, two opposite S6/S6c helices (residues 4910-4956) are represented as Cα traces viewed orthogonal to the channel axis for both the closed state (dark gray) and the open state (light gray), with several marker residues colored on both models. (C) A side view of the transmembrane pore (residues 4820-5037) is depicted in ribbon representation with the hydrophobic gate residue, I4937, highlighted in magenta for the closed state (left, Ca2+/ATP/Caf. Class 3&4) and the open state (right, Ca2+/ATP/Caf. Class 1&2). A dotted representation of the accessible inner surface of the channel as calculated by HOLE (Smart et al., 1996) is colored green (where accessible to a sphere of greater than 4 Å radius) and red (where inaccessible to such a sphere) (D) 1D graph of the pore radius in the Ca2+-free closed state (dark red), activated closed (orange) and activated open (dark blue) structures. (E) Superposition of the rigid units (residues 1657-4253 and residues 4934-5037) between the closed (orange) and open (dark blue) activated structures.
The dilated-pore and constricted-pore classes each independently segregate into classes in which the cytosolic assembly is either up or down with respect to the plane of the SR membrane, as also occurs in other states (Figure S4A-C & Data File S2-3), without accompanying changes in the channel core. Moreover, the two constricted-pore classes are similar to the up and down states from the Ca2+-alone and ATP/caffeine conditions, respectively.
The conformational change between open and closed classes involves concerted movement of the channel core and the cytosolic shell in which a rigid unit comprising shell residues 1657-3613 (JSol and BSol-RY3/4) plus the primed activation module (core residues 3614-4253, 4666-4786, and 4938-5037) tilts outward from the vertical axis of the channel, resulting in dilation of the pore (Figure 4A). The rigid units responsible for pore dilation can be superposed very closely, with an R.M.S.D. of 0.93 Å over 2300 aligned Cα positions (Figure 4E). Concomitantly, the remainder of the cytosolic shell (residues 1-1656) rotates about an axis parallel to the vertical axis of the channel to accommodate the tilting of the rest of the protomer. This movement disrupts interprotomer contacts between NTD-A and NTD-B (Data File S1-8) at a previously identified ‘hotspot’ for malignant hyperthermia (MH) mutations (Kimlicka et al., 2013). Dilation of the pore also disrupts the inter-protomer contact that forms between EF1&2 and S2S3′ upon binding of Ca2+ or addition of ATP/caffeine (Data File S2-25).
Dilation of the pore does not occur via bending of S6 at a single ‘hinge’ alone, but rather by bowing and distortion of the S6 helix from a point midway through the membrane (Figure 4B), near the position of G4934 hinge, previously identified to influence gating of both RyR1 and RyR2 (Euden et al., 2013; Mei et al., 2015). Three other notable changes occur in the transmembrane region coincident with pore dilation. Firstly, the ‘cinch’ formed by the four S4S5L helices of the tetramer loosens, with S4S5L straightening and its contacts with S6 altering. Most notably, F4940 from S6c switches from interacting with L4935 on S6 of the adjacent protomer to interacting with T4825, I4826 and S4829 from S4S5L on the same protomer (Figure 1C). Secondly, the displacement of S4S5L away from the channel verticalaxis in turn pushes on the cytosolic end of S4, which is displaced internally within the pVSD, resulting in the apparent formation of a salt bridge between R4563 on S1 and D4815 on S4 (Figure 5D), and the reorientation of S4 by 3.7°. A network of hydrogen bonds involving two histidines in pVSD, H4650 from S2 and H4812 from S4, also rearranges during this process. Both R4563 and D4815 are conserved in the IP3R family, highlighting the potential importance of this interaction. Lastly, the interactions between S6c and S6c′ are altered. R4944 shifts from forming an apparent salt bridge with E4942 on S6c′ in the closed state (Figure 5A) to interacting with D4938 on S6c′ when opened (Figure 5B), one turn closer to the membrane. All three residues involved in this interaction are highly conserved in both RyR and IP3R families (Figure 5C). Concomitantly, R4944 rotates outward to leave an electronegative ion conduction pathway lined by E4942 and other carboxylate residues.
Figure 5. A conserved network of interactions between S6c and S6c′ rearranges during gating.
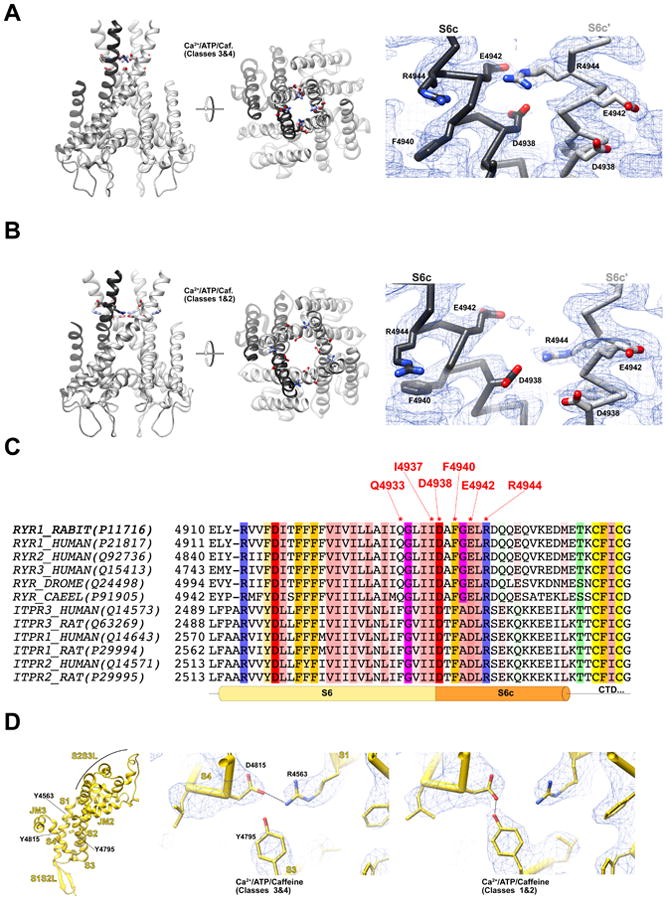
(A) In the closed state (here Ca2+/ATP/caffeine Classes 3&4), R4944 is in close contact with E4942 of the adjacent protomer, and the guanadino moiety of the arginine side-chain lines the conduction pathway of the pore. Density map is shown at right (blue mesh, contoured at 2.5σ) with fitted model (Cα trace with selected sidechains shown and labeled). (B) In the open state (here Ca2+/ATP/caffeine Classes 1&2), S6 splays out away from the channel axis, and R4944 rotates out of the conduction pathway, approaching the sidechain of D4938 from the adjacent protomer. Density map is shown at right (blue mesh, contoured at 2.5σ) with fitted model (Cα trace with selected sidechains shown and labeled). (C) Portion of a sequence alignment of representative RyR and IP3R genes, prepared using MUSCLE (Edgar, 2004) with default parameters, visualized using Jalview (Waterhouse et al., 2009). The alignment is colored by conservation using a modified version of the Zappo coloring scheme, with a conservation cutoff of 60%. Residues of particular interest are asterisked and labeled, including R4944, D4938 and E4942. The Uniprot accession numbers of the relevant sequences are given in parentheses. (D) An apparent salt bridge between R4563 and D4815, in the core of the pVSD (left), rearranges during channel opening, due to the conformational change of S4S5L and the rotation of S5. Here, the fitted atomic models of the Ca2+/ATP/caffeine closed (center, classes 3&4) and open (right, classes 1&2) structures are shown with the density maps (blue mesh, contoured at 3×r.m.s.
Ryanodine Locks the Pore in the Dilated State
In contrast to the closed state that predominates in the Ca2+-only dataset, the addition of 10 μM ryanodine together with 300 μM Ca2+ results in a map in which both major conformational classes have the pore maximally opened (Figure 1A, 1C & 6A). The ryanodine-modified pore structure is very similar to that of the open-pore reconstruction (Classes 1&2) of the Ca2+/ATP/caffeine-activated state (Figure 1A), except for extra density features in the ryanodine-modified pore (Figure 6B). We attribute these features to one ryanodine molecule averaged over four symmetrically equivalent binding sites. Besides locking the channel in a partially open state, even a single ryanodine molecule is likely to occlude part of the pore, consistent with reduced conductance in the presence of ryanodine (Nagasaki and Fleischer, 1988). This putative ryanodine position is directly adjacent to Q4933 (Figure 6B), mutation of which specifically reduces ryanodine binding without affecting channel function (Fessenden et al., 2001). The equivalent residue in IP3R channels, which have a similar transmembrane pore but do not respond to ryanodine, is a phenylalanine (Figure 5C).
Figure 6. Ryanodine binding site and mechanism of channel modulation.
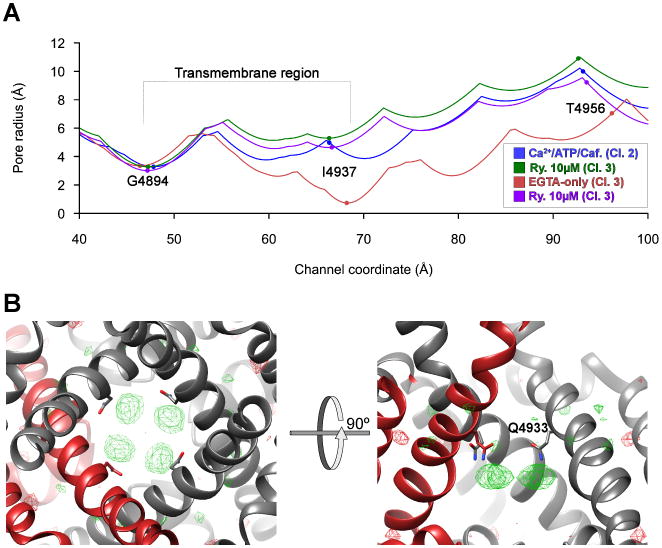
(A) 1D graph of the pore radius in the EGTA-only (red), Ca2+/ATP/caffeine- activated open (dark blue), and ryanodine-locked (purple and green) structures, prepared using the HOLE software package. Classes as labeled. (B) Difference map between Ca2+/ATP/caffeine-activated open-pore reconstruction and 10 μM ryanodine-locked open-pore reconstruction, superimposed onto the 10 μM ryanodine-locked open model (gray ribbons; one protomer colored dark red) and shown in two orientations (left, from the cytosol; right, parallel to the membrane). The difference map is represented as green/red mesh and contoured at 5σ. Q4933 is depicted in stick representation.
Discussion
Mechanism of Ryanodine Receptor Activation
We have developed a structural description of RyR1 activation and gating (Figure S5). In the absence of Ca2+, ATP or caffeine, the RyR1 core as a whole is extremely rigid and non-permissive of pore dilation. The minor displacements of the CTD and CSol that do occur in Classes 1 & 3 (Data File S2-1) are not accompanied by pore dilation. In contrast, the interfaces between the core and the shell are flexible allowing for a large range of cytosolic shell conformations without affecting dilation of the pore (Data File S2-1). Binding of activators (Ca2+, ATP and caffeine) at the CTD interfaces initiates a rearrangement of the RyR1 core (‘priming’), involving a reorientation of the CTD and flexible changes in the activation module (CSol-EF1&2, TaF, and CTD from the cytoplasmic core and S2S3 and S6c from the 6TM channel pore) (Data File S2-6). The activation module now adopts a conformation and orientation poised for pore dilation. In the presence of a single activator (either Ca2+ or ATP), the population of open channels is too low to isolate by our 3D classification; addition of ryanodine prevents these channels from closing, by intercalating between the pore-lining S6 helices and locking the channel in the open state. Multiple activators (Ca2+, ATP and caffeine), binding at different sites on the CTD, increase the channel open probability such that open channels are observed (Data File S2-3). For our Ca2+/ATP/caffeine sample, the populations with open and closed pores are nearly equal.
The comparison of open and closed states under activating conditions allows us to describe the structural changes that occur in the channel upon stochastic channel gating at equilibrium. These closed-state structures are essentially the same as those from the single-activator conditions. Cytoplasmic portions of the channel protomers, including the activation modules, move as rigid bodies and interface with the 6TM pore domain from this closed state to open the pore. An unanswered question concerns the activation barrier that restricts channels from opening freely whenever activators are present.
The dilation of the opened transmembrane pore is particularly evident at the S6 helix-bundle crossing, where I4937 forms a hydrophobic seal in the closed state (Data File S1-9). Pore dilation occurs due to outward bowing and rotation of the transmembrane S6 helices such that I4937 no longer points directly towards the channel axis. S6 bowing is also coincident with a conformational change in which S4S5L which otherwise curves inwards towards the outer edge of S6, straightens (Figure 1C & Data File S1-9), accommodating both the increased aperture of the open channel and associated changes in pVSD and in the interfaces between the S6c pore helices. The structurally equivalent S4-S5 linker of Kv channels plays a role in coupling conformational changes at the activation gate to those in the voltage sensor (Labro et al., 2008), and mutations in this region of RyR1 have been functionally characterized (Murayama et al., 2011). The bowing in S6 coincides with displacement of a region that encompasses CTD and the BSol, JSol and CSol solenoids away from the pore axis.
Ca2+-binding Site
Previous work on Ca2+-dependent activation of RyR1 has suggested that the channel contains a moderate-affinity Ca2+-dependent activation binding site (μM KD), and a lower-affinity Ca2+-dependent inhibition site at high (millimolar) Ca2+ (Bezprozvanny et al., 1991). Here, we have identified a Ca2+ binding site that is highly occupied at a concentration of 30 μM Ca2+ consistent with a Ca2+-activation site. All residues involved in the Ca2+-coordination are conserved in both RyRs and IP3Rs (Figure S3B). Moreover, the putative Ca2+-activation site is located in proximity to two other channel activators (ATP and caffeine). Finally, this site is directly adjacent to an interface involving E4032, mutation of which inhibits Ca2+-dependent activation of the channel. Although E4032 has previously been proposed to form part of the activating Ca2+-binding site (Fessenden et al., 2001), this not so in the Ca2+-bound structures. Rather E4032 forms an interface with CTD in the Ca2+-bound state, most likely supporting a conformation that permits Ca2+ binding.
ATP Binding Site
Adenine-derived nucleosides and nucleotides are competitive partial agonists of both skeletal (Laver et al., 2001) and cardiac (Kermode et al., 1998) RyRs. ATP increases the maximal PO of the channel, without altering conductance (Kermode et al., 1998). Given that the cytosolic concentration of ATP in amyocyteis ∼5 mM (Kushmerick et al., 1992), ATP or one of its analogs will likely be constitutively bound to the channel. The adenine ring appears to be critically important for affinity, as neither GTP (Chan et al., 2000) nor inosine monophosphate (Laver et al., 2001) activate RyRs. Channel activation as assessed by the maximal PO in the presence of varying [Ca2+] correlates with the number of phosphates attached to the ribose ring (ATP>ADP>AMP>adenosine≈adenine), with adenine and adenosine exhibiting minimal effects on the open probability of RyR2, whereas ATP increases the maximal open probability of RyR2 to >0.9 (Chan et al., 2000).
Studies using spin-labeled ATP analogs have shown that each protomer of RyR1 possesses one to two ATP binding sites (Dias and Vogel, 2009). It has been suggested that the last 100 residues of the C-terminal ‘tail’ contribute to the ATP-binding pocket (Popova et al., 2012). We now confirm that this C-terminal segment contains the ATP-binding pocket of RyR1.
Caffeine Binding Site
The possibility that natural nucleotide metabolites may regulate the caffeine site in vivo remains to be explored. The putative caffeine-binding site identified here is at the interface of the CTD, S2S3 and TaF domains. The co-localization of binding sites for ATP, caffeine and Ca2+ at interfaces of the CTD with other channel domains highlights the importance of this region in channel activation.
Ryanodine Binding Site
At low (nM-μM) concentrations ryanodine binds to the open channel and locks it in a partial conductance state of exceptionally long duration, the fractional conductance of which varies with the structural features of the ryanoid (Welch et al., 1997). Increased ryanoid concentrations (∼100 μM) lock the channel in a non-conducting state (Zimányi et al., 1992). RyRs possess one high-affinity ryanodine binding site per tetramer, and up to three lower-affinity sites (Carroll et al., 1991). Several lines of evidence suggest that the high-affinity ryanodine binding site is located within the transmembrane pore of the channel. First, in photo affinity labeling experiments an azido derivative of ryanodine specifically labels the C-terminal 76 kDa fragment of the channel (Witcher et al., 1994). Concordant results were obtained by proteolysis of the channel labeled with tritiated ryanodine (Callaway et al., 1994). Second, ryanodine and the impermeant cation tetraethylammonium (TEA) compete for the same binding site, and ryanodine binding results in relocation of the TEA binding site towards the cytoplasm (Tanna et al., 2006). Third, mutation of a pore-lining glutamine residue (Q4863 in RyR2; Q4933 in RyR1) located just luminal to the cytoplasmic gate (I4937) dramatically affects ryanoid-binding kinetics (Ranatunga et al., 2007). Comparison of RyR1 with structures of potassium and sodium channels complexed with quaternary ammonium ions demonstrates that the expected TEA binding site is directly adjacent to Q4933, consistent with the aforementioned report that ryanoids and quaternary alkyl ammonium ions compete for the same binding site (Tinker and Williams, 1993). Lastly, ryanoid binding kinetics are positively correlated with prior open probability, such that RyR activators, including Ca2+ and ATP, increase the rate of ryanoid association, while ryanoids do not bind when the pore is closed (Chu et al., 1990).
We show here that a ryanodine binding site is located within the pore, proximal to Q4933, and that binding of ryanodine to this site locks the transmembrane pore in a dilated state. The structure of ryanodine-modified RyR1 has several implications for understanding the mechanism of ryanodine binding and its modulation of RyR channel activity. The four equivalent ryanodine binding sites within the pore are sufficiently close to one another that the association of a ryanodine molecule to any one of them likely adversely impacts ryanodine binding at the three remaining sites, consistent with the observation that ryanodine binds to a single site with high affinity, followed by slow association at lower-affinity sites. The ryanodine binding site is located within the permeation pathway of the channel, but displaced from the vertical axis of the channel, suggesting that association of a single ryanoid may lock the channel in a fractional conductance state by partially occluding the permeation pathway, while association of multiple ryanoids would likely result in complete occlusion of Ca2+ permeation. One implication of these observations is that the high- and low-affinity ryanodine binding sites may be one and the same, as previously proposed (Callaway et al., 1994).
Conservation of Shared Functional Motifs in RyRs and IP3Rs
IP3Rs are the other members of the intracellular Ca2+ release channel family and are also tetrameric, Ca2+-modulated channels. IP3Rs share significant sequence and structural similarity with RyR1 in the TM, CSol and NTD-A/B/C regions (Fan et al., 2015). IP3Rs also exhibit a biphasic response to Ca2+ and sensitivity to ATP, but lack sensitivity to ryanodine (Bezprozvanny et al., 1991). Mapping sequence conservation across the RyR/IP3R family onto our structure, we see that the residues comprising the putative Ca2+-binding site at the CTD-CSol interface are conserved, as are many residues in the ATP binding site. Notably, both the EF1&2 and S2S3 domains are absent in IP3R, suggesting that any function these two domains (and the interactions between them) have is likely to be of a regulatory, RyR-specific nature, rather than due to involvement in Ca2+-activation. This hypothesis is supported by the observations that the EF1&2 pair can be deleted without substantially affecting RyR2 activation by Ca2+ (Guo et al., 2016), and that the EF-hands of the equivalent region in RyR1 can be sequence-scrambled without abolishing Ca2+-activation of the channel (Fessenden et al., 2004). Lastly, the key residues in the ryanodine binding-site, most notably Q4933, are not conserved in IP3Rs, supporting the hypothesis that this is the functionally relevant ryanodine binding-site.
Methods and Resources
Contact for Reagent and Resource Sharing
Further information and requests for reagents may be directed to, and will be fulfilled by the Lead Contact Joachim Frank (jf2192@cumc.columbia.edu).
Method Details
Rabbit Skeletal Muscle RyR1 Purification
To obtain highly purified RyR1, 100 grams of rabbit skeletal muscle flash frozen in liquid nitrogen were blended for 90 seconds in 500 ml of cold buffer containing 10 mM Tris-Maleate pH 6.8, 1 mM DTT, 1 mM EDTA, 150 μM PMSF and 1 mM benzamidine. The mixture was then centrifuged for 10 min at 8000 × g. The supernatant was centrifuged for 20 min at 40,000 × g. Pellets were solubilized in 50 ml buffer containing 10 mM HEPES pH=7.5, 1% CHAPS, 1 M NaCl, 2 mM EGTA, 2 mM TCEP, and a protease inhibitors cocktail (Roche). The solubilized membranes were diluted 1:1 in the same buffer without NaCl and centrifuged for 30 min at 100,000 × g. The supernatant was filtered using a 0.2 μm filter and bound overnight at 4 °C to a pre-equilibrated 5 ml GSTrap (GE healthcare) column with bound GST-Calstabin1. The column was washed with 10 column volumes of cold modified solubilization buffer (0.5% CHAPS and 0.5 M NaCl) and RyR1 was eluted with two column volumes of 10 μM Calstabin2 in the same buffer (expressed in 1L IPTG induced transformed BL-21 cells; Calstabin2 was used for elution because of its higher affinity to RyR1 compared to Calstabin1). The eluent was pre-cleared with 0.5 ml glutathione beads to remove traces of GST-calstabin and treated with calf intestinal alkaline phosphatase (CIP, NEB, 100 U/ml) for 3 hr at room temperature to dephosphorylate RyR1 (Zalk et al., 2015). The CIP-treated RyR1 was concentrated on a 100,000 kDa cut-off centrifugation filter (Millipore) to less than 500 μl, loaded on a size-exclusion column (Tosoh TSKgel G4SWxl) and washed in modified solubilization buffer with 1 mM EGTA, 0.25% CHAPS and 0.001% (w/v) DOPC (Avanti). The peak fractions were combined and modulating regents were added and allowed to bind (Ca2+, ATP and caffeine for 10 minutes at room temperature, ryanodine for 2 hours at room temperature followed by overnight at 4 °C, See Table S1 for a summary of the final solution conditions of each sample). Final free [Ca2+] was determined using MaxChelator (Schoenmakers et al., 1992). The samples were concentrated on a 100,000 kDa cut-off centrifugation filter (Millipore) to 5-10 mg/ml determined using spectroscopy (Abs at 280 nm, 1% (w/v) =1.018).
Single-Channel Recordings
To prepare SR vesicles, muscles were homogenized using a tissue vitamizer (Fisher Scientific) at the highest speed for 1 minute with 2 volumes of 20 mM Tris-maleate (pH 7.4), 1 mM EDTA and protease inhibitors (Roche). Homogenate was centrifuged at 4,000 × g for 15 min at 4°C and the supernatant was centrifuged at 40,000 × g for 30 min at 4°C. The final pellet, containing the SR fractions, was resuspended and aliquoted in: 250 mM sucrose, 10 mM MOPS (pH 7.4), 1 mM EDTA and protease inhibitors. Samples were frozen in liquid nitrogen and stored at -80°C.
Purified RyR1/Cs2 was reconstituted into microsomes as described by (Lee et al., 1994). Briefly, the purified receptor complex was brought to 5 mg/mL of lipid mixture (5:3 DOPC:DOPE) and placed in a dialysis bag. The sample was dialyzed overnight in 1L of buffer containing 0.5 M NaCl, 0.1 mM EGTA, 0.2 mM Ca, 150 uM PMSF, 1 mM DTT, and 10 mM HEPES (pH 7.4) to remove free detergent. Following dialysis, samples were snap frozen in liquid nitrogen and stored at -80C until use.
SR vesicles containing RyR1 or microsomes reconstituted with purified RyR1/Cs2 complex were fused to planar lipid bilayers formed by painting a lipid mixture of phosphatidylethanolamine and phosphatidylcholine (Avanti Polar Lipids) in a 5:3 ratio in decane across a 200-μm hole in polysulfonate cups (Warner Instruments) separating 2 chambers. The trans chamber (1.0 ml), representing the intra-SR (luminal) compartment, was connected to the head stage input of a bilayer voltage clamp amplifier. The cis chamber (1.0 ml), representing the cytoplasmic compartment, was held at virtual ground. Symmetrical solutions used were as follows (in mM): 1 mM EGTA, 250/125 mM Hepes/Tris, 50 mM KCl, pH 7.35 as cis solution and 53 mM Ca(OH)2, 50 mM KCl, 250 mM Hepes, pH 7.35 as trans solution. The concentration of free Ca2+ in the cis chamber was calculated with WinMaxC program (version 2.50; www.stanford.edu/∼cpatton/maxc.html). SR vesicles or microsomes containing purified RyR1/Cs2 complex were added to the cis side and fusion with the lipid bilayer was induced by making the cis side hyperosmotic by the addition of 400-500 mM KCl. After the appearance of potassium and chloride currents, the cis side was perfused with the cis solution. For the nominally Ca2+-free condition, EGTA was added to the cis side to get a final concentration of 5 mM EGTA. For the EGTA/ATP/Caffeine condition, ATP (2 mM final concentration) and caffeine (5 mM final concentration) were added to the cis side. Ca2+ (30 μM final concentration) was added to cis side to get the Ca2+-only condition. ATP (2 mM final concentration), caffeine (5 mM final concentration) and Ca2+ (30 μM final concentration) was added to cis side to get the ATP/Caffeine/Ca2+ condition. All ligands and additives were prepared as 100× stock solutions solubilized in cis buffer. At the end of each experiment, Ryanodine (10 μM) was added to block the RyR channel. Single-channel currents were recorded at 0 mV using a Bilayer Clamp BC-525C (Warner Instruments), filtered at 1 kHz using a Low-Pass Bessel Filter 8 Pole (Warner Instruments), and digitized at 4 kHz. All experiments were performed at room temperature (23°C). Data acquisition was performed using Digidata 1322A and Axoscope 10.1 software (Axon Instruments). The recordings were analyzed by using Clampfit 10.1 (Molecular Devices) and Graphpad Prism software.
Electron Microscopy
Grid preparation and imaging were carried out in two different ways (see Table S1). For grid preparation, early samples (RyR-EGTA and RyR-30 μM Ca2+-ATP-caffeine) were prepared as previously described (Zalk et al., 2015). Briefly, 3 μL of each sample was applied to holey carbon grids (C-flat CF-1.2/1.3-2C-T, Protochips Inc, NC) without prior plasma cleaning. Grids were blotted for ∼1 second and vitrified by rapidly plunging into liquid ethane at −180°C (Dubochet et al., 1988; Wagenknecht et al., 1988) with a Vitrobot (FEI).
For subsequent experiments (RyR-30 μM Ca2+, RyR-300 μM Ca2+-10 μM Ryanodine), holey-gold grids were prepared as described by Russo and Passmore (Russo and Passmore, 2014) from either C-flat CF-1.2/1.3-2C-T (Protochips Inc, NC) or from Quantifoil R1.2/1.3 (Quantifoil Micro Tools GmbH, Germany). 3 μL of each sample was applied to the holey-gold grids after plasma cleaning with a mixture of H2 and O2. Grids were blotted for 3.5 to 4 seconds and vitrified by rapidly plunging into liquid ethane at −180°C (Dubochet et al., 1988) (Wagenknecht et al., 1988) with a Vitrobot (FEI). Optimal blotting time was reproducible across samples.
Data acquisition was done on a FEI Tecnai F30 Polara (FEI, Eindhoven) operating at 300 kV. Datasets were collected with the automated data collection system Leginon (Suloway et al., 2005) at a nominal magnification of 31,000× in electron counting mode on a K2 Summit direct electron detector camera (Gatan, Pleasanton, CA) with a camera pixel size of 5 μm, corresponding to a calibrated pixel size of 1.255 Å on the specimen scale.
The pixel size was calibrated after obtaining the initial reconstruction by real-space correlation with a crystal structure of the N-terminal 3 domains of the receptor (PDB ID: 2XOA). A clear maximum was observed at 1.255 Å, and this value was found to be consistent across multiple different reconstructions, data collections and samples, and the same value is obtained using crystal structures of other fragments of the receptor. Applying the same approach to the reconstructions reported in (Yan et al., 2015) gave a calibrated pixel size for that map of 1.38 Å, compared to the nominal pixel size of 1.34 Å. This discrepancy highlights the importance of always performing pixel size calibration with a known crystal structure – a 3% difference in magnification could easily stymie interpretation of global conformational changes between structures determined of the same molecule by different groups. This does not have to be carried out on the molecule under investigation – the value determined here proved accurate for other datasets collected at this magnification on the same microscope for structurally unrelated assemblies.
For the early experiments on holey carbon grids, images were recorded in dose-fractionated mode, each image being fractionated into 20 frames, with an accumulation time of 0.2 s per frame. The dose rate on the camera was set to 8 counts per physical pixel per second. The total exposure time was 4 s, leading to a total accumulated dose of 25 electrons per Å2 on the specimen. The beam diameter was set at approximately 500 nm in order to capture 2 images per hole using image shift.
For subsequent experiments using holey gold grids, data acquisition was done under the same conditions except that the dose was increased from 25 to 50 e−/Å2 (100 e-/Å2 for the 10 μM ryanodine dataset) (Supplemental Figure S6), with images fractionated into 34 frames with 1.5 e−/Å2/frame. The total exposure time was 8 s (16 s for the 10 μm ryanodine dataset). The additional contrast provided by the doubling of the electron dose improved the accuracy of orientation assignment estimated by RELION (Scheres, 2012) from 1.3 degrees to 1 degree. The beam diameter was also increased from ∼0.5 μm to approximately 2 μm in order to obtain more parallel illumination and to expose gold on all sides of the hole to further limit beam-induced movement of the ice layer (Russo and Passmore, 2014). Therefore, only one image per hole was collected. Images were collected every second hole to reduce the possibility of double exposure of the imaged areas, which we found to be frequent with automated acquisition procedures.
Image Processing
The image processing followed the same protocol for every dataset. Datasets initially processed with a different protocol were subsequently reprocessed to match the established protocol. Dose-fractionated image stacks were aligned using either MotionCorr (Li et al., 2013) or UNBLUR (Grant and Grigorieff, 2015a), depending on the dataset, and the sum of aligned frames was used for further preprocessing. The first two frames were omitted in the early datasets collected on C-flat holey-carbon grids, as they contained motions greater than 4Å per frame on average. For the datasets collected on holey-gold grids all frames were kept, as the motion was measured as less than 1Å per frame in the first frames. The particles were picked with the reference-based automated particle picking procedure implemented in RELION 1.3 (Scheres, 2015) and their defocus values estimated by CTFFIND4 (Rohou and Grigorieff, 2015). Those particles were subjected to 3D classification using RELION with the number of classes, K, varying between 8 and 10. The particles from the highest-resolution classes were then pooled and their frame stacks realigned using alignparts_lmbfgs (Rubinstein and Brubaker, 2015) with optimized alignment parameters (See Figure S7). Radiation-damage weighting following the procedure described by Grant and Grigorieff (Grant and Grigorieff, 2015a) was used as incorporated in alignparts_lmbfgs, as follows. For each dataset, individual frames were filtered according to the optimal exposure curve calculated for Rotavirus VP6 (Grant & Grigorieff, 2015), modulated by a constant (the effective dose per frame), chosen based on the overall resolution of the resulting reconstruction. The resulting per frame resolution dependent weighting factors are presented in Table S2. In the case of the ryanodine dataset, for which the largest dose of 100e-/Å2 was collected, the dose-filtered reconstruction using the full 100e-/Å2 dose reached a resolution of 4.7 Å for the core, while a resolution of 4.2 Å in the core was obtained for a reconstruction using only the frames up to 60e-/Å2 total dose.
All pooled particles were then refined using RELION 1.4 with C4 symmetry imposed. The combination of per-particle frame alignment, radiation-damage weighting and use of CTFFIND4 brought the resolution of our RyR-EGTA closed-state dataset (Zalk et al., 2015) from 4.8 to 4.4 Å in the core (see Table S1 and S4). This procedure was applied to all subsequent datasets. On the later datasets, the combination of holey-gold grids, higher total electron dose and parallel illumination greatly reduced the number of particles needed to reach resolutions of 4.5 Å and better (see Table S4 and Figure S8) with the highest resolution dataset reaching 3.6 Å resolution in the core from only 65,000 particles. Plots of the orientation distribution for each dataset reveals that preferred orientations of the particles vary between datasets, independently of the grid support material (Figure S6B).
3D Classification
The first one or two rounds of 2D/3D classification were used to remove low-quality particles or non-particle contaminants from the dataset and to obtain a first assessment of the conformational variability in the dataset. The particles retained were then classified following the same protocol for each dataset in order to minimize bias in the classification results. The results were also compared with those of earlier non-standardized rounds of classification and showed consistent results. The remaining factors varying between experiments were the number of particles and the imaging conditions.
For each dataset, the orientation parameters of the retained particles were first refined together as a single class (Table S4). The orientations were then kept for a classification with C4 symmetry imposed, a local orientation search and a number of classes set at K=4. Maps from the previous combined refinement filtered at 25 Å served as initial references. Orientational sampling was set to 0.9 degrees until convergence (∼5 to 10% of particles changing class assignment) and then to 0.5 degrees for an additional 10 iterations (1% or less particles changing class assignment). Each class was then refined using RELION autorefine procedure with C4 symmetry imposed (Table S4). Deviations from C4 symmetry were observed in non-symmetrized reconstructions. These deviations were difficult to interpret due to the constraints introduced by the local nature of the particles alignment search at fine increment, which prevents particles from changing between the 4 possible orientations about the symmetry axis. This prompted us to impose C4 symmetry during classification to simplify interpretation of the results and to maintain consistency with subsequent refinements, which reached higher resolution when carried out with C4 symmetry imposed.
Those classifications gave very similar ranges of motions of the shell and similar radius for the maximally open and the maximally closed pores as in the primary classifications. To measure the reproducibility of those classifications, we ran the Ca2+/ATP/Caffeine dataset classification 3 times with the number of classes constant at k=4, but with randomly-generated seeds and with slightly different initial model resolutions (25, 30 and 35 Å resolution) to introduce more variability in the starting parameters. The resulting classifications gave similar results with differences in min and max shell height between each run of 0.9 and 1.2 Å standard deviation, respectively. We have not refined the classes of all the runs to high resolution; we therefore cannot estimate the standard deviation in pore aperture as precisely as for the shell height. But based on the ∼7 Å resolution classification volumes obtained, we do not see significant differences in pore aperture among the maximally open classes nor among the maximally closed classes of the Ca2+/ATP-Caffeine dataset.
Resolution Estimation
Reported resolutions are based on the ‘gold-standard’ protocol with the FSC=0.143 criterion using soft masks with a 8 pixel soft edge, and were corrected for the effects of the mask on the FSC curve using high-resolution noise substitution (Chen et al., 2013).
Because of the intrinsic flexibility of the channel, in particular the cytosolic assembly, resolution varies substantially between domains (Figure S6C). To characterize the local resolution on a domain level, FSCs were calculated domain by domain. The fitted atomic model of the ryanodine receptor was segmented following domain boundaries described in Figure S2 to create simulated density maps for each domain in UCSF Chimera (Pettersen et al., 2004). These simulated maps were then used to create masks for each domain of the receptor using relion_mask_create, with mask edges having an 8 pixel falloff. The FSC of each domain was then calculated by masking the two half-sets with the corresponding masks using relion_postprocess. Domain-by-domain resolutions are reported in Table S3 for each dataset. For the highest-resolution dataset (30 μM Ca2+) resolutions vary among domains from 8.5 Å (RY34) to 3.5 Å (CTD). Resolutions reported by this method agreed well, in most cases, with features visible in the corresponding domains and with the local resolution estimated by ResMap (Kucukelbir et al., 2014) (Figure S6B). However, in lower-resolution domains, local resolutions determined by domain masking tend to be artificially high due to the width of the mask edge overlapping with neighboring domains of much higher local resolution.
For each class of every dataset, average resolution for the whole protomer, the core and shell regions, as well as class distributions and shell height are reported in Table S4. Shell height is measured from the membrane plane (approximated by a plane intersecting with the S4-S5 linkers) to the centers of mass of the RY1&2 domains at the corner of the shell.
Prior to visualization, all density maps were sharpened by applying a negative B-factor estimated according to Rosenthal and Henderson (Rosenthal and Henderson, 2003) on solvent-flattened maps. An additional negative B-factor, empirically determined based on visual inspection of the map in Coot, was applied for visualization and model fitting in areas of particularly good local resolution within the core of the molecule. In preparation for model building and map comparison, all maps were aligned (using the ‘fitmap’ module of UCSF Chimera) to a single reference map, comprised of the transmembrane region of the highest-resolution map, such that all maps are effectively aligned on the transmembrane pore.
Model building
Model building proceeded in two stages. First, an optimal model was built and refined into the highest-resolution map (Ca2+-only classes 1-4 combined), and then, in the second stage, this model was used for fitting the other states – Ca2+/ATP/Caffeine (classes 1&2 and classes 3&4), EGTA/ATP/Caffeine (all classes combined), EGTA-only (all classes combined), and Ca2+/ryanodine (all classes combined). The Ca2+-only model had an EM ringer score of 2.3, consistent with a well-refined model.
In the first stage, fitting began by placing the highest-resolution model of the Ca2+-free closed state (PDB ID: 3J8H; (Yan et al., 2015)), into the Ca2+-30 μM map and rigid-body fitting each domain using a Python script launched from within Coot (Emsley et al., 2010). Recently determined crystal structures of SPRY1 (PDB ID: 5C33), SPRY2 (PDB ID: 4P9J) and the RY1&2 tandem repeat (PDB ID: 5C30) were incorporated into the model (Lau and Van Petegem, 2014; Yuchi et al., 2015), and the model was manually rebuilt and corrected in Coot based on the improved map, resulting in an increase in the overall number of placed and sequenced residues and a reduced number of chain breaks. Global real-space refinement of the model was carried out using phenix.real_space_refine, and this model became the initial model for subsequent fits to maps of other samples and conformational classes. Two regions of unassigned sequence remain. These are a large portion of SPRY3 (1275-1573) and the second half of BSol (2487-2735 & 2939-3613). Either a cryoEM reconstruction of substantially improved resolution, or in the case of SPRY3 a crystal structure of this region, will be required to address these remaining ambiguities in the structure. Residues with uncertain sequence assignment have been given the residue name “UNK”; The sequence numbering still reflects our best guess at the most likely assignment of the residue in question. All maps used in model fitting were first aligned to a reference map consisting of density corresponding to the RyR1 transmembrane region alone, and resampled upon the grid of this map, allowing facile comparison of the different structures. The map used in model fitting has had B-factor sharpening applied to a level appropriate for optimal visualization of high resolution features such as sidechains in the best resolved parts of the structure. In order to have a better view of connectivity, model fitting and interpretation used two maps in conjunction – the aforementioned map, and a version of it that had been filtered at 4.5 Å, for which connectivity and low resolution features are more clearly defined due to the absence of high resolution noise.
In the second stage, the refined atomic model was rigid-body fit to each map, domain by domain, using the same Python script as used in the initial stages of building the reference model. The entire chain was manually inspected, and sections of the main chain that were flexibly deformed (for example, S6/S6c and interdomain interfaces) were corrected using the real-space refine zone tool within Coot, with Ramachandran restraints applied. In the case of the EGTA-only, Ca2+/ATP/Cafeine and ryanodine sets, the putative transmembrane helix hairpin was modeled in, although sequence was not assigned. After identification and placement of ligands (see below), a final round of global real space refinement was carried out using phenix.real_space_refine.
Ligand identification and assignment
Candidate ligand binding sites (for ATP, caffeine, ryanodine and Ca2+) were identified using locally aligned difference maps, calculated as follows for each domain (implemented as a command line alias in UCSF Chimera). First, the density within a 5 Å border of the atoms is extracted (using the ‘vop zone’ command) for both map/model pairs. Then, the two map fragments are aligned, using the ‘fitmap’ command, and a difference map is calculated in real space using the ‘vop subtract’ command. This procedure allows for the identification of ligand densities from a difference map in the presence of domain reorientation, which would otherwise introduce too much noise into the difference map to visualize the bound ligands. Most of the locally aligned difference maps generated by this procedure were essentially featureless, but two had strong peaks of distinctive shape – those calculated around the CTD, and those calculated around the transmembrane region. The presence and morphology of these density features corresponded to the addition of specific ligands – a spherical density feature at the interface of the CTD and CSol was exclusively present when Ca2+ was included, while two larger, more elongated features at the S6c/CTD boundary and the CTD-S2S3 interface only appeared in the presence of ATP and caffeine. The larger of the pair of features correlating with the addition of ATP/caffeine could readily be assigned as ATP based on density fit considerations and the local chemical environment at the S6c/CTD/TaF interface. The smaller feature was more ambiguous, but was assigned as caffeine based on size, shape, and the fact that it was not present in a map obtained in the presence of Ca2+ and ATP alone (unpublished data). A peak was also located within the transmembrane pore, above Q4933 specifically in the presence of ryanodine, and was assigned as such.
Pore radius calculations
Pore radius calculations were carried out on the final atomic models of the core region of the channel (residues 3639-5037) using HOLE (Smart et al., 1996), with the default set of atomic radii; graphs of the pore radius vs the channel coordinate were generated using gnuplot. Prior to HOLE, incomplete sidechains were replaced using the DOCK-PREP tool as implemented in UCSF Chimera, which picks the most probable rotamer from the Dunbrack rotamer library that introduces the least severe steric clashes. HOLE calculations carried out on models with sidechains omitted yielded qualitatively consistent results (in terms of the differences between conformational classes) to those performed using the full atomic models with sidechains intact.
Movie preparation
The movies included in Data File S1, which are intended to convey the nature of the conformational changes observed between different states of the channel based solely on linear interpolation of the aligned density maps, were prepared in UCSF Chimera as follows: The “Morph Map” tool in UCSF Chimera was used to create a trajectory consisting of a linear interpolation at each voxel of the density values of the pair of maps being compared. Each resulting trajectory was displayed as a maximum intensity projection using the following command line alias, alias ^volume_project background solid black; volume $1 step 1 sd level 0,0 color white sd level 20,1 color white style solid projection Mode 3d maximum Intensity Projection true btCorrection true linear Interpolation true; unset depthCue such that the intensity of each voxel is related to the density values (higher density is white, while zero density is black. In each case, five key density slabs of 5-10 Å thickness were chosen to best represent the observed conformational changes. These consisted of two slabs parallel to and intersecting with the channel axis, spaced 45 degrees apart about the channel axis; and three slabs orthogonal to the channel axis, one of which is at the putative activation gate (I4937), the second of which is at the height of the EF1&2 domains, and the third of which is in the plane of NTD-A&B. The location and thickness of each slab is depicted on an orthogonal view of the entire volume as a thumbnail in the lower right-hand corner of the movie frame. Movies were saved in UCSF Chimera as MP4, converted to Graphics Interchange Format (GIF) using VideoBlend, labeled and annotated frame-by-frame using Affinity Designer and Gnu Image Manipulation Program (GIMP), and finally converted back to MP4 format.
Supplementary Material
Supplemental Data File 1, related to Figure 3: This dataset contains movies depicting morphs of the coordinates of the atomic models of several key structural states. A detailed description of the contents is provided as a separate document within the archive.
Supplemental Data File 2, related to Figure 3: This dataset contains movies depicting morphs of the density maps of several key class comparisons. A detailed description of the contents is provided as a separate document within the archive.
Figure S1. Functional characterization of RyR1/Cs2 complex, related to Figure 1.
(A) Representative single-channel current traces of purified RyR1 channels measured under the following conditions: 1) cis chamber, 5 mM EGTA and no added Ca2+ (nominally Ca2+ free) [Ca2+]; 2) cis chamber, nominally Ca2+ free plus 2 mM ATP and 5 mM caffeine; 3) cis chamber, 30 μM cytosolic [Ca2+]; 4) cis chamber, 30 μM cytosolic [Ca2+] plus 2 mM ATP and 5 mM caffeine. The trans chamber contained [Ca2+] = 53 mM. Channel openings are shown as upward deflections; the closed (C−) state of the channel is indicated by horizontal bars at the beginning of each trace. Representative traces are displayed with two different time scales (100 ms for upper trace and 1000 ms for lower trace) as indicated by scale bars, and the PO, TO (average open time) and TC (average closed time) are shown above each trace. Specific modification by 10 μM ryanodine is displayed. Bar graph summarizing single channel PO (B), TO (C) and TC (D) nominally Ca2+-free with 2 mM ATP and 5 mM caffeine, 30 μM cytosolic [Ca2+] and 30 μM cytosolic [Ca2+] with 2 mM ATP and 5 mM caffeine (n = 6) channels.
Figure S2. Domain organization of RyR1, related to Figure 1.
(A) Revised domain boundaries are colored both on a 2D schematic of the sequence (with the length of each box proportional to the number of residues it contains) and (B) on a low-pass filtered surface representation of the model, using the same coloring scheme. The hypothetical location of the TM×2 transmembrane hairpin is indicated in (A) with a black box. (C) The junctional solenoid (JSol, here shown as a khaki molecular surface) forms the attachment site for the three other solenoid domains – NTD-C (dark blue ribbon), BSol (green ribbon) and CSol (purple ribbon), as well as for calstabin2 (gold). In the case of each of the interacting solenoids, only the first repeat of the solenoid is represented here for clarity. Both opposed views represented here are parallel to the membrane plane.
Figure S3. Coordination of bound Ca2+ at a conserved site at the CTD-CSol interface, related to Figure 1.
(A) Region of the density map and model around the Ca2+-binding site in the Ca2+-only structure, with fitted model. Residues believed to participate in Ca2+-coordination either directly (E3893, E3967, T5001) or indirectly (Q3970, H3895) are labeled, as is the auxiliary interaction at the same interface formed by E4032 and Q5006. Labeled residues are depicted in full; all others are shown as a Cα trace only. Core solenoid residues are colored sea-green here, and CTD residues depicted in dark red. The Ca2+ ion is represented by a green sphere. The density map is contoured at 2 σ. (B) Portion of a sequence alignment of representative RyR and IP3R genes, prepared using MUSCLE (Edgar, 2004) with default parameters, visualized using Jalview (Waterhouse et al., 2009). The alignment is colored by conservation using a modified version of the Zappo coloring scheme, with a conservation cutoff of 60%. Residues involved in Ca2+ coordination (E3893, H3895, E3967 and Q3970) in addition to a residue (E4032) previously identified as playing a role in Ca2+-activation of RyR1 and RyR2 are labeled. The first sequence in the alignment is that of rabbit RyR1; the Uniprot accession number of each sequence is given in parentheses.
Figure S4. Gating-independent motions of the cytoplasmic assembly, related to Figure 2.
A comparison of the cytoplasmic assembly conformations for each class of four different datasets: (A), nominally Ca2+-free, (B), Ca2+-only, (C), ATP/caffeine only, (D), Ca2+/ATP/caffeine, and (E), all of (A-D) together, in two orientations (left, in the membrane plane, and right, from the cytoplasm). In all panels (A-E), the structure of one EGTA class is shown for reference in dark gray, and a single rigid unit, comprising the bridging solenoid of one protomer and NTD-A-JSol of the adjacent protomer, is shown in colored ribbon for each of the four classes in the dataset. Pairwise comparisons between the rigid units from each class give the rotation axes, which are depicted as sticks in each panel. (F) An alternative visualization of the conformational variability in each dataset, displaying the height of the RY1&2 domain above the SR membrane plane for each class of each dataset as a box and whisker plot. The fraction of particles in each dataset belonging to each class is represented by the size of each circular marker; the class number is given by the color of the marker.
Figure S5. Schematic representation of ryanodine receptor activation and pore opening, related to Figure 4.
Boxes represent datasets in particular conditions: (1) EGTA, (2) EGTA/caffeine/ATP, (3) Ca2+, (4) Ca2+/ATP/caffeine, (5) ryanodine-activated, (6) ryanodine-blocked. Square activation module: Apo state; rounded activation module: primed state. Dotted borders: state not structurally characterized, but known to exist from bilayer recordings.
Figure S6. Representative data and local resolution, related to Figure 1.
(A) Motion-corrected micrograph from the EGTA dataset collected on holey-carbon grids, compared with micrographs from the 30 μM Ca2+ and 10 μM ryanodine datasets collected on holey-gold grids at different electron doses. (B) Euler angle distribution of all particles used for calculating the 3D reconstruction of each dataset with c4 symmetry imposed. The Euler angle distributions displayed here correspond to reconstructions of all the particles of a given dataset; the reconstructed volume is displayed at the center of the distribution as a reference for orientation. The height and color of cylinders indicate the number of particles in each view. Dark blue: Orientation with the smallest number of particles, Red: orientation with the largest number of particles. The plot is calculated with an angular sampling of 3.7 degrees between views. Datasets from left to right: EGTA, Ca2+/ATP/Caffeine, EGTA/ATP/Caffeine, Ca2+, Ryanodine. (C) Reconstruction colored by local resolution (from 3.0 Å, blue, to 6.0 Å, red) using ResMap (Kucukelbir et al., 2014), viewed from the membrane plane, together with an orthogonal slab through the reconstruction. (D) Density quality in the core of the assembly (centered on the CTD) with fitted atomic model displayed as a Cα-trace with sidechains. Many bulky residues that are well defined in the density.
Figure S7. Particle polishing optimization, related to Figure 1.
Plots of local motion correction with individual particle trajectories obtained from alignparts_lmbfgs. The color of each line represents the frame number, from 0 (black) to 35 (blue). All plots are exaggerated 25 times. The smoothing parameter is used as a penalty for having abrupt changes in direction during a trajectory. This is based on the assumption that particles are unlikely to have sudden changes in direction. The “neiflag” parameter determines the extent of local averaging for the smoothing of particle trajectories. This parameter is derived from the assumption that nearby particles have correlated trajectories. (A) Series of plots showing the change in individual particle trajectories as the rmax2 (maximum resolution used during alignment; in Å) variable is decreased. (B) The original particle trajectories determined with the default parameters (left) and the new particle trajectories determined with the optimized parameters (right).
Figure S8. Fourier Shell Correlation curves and central slices for each dataset, related to Figure 1.
Dataset: (A) EGTA, (B) Ca2+-ATP-Caffeine, (C) EGTA-ATP-Caffeine, (D) Ca2+, (E) Ryanodine. (Top) FSC curves using the ‘gold standard’ protocol for the complete protomer (gray), the shell (light blue) and the core (light red), with the respective resolutions derived from FSC=0.143. Resolutions for the shell, the core, and the entire protomer were calculated using relion_postprocess, using masks created from the molecular model of the respective regions and shown as thumbnails in (A). (Bottom) Central slices from the 3D reconstruction in the y and z axis with (left) and without (right) B-factor sharpening.
Table S1: Solution conditions, data collection and reconstruction statistics, related to Figure 1.
Table S2. Electron dose-dependent per frame low-pass resolution cutoff, related to Figure 1.
The program alignparts_lmbfgs outputs a weighted sum of the movie frames that maximizes the signal to noise ratio in each resolution shell according to the critical dose calculated by Grant & Grigorieff (Grant and Grigorieff, 2015b). The table describes the effective resolution cutoff produced by the scheme when our ad-hoc exposure factor is applied.
Table S3. Dataset size and resolution by domain, related to Figure 1
Resolutions after refinement of all particles of each dataset are described, unless otherwise specified. Domain boundaries are described in supplemental figure. Resolution measurements are described in Methods. The highest resolution domains for each reconstruction are highlighted in bold type, while the lowest resolution domains are indicated in italics.
Table S4. Classification summary for each dataset, related to Figure 2.
For each dataset, classes are described in terms of number of particles, particle distribution, overall shell conformation and resolution. Resolution measurements are described in Methods. Shell height is measured in Å from the top of the membrane plane to the center of mass of RY12 domain (also plotted in Figure S4F).
Key Resources Table.
| Reagent or Resource | Source | Identifier |
|---|---|---|
| Chemicals, Peptides, and Recombinant Proteins | ||
| GST-Calstabin1 | Zalk et al. 2015 | |
| GST-Calstabin2 | Zalk et al. 2015 | |
| Calf intestinal alkaline phosphatase | NEB | Cat# M0290L |
| CHAPS | Anatrace | Cat# C316S; CAS: 75621-03-3 |
| DOPC | Avanti Polar Lipids | Cat# 850375C; CAS:4235-95-4 |
| DOPE | Avanti Polar Lipids | Cat# 850725C; CAS:4004-05-1 |
| Na2ATP | Sigma-Aldrich | Cat# A2383; CAS:34369-07-8 |
| Ryanodine | Abcam | Cat# ab120083; CAS:15662-33-6 |
| Caffeine | Sigma-Aldrich | Cat# C0750; CAS:58-08-2 |
| Critical Commercial Assays | ||
| GSTrap | GE Healthcare | Cat# 17-5281-01 |
| Ni Sepharose 6 fast flow | GE Healthcare | Cat# 17-5318-01 |
| Thrombin | Biopharm | Cat# 91-035 |
| TSK-GEL G4SWxl | Tosoh | Cat# 20025 |
| Deposited Data | ||
| N-terminal 3 domain of ryr1 | Tung et al., 2010 | PDB: 2XOA |
| RyR1-Cs2 EGTA closed-state | Zalk et al., 2015 | PDB: 3J8E |
| RyR1-Cs2 EGTA closed-state | Yan et al., 2015 | PDB: 3J8H |
| SPRY1 domain of RyR2 | Yushi & Van Petegem, 2015 | PDB: 5C33 |
| SPRY2 domain of RyR1 | Lau & Van Petegem, 2014 | PDB: 4P9J |
| RY1&2 domain of RyR1 | Yuchi & Van Petegem | PDB: 5C30 |
| RyR1-Cs2, Ca2+ only (all particles) | This study | PDB:5T15 EMDB:EMD-8342 |
| RyR1-Cs2, Ca2+ only (Class 1) | This study | PDB:TBA EMDB:TBA |
| RyR1-Cs2, Ca2+ only (Class 2) | This study | PDB:TBA EMDB:TBA |
| RyR1-Cs2, Ca2+ only (Class 3) | This study | PDB:TBA EMDB:TBA |
| RyR1-Cs2, Ca2+ only (Class 4) | This study | PDB:TBA EMDB:TBA |
| RyR1-Cs2, EGTA (all particles) | This study | PDB:TBA EMDB:TBA |
| RyR1-Cs2, EGTA (Class 1) | This study | PDB:TBA EMDB:TBA |
| RyR1-Cs2, EGTA (Class 2) | This study | PDB:TBA EMDB:TBA |
| RyR1-Cs2, EGTA (Class 3) | This study | PDB:TBA EMDB:TBA |
| RyR1-Cs2, EGTA (Class 4) | This study | PDB:TBA EMDB:TBA |
| RyR1-Cs2, ATP/Caf. (all particles) | This study | PDB:TBA EMDB:TBA |
| RyR1-Cs2, ATP/Caf. (Class 1) | This study | PDB:TBA EMDB:TBA |
| RyR1-Cs2, ATP/Caf. (Class 2) | This study | PDB:TBA EMDB:TBA |
| RyR1-Cs2, ATP/Caf. (Class 3) | This study | PDB:TBA EMDB:TBA |
| RyR1-Cs2, ATP/Caf. (Class 4) | This study | PDB:TBA EMDB:TBA |
| RyR1-Cs2, Ca2+/ATP/Caf. (all particles) | This study | PDB:TBA EMDB:TBA |
| RyR1-Cs2, Ca2+/ATP/Caf. (Class 1) | This study | PDB:TBA EMDB:TBA |
| RyR1-Cs2, Ca2+/ATP/Caf. (Class 2) | This study | PDB:TBA EMDB:TBA |
| RyR1-Cs2, Ca2+/ATP/Caf. (Class 1&2) | This study | PDB:TBA EMDB:TBA |
| RyR1-Cs2, Ca2+/ATP/Caf. (Class 3) | This study | PDB:TBA EMDB:TBA |
| RyR1-Cs2, Ca2+/ATP/Caf. (Class 4) | This study | PDB:TBA EMDB:TBA |
| RyR1-Cs2, Ca2+/ATP/Caf. (Class 3&4) | This study | PDB:TBA EMDB:TBA |
| RyR1-Cs2, ryanodine (all particles) | This study | PDB:TBA EMDB:TBA |
| RyR1-Cs2, ryanodine (Class 1) | This study | PDB:TBA EMDB:TBA |
| RyR1-Cs2, ryanodine (Class 2) | This study | PDB:TBA EMDB:TBA |
| RyR1-Cs2, ryanodine (Class 3) | This study | PDB:TBA EMDB:TBA |
| RyR1-Cs2, ryanodine (Class 4) | This study | PDB:TBA EMDB:TBA |
| Experimental Models: Organisms/Strains | ||
| Male New Zealand rabbits skeletal muscle | Sierra for Medical Science | By request |
| Software and Algorithms | ||
| MaxChelator | Bers et al., 2010 | version 2.50; http://maxchelator.stanford.edu |
| Axoscope 10.1 | Axon Instruments | http://mdc.custhelp.com/app/answers/detail/a_id/18779 |
| Clampfit 10.1 | Molecular Devices | http://mdc.custhelp.com/app/answers/detail/a_id/18779 |
| MotionCorr | Li et al., 2013 | http://cryoem.ucsf.edu/software/driftcorr.html |
| UNBLUR | Grant and Grigorieff, 2015 | http://grigoriefflab.janelia.org/unblur |
| RELION 1.4 | Scheres, 2015 | http://www2.mrc-lmb.cam.ac.uk/relion |
| CTFFIND4 | Rohou and Grigorieff, 2015 | http://grigoriefflab.janelia.org/ctf |
| alignparts_lmbfgs | Rubinstein and Brubaker, 2015 | https://sites.google.com/site/rubinsteingroup/direct-detector-align_lmbfgs |
| UCSF Chimera | Pettersen et al., 2004 | https://www.cgl.ucsf.edu/chimera/ |
| ResMap | Kucukelbir et al., 2014 | http://resmap.sourceforge.net |
| EMringer | Barad et al., 2015 | http://fraserlab.com/2015/02/18/EMringer |
| Coot | Emsley et al., 2010 | http://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| phenix.real_space_refine | Afonine et al., 2013 | https://www.phenix-online.org/documentation/reference/real_space_refine.html |
| HOLE | Smart et al., 1996 | http://www.smartsci.uk/hole/ |
| DOCK-PREP | Lang et al., 2009 | https://www.cgl.ucsf.edu/chimera/docs/ContributedSoftware/dockprep/dockprep.html |
| VideoBlend | Videoblend | http://www.videoblend.org |
| Affinity Designer | Serif | https://affinity.serif.com |
| Gnu Image Manipulation Program | GIMP | https://www.gimp.org |
| Other | ||
| C-flat CF-1.2/1.3-2C-T grids | Protochips Inc, NC | CF-1.2/1.3-2C-T |
| Quantifoil R1.2/1.3 grids | Quantifoil Micro Tools GmbH, Germany | R1.2/1.3 |
Acknowledgments
This work was supported by grants from the National Institutes of Health (R01AR060037 and R01HL061503 to A.R.M, U54GM095315 to W.A.H., R01GM29169 to J.F.), by HHMI (to J.F.). R.Z. was a fellow of the American Heart Association (0625919T), K.C. was supported by the HHMI EXROP-Capstone Program and O.B.C. was supported by a Charles H. Revson Senior Fellowship.
Footnotes
Author Contributions: Conceptualization, A.G, O.B.C, R.Z., W.A.H., A.R.M. and J.F.; Investigation, A.G, O.B.C, R.Z., Q.Y, K.C. and R.G.; Writing, A.G., O.B.C., R.Z., K.C., R.G., W.A.H., A.R.M. and J.F.
Accession codes: PDB, EMDB and EMPIAR accession codes TBA.
References
- Bezprozvanny I, Watras J, Ehrlich BE. Bell-shaped calcium-response curves of Ins(1,4,5)P3- and calcium-gated channels from endoplasmic reticulum of cerebellum. Nature. 1991;351:751–754. doi: 10.1038/351751a0. [DOI] [PubMed] [Google Scholar]
- Brillantes AB, Ondrias K, Scott A, Kobrinsky E, Ondriasová E, Moschella MC, Jayaraman T, Landers M, Ehrlich BE, Marks AR. Stabilization of calcium release channel (ryanodine receptor) function by FK506-binding protein. Cell. 1994;77:513–523. doi: 10.1016/0092-8674(94)90214-3. [DOI] [PubMed] [Google Scholar]
- Callaway C, Seryshev A, Wang JP, Slavik KJ, Needleman DH, Cantu C, Wu Y, Jayaraman T, Marks AR, Hamilton SL. Localization of the high and low affinity [3H] ryanodine binding sites on the skeletal muscle Ca2+ release channel. J Biol Chem. 1994;269:15876–15884. [PubMed] [Google Scholar]
- Carroll S, Skarmeta JG, Yu X, Collins KD, Inesi G. Interdependence of ryanodine binding, oligomeric receptor interactions, and Ca2+ release regulation in junctional sarcoplasmic reticulum. Arch Biochem Biophys. 1991;290:239–247. doi: 10.1016/0003-9861(91)90615-p. [DOI] [PubMed] [Google Scholar]
- Chan WM, Welch W, Sitsapesan R. Structural factors that determine the ability of adenosine and related compounds to activate the cardiac ryanodine receptor. Br J Pharmacol. 2000;130:1618–1626. doi: 10.1038/sj.bjp.0703459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, McMullan G, Faruqi AR, Murshudov GN, Short JM, Scheres SHW, Henderson R. High-resolution noise substitution to measure overfitting and validate resolution in 3D structure determination by single particle electron cryomicroscopy. Ultramicroscopy. 2013;135:24–35. doi: 10.1016/j.ultramic.2013.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu A, Díaz-Muñoz M, Hawkes MJ, Brush K, Hamilton SL. Ryanodine as a probe for the functional state of the skeletal muscle sarcoplasmic reticulum calcium release channel. Mol Pharmacol. 1990;37:735–741. [PubMed] [Google Scholar]
- Dias JM, Vogel PD. Effects of Small Molecule Modulators on ATP Binding to Skeletal Ryanodine Receptor. Protein J. 2009;28:240–246. doi: 10.1007/s10930-009-9189-9. [DOI] [PubMed] [Google Scholar]
- Du GG, Sandhu B, Khanna VK, Guo XH, MacLennan DH. Topology of the Ca2+ release channel of skeletal muscle sarcoplasmic reticulum (RyR1) Proc Natl Acad Sci USA. 2002;99:16725–16730. doi: 10.1073/pnas.012688999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubochet J, Adrian M, Chang JJ, Homo JC, Lepault J, McDowall AW, Schultz P. Cryo-electron microscopy of vitrified specimens. Quart Rev Biophys. 1988;21:129–228. doi: 10.1017/s0033583500004297. [DOI] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucl Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efremov RG, Leitner A, Aebersold R, Raunser S. Architecture and conformational switch mechanism of the ryanodine receptor. Nature. 2015;517:39–43. doi: 10.1038/nature13916. [DOI] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Euden J, Mason SA, Williams AJ. Functional Characterization of the Cardiac Ryanodine Receptor Pore-Forming Region. PLoS ONE. 2013;8:e66542. doi: 10.1371/journal.pone.0066542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan G, Baker ML, Wang Z, Baker MR, Sinyagovskiy PA, Chiu W, Ludtke SJ, Serysheva II. Gating machinery of InsP3R channels revealed by electron cryomicroscopy. Nature. 2015;527:336–341. doi: 10.1038/nature15249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fessenden JD, Chen L, Wang Y, Paolini C, Franzini-Armstrong C, Allen PD, Pessah IN. Ryanodine receptor point mutant E4032A reveals an allosteric interaction with ryanodine. Proc Natl Acad Sci USA. 2001;98:2865–2870. doi: 10.1073/pnas.041608898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fessenden JD, Feng W, Pessah IN, Allen PD. Mutational analysis of putative calcium binding motifs within the skeletal ryanodine receptor isoform, RyR1. J Biol Chem. 2004;279:53028–53035. doi: 10.1074/jbc.M411136200. [DOI] [PubMed] [Google Scholar]
- Franzini-Armstrong C, Protasi F, Ramesh V. Shape, size, and distribution of Ca(2+) release units and couplons in skeletal and cardiac muscles. Biophys J. 1999;77:1528–1539. doi: 10.1016/S0006-3495(99)77000-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant T, Grigorieff N. Measuring the optimal exposure for single particle cryo-EM using a 2.6 Å reconstruction of rotavirus VP6. eLife. 2015a;4:e06980. doi: 10.7554/eLife.06980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant T, Grigorieff N. Measuring the optimal exposure for single particle cryo-EM using a 2.6 Å reconstruction of rotavirus VP6. eLife. 2015b;4:e06980. doi: 10.7554/eLife.06980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Sun B, Xiao Z, Liu Y, Wang Y, Zhang L, Wang R, Chen SRW. The EF-hand Ca2+ Binding Domain Is Not Required for Cytosolic Ca2+ Activation of the Cardiac Ryanodine Receptor. J Biol Chem. 2016;291:2150–2160. doi: 10.1074/jbc.M115.693325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton SL, Reid MB. RyR1 modulation by oxidation and calmodulin. Antioxid Redox Signal. 2000;2:41–45. doi: 10.1089/ars.2000.2.1-41. [DOI] [PubMed] [Google Scholar]
- Imagawa T, Smith JS, Coronado R, Campbell KP. Purified ryanodine receptor from skeletal muscle sarcoplasmic reticulum is the Ca2+-permeable pore of the calcium release channel. J Biol Chem. 1987;262:16636–16643. [PubMed] [Google Scholar]
- Kaftan E, Marks AR, Ehrlich BE. Effects of rapamycin on ryanodine receptor/Ca2+-release channels from cardiac muscle. Circ Res. 1996;78:990–997. doi: 10.1161/01.res.78.6.990. [DOI] [PubMed] [Google Scholar]
- Kermode H, Williams AJ, Sitsapesan R. The Interactions of ATP, ADP, and Inorganic Phosphate with the Sheep Cardiac Ryanodine Receptor. Biophys J. 1998;74:1296–1304. doi: 10.1016/S0006-3495(98)77843-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimlicka L, Lau K, Tung CC, Van Petegem F. Disease mutations in the ryanodine receptor N-terminal region couple to a mobile intersubunit interface. Nat Comms. 2013;4:1506. doi: 10.1038/ncomms2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucukelbir A, Sigworth FJ, Tagare HD. Quantifying the local resolution of cryo-EM density maps. Nat Meth. 2014;11:63–65. doi: 10.1038/nmeth.2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushmerick MJ, Moerland TS, Wiseman RW. Mammalian skeletal muscle fibers distinguished by contents of phosphocreatine, ATP, and Pi. Proc Natl Acad Sci USA. 1992;89:7521–7525. doi: 10.1073/pnas.89.16.7521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labro AJ, Raes AL, Grottesi A, Van Hoorick D, Sansom MSP, Snyders DJ. Kv channel gating requires a compatible S4-S5 linker and bottom part of S6, constrained by non-interacting residues. J Gen Physiol. 2008;132:667–680. doi: 10.1085/jgp.200810048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau K, Van Petegem F. Crystal structures of wild type and disease mutant forms of the ryanodine receptor SPRY2 domain. Nat Comms. 2014;5:1–11. doi: 10.1038/ncomms6397. [DOI] [PubMed] [Google Scholar]
- Laver DR, Lenz GK, Lamb GD. Regulation of the calcium release channel from rabbit skeletal muscle by the nucleotides ATP, AMP, IMP and adenosine. J Physiol. 2001;537:763–778. doi: 10.1111/j.1469-7793.2001.00763.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HB, Xu L, Meissner G. Reconstitution of the skeletal muscle ryanodine receptor-Ca2+ release channel protein complex into proteoliposomes. J Biol Chem. 1994;269:13305–13312. [PubMed] [Google Scholar]
- Li P, Chen SR. Molecular basis of Ca(2)+ activation of the mouse cardiac Ca(2)+ release channel (ryanodine receptor) J Gen Physiol. 2001;118:33–44. doi: 10.1085/jgp.118.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Mooney P, Zheng S, Booth CR, Braunfeld MB, Gubbens S, Agard DA, Cheng Y. Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nat Meth. 2013;10:584–590. doi: 10.1038/nmeth.2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx SO, Ondrias K, Marks AR. Coupled gating between individual skeletal muscle Ca2+ release channels (ryanodine receptors) Science. 1998;281:818–821. doi: 10.1126/science.281.5378.818. [DOI] [PubMed] [Google Scholar]
- Mei Y, Xu L, Mowrey DD, Mendez Giraldez R, Wang Y, Pasek DA, Dokholyan NV, Meissner G. Channel Gating Dependence on Pore Lining Helix Glycine Residues in Skeletal Muscle Ryanodine Receptor. J Biol Chem. 2015;290:17535–17545. doi: 10.1074/jbc.M115.659672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murayama T, Kurebayashi N, Oba T, Oyamada H, Oguchi K, Sakurai T, Ogawa Y. Role of amino-terminal half of the S4-S5 linker in type 1 ryanodine receptor (RyR1) channel gating. J Biol Chem. 2011;286:35571–35577. doi: 10.1074/jbc.M111.255240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagasaki K, Fleischer S. Ryanodine sensitivity of the calcium release channel of sarcoplasmic reticulum. Cell Calcium. 1988;9:1–7. doi: 10.1016/0143-4160(88)90032-2. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Popova OB, Baker MR, Tran TP, Le T, Serysheva II. Identification of ATP-Binding Regions in the RyR1 Ca2+ Release Channel. PLoS ONE. 2012;7:e48725. doi: 10.1371/journal.pone.0048725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranatunga KM, Chen SRW, Ruest L, Welch W, Williams AJ. Quantification of the effects of a ryanodine receptor channel mutation on interaction with a ryanoid. Mol Membr Biol. 2007;24:185–193. doi: 10.1080/09687860601076522. [DOI] [PubMed] [Google Scholar]
- Rohou A, Grigorieff N. CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J Struct Biol. 2015;192:216–221. doi: 10.1016/j.jsb.2015.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal PB, Henderson R. Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J Mol Biol. 2003;333:721–745. doi: 10.1016/j.jmb.2003.07.013. [DOI] [PubMed] [Google Scholar]
- Rubinstein JL, Brubaker MA. Accepted Manuscript. J Struct Biol. 2015:1–12. [Google Scholar]
- Russo CJ, Passmore LA. Ultrastable gold substrates for electron cryomicroscopy. Science. 2014;346:1377–1380. doi: 10.1126/science.1259530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samsó M, Feng W, Pessah IN, Allen PD. Coordinated Movement of Cytoplasmic and Transmembrane Domains of RyR1 upon Gating. PLoS Biol. 2009;7:e85. doi: 10.1371/journal.pbio.1000085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres SHW. Journal of Structural Biology. J Struct Biol. 2012;180:519–530. doi: 10.1016/j.jsb.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres SHW. Semi-automated selection of cryo-EM particles in RELION-1.3. J Struct Biol. 2015;189:114–122. doi: 10.1016/j.jsb.2014.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenmakers TJ, Visser GJ, Flik G, Theuvenet AP. CHELATOR: an improved method for computing metal ion concentrations in physiological solutions. BioTechniques. 1992;12:870–4–876–9. [PubMed] [Google Scholar]
- Serysheva II, Schatz M, van Heel M, Chiu W, Hamilton SL. Structure of the skeletal muscle calcium release channel activated with Ca2+ and AMP-PCP. Biophys J. 1999;77:1936–1944. doi: 10.1016/S0006-3495(99)77035-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P, Ishiyama N, Nair U, Li W, Dong A, Miyake T, Wilson A, Ryan T, MacLennan DH, Kislinger T, et al. Structural determination of the phosphorylation domain of the ryanodine receptor. FEBS J. 2012;279:3952–3964. doi: 10.1111/j.1742-4658.2012.08755.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smart OS, Neduvelil JG, Wang X, Wallace BA, Sansom MS. HOLE: a program for the analysis of the pore dimensions of ion channel structural models. J Mol Graph. 1996;14:354–60–376. doi: 10.1016/s0263-7855(97)00009-x. [DOI] [PubMed] [Google Scholar]
- Suloway C, Pulokas J, Fellmann D, Cheng A, Guerra F, Quispe J, Stagg S, Potter CS, Carragher B. Automated molecular microscopy: the new Leginon system. J Struct Biol. 2005;151:41–60. doi: 10.1016/j.jsb.2005.03.010. [DOI] [PubMed] [Google Scholar]
- Tanna B, Welch W, Ruest L, Sutko JL, Williams AJ. The Interaction of an Impermeant Cation with the Sheep Cardiac RyR Channel Alters Ryanoid Association. Mol Pharmacol. 2006;69:1990–1997. doi: 10.1124/mol.105.021659. [DOI] [PubMed] [Google Scholar]
- Tung CC, Lobo PA, Kimlicka L, Van Petegem F. The amino-terminal disease hotspot of ryanodine receptors forms a cytoplasmic vestibule. Nature. 2010;468:585–588. doi: 10.1038/nature09471. [DOI] [PubMed] [Google Scholar]
- Wagenknecht T, Grassucci R, Frank J. Electron microscopy and computer image averaging of ice-embedded large ribosomal subunits from Escherichia coli. J Mol Biol. 1988;199:137–147. doi: 10.1016/0022-2836(88)90384-1. [DOI] [PubMed] [Google Scholar]
- Waterhouse AM, Procter JB, Martin DMA, Clamp M, Barton GJ. Jalview Version 2--a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25:1189–1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch W, Williams AJ, Tinker A, Mitchell KE, Deslongchamps P, Lamothe J, Gerzon K, Bidasee KR, Besch HR, Airey JA, et al. Structural components of ryanodine responsible for modulation of sarcoplasmic reticulum calcium channel function. Biochemistry. 1997;36:2939–2950. doi: 10.1021/bi9623901. [DOI] [PubMed] [Google Scholar]
- Witcher DR, McPherson PS, Kahl SD, Lewis T, Bentley P, Mullinnix MJ, Windass JD, Campbell KP. Photoaffinity labeling of the ryanodine receptor/Ca2+ release channel with an azido derivative of ryanodine. J Biol Chem. 1994;269:13076–13079. [PubMed] [Google Scholar]
- Xiong L, Zhang JZ, He R, Hamilton SL. A Ca2+-binding domain in RyR1 that interacts with the calmodulin binding site and modulates channel activity. Biophys J. 2006;90:173–182. doi: 10.1529/biophysj.105.066092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto T, El-Hayek R, Ikemoto N. Postulated role of interdomain interaction within the ryanodine receptor in Ca(2+) channel regulation. J Biol Chem. 2000;275:11618–11625. doi: 10.1074/jbc.275.16.11618. [DOI] [PubMed] [Google Scholar]
- Yan Z, Bai XC, Yan C, Wu J, Li Z, Xie T, Peng W, Yin CC, Li X, Scheres SHW, et al. Structure of the rabbit ryanodine receptor RyR1 at near-atomic resolution. Nature. 2015;517:50–55. doi: 10.1038/nature14063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuchi Z, Yuen SMWK, Lau K, Underhill AQ, Cornea RL, Fessenden JD, Van Petegem F. Crystal structures of ryanodine receptor SPRY1 and tandem-repeat domains reveal a critical FKBP12 binding determinant. Nat Comms. 2015;6:7947. doi: 10.1038/ncomms8947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalk R, Clarke OB, des Georges A, Grassucci RA, Reiken S, Mancia F, Hendrickson WA, Frank J, Marks AR. Structure of a mammalian ryanodine receptor. Nature. 2015;517:44–49. doi: 10.1038/nature13950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimányi I, Buck E, Abramson JJ, Mack MM, Pessah IN. Ryanodine induces persistent inactivation of the Ca2+ release channel from skeletal muscle sarcoplasmic reticulum. Mol Pharmacol. 1992;42:1049–1057. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Data File 1, related to Figure 3: This dataset contains movies depicting morphs of the coordinates of the atomic models of several key structural states. A detailed description of the contents is provided as a separate document within the archive.
Supplemental Data File 2, related to Figure 3: This dataset contains movies depicting morphs of the density maps of several key class comparisons. A detailed description of the contents is provided as a separate document within the archive.
Figure S1. Functional characterization of RyR1/Cs2 complex, related to Figure 1.
(A) Representative single-channel current traces of purified RyR1 channels measured under the following conditions: 1) cis chamber, 5 mM EGTA and no added Ca2+ (nominally Ca2+ free) [Ca2+]; 2) cis chamber, nominally Ca2+ free plus 2 mM ATP and 5 mM caffeine; 3) cis chamber, 30 μM cytosolic [Ca2+]; 4) cis chamber, 30 μM cytosolic [Ca2+] plus 2 mM ATP and 5 mM caffeine. The trans chamber contained [Ca2+] = 53 mM. Channel openings are shown as upward deflections; the closed (C−) state of the channel is indicated by horizontal bars at the beginning of each trace. Representative traces are displayed with two different time scales (100 ms for upper trace and 1000 ms for lower trace) as indicated by scale bars, and the PO, TO (average open time) and TC (average closed time) are shown above each trace. Specific modification by 10 μM ryanodine is displayed. Bar graph summarizing single channel PO (B), TO (C) and TC (D) nominally Ca2+-free with 2 mM ATP and 5 mM caffeine, 30 μM cytosolic [Ca2+] and 30 μM cytosolic [Ca2+] with 2 mM ATP and 5 mM caffeine (n = 6) channels.
Figure S2. Domain organization of RyR1, related to Figure 1.
(A) Revised domain boundaries are colored both on a 2D schematic of the sequence (with the length of each box proportional to the number of residues it contains) and (B) on a low-pass filtered surface representation of the model, using the same coloring scheme. The hypothetical location of the TM×2 transmembrane hairpin is indicated in (A) with a black box. (C) The junctional solenoid (JSol, here shown as a khaki molecular surface) forms the attachment site for the three other solenoid domains – NTD-C (dark blue ribbon), BSol (green ribbon) and CSol (purple ribbon), as well as for calstabin2 (gold). In the case of each of the interacting solenoids, only the first repeat of the solenoid is represented here for clarity. Both opposed views represented here are parallel to the membrane plane.
Figure S3. Coordination of bound Ca2+ at a conserved site at the CTD-CSol interface, related to Figure 1.
(A) Region of the density map and model around the Ca2+-binding site in the Ca2+-only structure, with fitted model. Residues believed to participate in Ca2+-coordination either directly (E3893, E3967, T5001) or indirectly (Q3970, H3895) are labeled, as is the auxiliary interaction at the same interface formed by E4032 and Q5006. Labeled residues are depicted in full; all others are shown as a Cα trace only. Core solenoid residues are colored sea-green here, and CTD residues depicted in dark red. The Ca2+ ion is represented by a green sphere. The density map is contoured at 2 σ. (B) Portion of a sequence alignment of representative RyR and IP3R genes, prepared using MUSCLE (Edgar, 2004) with default parameters, visualized using Jalview (Waterhouse et al., 2009). The alignment is colored by conservation using a modified version of the Zappo coloring scheme, with a conservation cutoff of 60%. Residues involved in Ca2+ coordination (E3893, H3895, E3967 and Q3970) in addition to a residue (E4032) previously identified as playing a role in Ca2+-activation of RyR1 and RyR2 are labeled. The first sequence in the alignment is that of rabbit RyR1; the Uniprot accession number of each sequence is given in parentheses.
Figure S4. Gating-independent motions of the cytoplasmic assembly, related to Figure 2.
A comparison of the cytoplasmic assembly conformations for each class of four different datasets: (A), nominally Ca2+-free, (B), Ca2+-only, (C), ATP/caffeine only, (D), Ca2+/ATP/caffeine, and (E), all of (A-D) together, in two orientations (left, in the membrane plane, and right, from the cytoplasm). In all panels (A-E), the structure of one EGTA class is shown for reference in dark gray, and a single rigid unit, comprising the bridging solenoid of one protomer and NTD-A-JSol of the adjacent protomer, is shown in colored ribbon for each of the four classes in the dataset. Pairwise comparisons between the rigid units from each class give the rotation axes, which are depicted as sticks in each panel. (F) An alternative visualization of the conformational variability in each dataset, displaying the height of the RY1&2 domain above the SR membrane plane for each class of each dataset as a box and whisker plot. The fraction of particles in each dataset belonging to each class is represented by the size of each circular marker; the class number is given by the color of the marker.
Figure S5. Schematic representation of ryanodine receptor activation and pore opening, related to Figure 4.
Boxes represent datasets in particular conditions: (1) EGTA, (2) EGTA/caffeine/ATP, (3) Ca2+, (4) Ca2+/ATP/caffeine, (5) ryanodine-activated, (6) ryanodine-blocked. Square activation module: Apo state; rounded activation module: primed state. Dotted borders: state not structurally characterized, but known to exist from bilayer recordings.
Figure S6. Representative data and local resolution, related to Figure 1.
(A) Motion-corrected micrograph from the EGTA dataset collected on holey-carbon grids, compared with micrographs from the 30 μM Ca2+ and 10 μM ryanodine datasets collected on holey-gold grids at different electron doses. (B) Euler angle distribution of all particles used for calculating the 3D reconstruction of each dataset with c4 symmetry imposed. The Euler angle distributions displayed here correspond to reconstructions of all the particles of a given dataset; the reconstructed volume is displayed at the center of the distribution as a reference for orientation. The height and color of cylinders indicate the number of particles in each view. Dark blue: Orientation with the smallest number of particles, Red: orientation with the largest number of particles. The plot is calculated with an angular sampling of 3.7 degrees between views. Datasets from left to right: EGTA, Ca2+/ATP/Caffeine, EGTA/ATP/Caffeine, Ca2+, Ryanodine. (C) Reconstruction colored by local resolution (from 3.0 Å, blue, to 6.0 Å, red) using ResMap (Kucukelbir et al., 2014), viewed from the membrane plane, together with an orthogonal slab through the reconstruction. (D) Density quality in the core of the assembly (centered on the CTD) with fitted atomic model displayed as a Cα-trace with sidechains. Many bulky residues that are well defined in the density.
Figure S7. Particle polishing optimization, related to Figure 1.
Plots of local motion correction with individual particle trajectories obtained from alignparts_lmbfgs. The color of each line represents the frame number, from 0 (black) to 35 (blue). All plots are exaggerated 25 times. The smoothing parameter is used as a penalty for having abrupt changes in direction during a trajectory. This is based on the assumption that particles are unlikely to have sudden changes in direction. The “neiflag” parameter determines the extent of local averaging for the smoothing of particle trajectories. This parameter is derived from the assumption that nearby particles have correlated trajectories. (A) Series of plots showing the change in individual particle trajectories as the rmax2 (maximum resolution used during alignment; in Å) variable is decreased. (B) The original particle trajectories determined with the default parameters (left) and the new particle trajectories determined with the optimized parameters (right).
Figure S8. Fourier Shell Correlation curves and central slices for each dataset, related to Figure 1.
Dataset: (A) EGTA, (B) Ca2+-ATP-Caffeine, (C) EGTA-ATP-Caffeine, (D) Ca2+, (E) Ryanodine. (Top) FSC curves using the ‘gold standard’ protocol for the complete protomer (gray), the shell (light blue) and the core (light red), with the respective resolutions derived from FSC=0.143. Resolutions for the shell, the core, and the entire protomer were calculated using relion_postprocess, using masks created from the molecular model of the respective regions and shown as thumbnails in (A). (Bottom) Central slices from the 3D reconstruction in the y and z axis with (left) and without (right) B-factor sharpening.
Table S1: Solution conditions, data collection and reconstruction statistics, related to Figure 1.
Table S2. Electron dose-dependent per frame low-pass resolution cutoff, related to Figure 1.
The program alignparts_lmbfgs outputs a weighted sum of the movie frames that maximizes the signal to noise ratio in each resolution shell according to the critical dose calculated by Grant & Grigorieff (Grant and Grigorieff, 2015b). The table describes the effective resolution cutoff produced by the scheme when our ad-hoc exposure factor is applied.
Table S3. Dataset size and resolution by domain, related to Figure 1
Resolutions after refinement of all particles of each dataset are described, unless otherwise specified. Domain boundaries are described in supplemental figure. Resolution measurements are described in Methods. The highest resolution domains for each reconstruction are highlighted in bold type, while the lowest resolution domains are indicated in italics.
Table S4. Classification summary for each dataset, related to Figure 2.
For each dataset, classes are described in terms of number of particles, particle distribution, overall shell conformation and resolution. Resolution measurements are described in Methods. Shell height is measured in Å from the top of the membrane plane to the center of mass of RY12 domain (also plotted in Figure S4F).