

Abstract
The nuclear export receptor, Exportin 1 (XPO1), mediates transport of growth-regulatory proteins including tumor suppressors and is overactive in many cancers, including chronic lymphocytic leukemia (CLL), acute myeloid leukemia (AML), and aggressive lymphomas. Oral Selective Inhibitor of Nuclear Export (SINE) compounds that block XPO1 function were recently identified and hold promise as a new therapeutic paradigm in many neoplasms. One of these compounds, KPT-330 (selinexor), has made progress in Phase I/II clinical trials, but systemic toxicities limit its administration to twice-per-week and requiring supportive care. We designed a new generation SINE compound, KPT-8602, with a similar mechanism of XPO1 inhibition and potency but considerably improved tolerability. Efficacy of KPT-8602 was evaluated in preclinical animal models of hematologic malignancies including CLL and AML. KPT-8602 shows similar in vitro potency compared to KPT-330 but lower central nervous system penetration which resulted in enhanced tolerability, even when dosed daily, and improved survival in CLL and AML murine models compared to KPT-330. KPT-8602 is a promising compound for further development in hematologic malignancies and other cancers in which upregulation of XPO1 is seen. The wider therapeutic window of KPT-8602 may also allow increased on-target efficacy leading to even more efficacious combinations with other targeted anticancer therapies.
Keywords: Leukemia, lymphoma, XPO1, nuclear export, SINE
Introduction
Nuclear-cytoplasmic transport of proteins is largely mediated by proteins of the karyopherin family of nuclear transport receptors. Exportin 1 (XPO1) is the best characterized exportin with >200 cargos, including tumor suppressor proteins (TSPs) such as p53, IκB and various RNAs1, 2. Proteins transported by XPO1 contain 10-15 residue nuclear export signals (NESs) that bind to a hydrophobic groove on XPO13-8. Overexpression of XPO1 has been detected in many hematologic9 and solid tumors10, and correlates with poor clinical outcomes. Increased levels of XPO1 in cancer cells promote egress of TSPs from the nucleus to the cytoplasm where they no longer regulate cell cycle, proliferation, and apoptosis. Subcellular mislocalization of TSPs (e.g. p53, BRCA1, and retinoblastoma) has been observed in cancer and has been linked to cancer progression and maintenance11.
The importance of the XPO1-dependent nuclear export pathway is highlighted in a subset of adult acute myeloid leukemia patients with normal cytogenetics (CN-AML) where mutations in the nucleophosmin (NPM1) gene (50-60% of cases) create a novel NES resulting in hyperactive XPO1-dependent export of NPM112. NPM1 relocation to the nucleus (thereby restoring TSP function) represents a potential targeted therapy for this frequent subtype of AML. Additionally, XPO1 overexpression in AML correlates with poor clinical outcome13. In chronic lymphocytic leukemia (CLL) recurrent XPO1 mutations have also been described14 although the impact of these mutations remains uncertain.
Selective Inhibitor of Nuclear Export (SINE) compounds, developed by Karyopharm Therapeutics Inc. (Newton, MA), are orally bioavailable small molecules that covalently bind to Cys528 in the NES-binding groove of XPO1 and prevent NES/cargo binding and export15. Our previous published work showed that XPO1 is a therapeutic target for CLL15 and AML16, 17, and has facilitated the translation of a SINE compound named KPT-330 (selinexor) to a Phase I clinical trial in advanced hematologic tumors (NCT01607892) and in multiple solid tumors (NCT01607905). Anti-tumor activity of selinexor has been observed in patients with diffuse large B cell lymphoma (DLBCL)18, CLL18, multiple myeloma19, and AML20. To date >1000 patients have been treated with selinexor in Phase I/II clinical trials. While constitutional symptoms (weight loss, fatigue, anorexia) were initially therapy-limiting, selinexor tolerability has been improved with supportive care, consisting of appetite stimulants (megesterol plus olanzapine) and anti-nausea agents (odansetron). Hovewer, despite these improvements, the admistration of selinexor is limited to twice per week at most. Therefore, continuous inhibition of XPO1 in cancer cells without unacceptable toxicity remains a challenge and new therapies are needed for the clinic.
In this report we describe the structural, biochemical and in vivo characterization of a new generation SINE compound, KPT-8602, which shows similar target binding properties as first generation SINE compounds (KPT-185 and KPT-330), but reduced brain penetration and greater tolerability in preclinical animal models of hematologic malignancies. The improved tolerability of KPT-8602 along with preserved target specificity suggest that it may have promising clinical efficacy in B-cell malignancies, AML, and a variety of other cancers where upregulation of XPO1 is seen.
Materials and Methods
Cloning, expression, and protein purification
ScXPO1 (residues 1-1058 with residues 377-413 removed and Thr539 mutated to Cys) was cloned into the previously described pGEX-Tev vector21, 22. Residues 62-201 of ScRanBP1 (Yrb1p) were also cloned into pGEX-TEV and full length human Ran was cloned into the pET-15b vector. Further details are in Supplemental Materials.
Assembly of the XPO1-Ran-Yrb1 complex, crystallization and X-ray data collection
KPT-8602-ScXPO1-Ran-Yrb1 complex was crystallized in conditions similar to those used by Koyama and Matsuura (17% PEG3350, 100mM Bis-Tris (pH 6.6) and 200mM ammonium nitrate). X-ray diffraction data was collected at Advanced Photon Source (APS) – Structural Biology Center (SBC) 19ID beamline. Further details are in Supplemental Materials.
In Vitro inhibition assays
For inhibition assays, 2μM purified XPO1 was incubated with the indicated inhibitor (0, 5, 10, 20μM) and 10μM RanGTP for 90mins at 4°C. The mixtures were then added to approximately 10μg of immobilized NESs and rotated for 2hrs at 4°C. Unbound proteins were washed extensively with buffer and remaining bound proteins were separated by SDS/PAGE and visualized by Coomassie Blue staining. For inhibition time-course experiments, incubation times of XPO1, RanGTP and inhibitors were varied and mixtures were rotated with immobilized GST-PKINES for 1hr before extensive washing. Reversibility assays were performed as previously described22. Further details are in Supplemental Materials.
In vivo XPO1 degradation by SINE compound treatment
Human fibrosarcoma HT1080 cells were cultured and grown to 80% confluence. KPT-185, KPT-330, or KPT-8602 were added to final concentrations of 25–1000nM and cells were grown for another 18hr. Treated and control cells were collected by centrifugation and lysed on ice for 30min in RIPA buffer. Further details are in Supplemental Materials.
Cell isolation and reagents
Human CLL and normal B cells were isolated and cultured as previously described15. Blood and frozen bone marrow aspirates from newly diagnosed untreated CLL and AML patients were obtained from the Ohio State University Leukemia Tissue Bank after getting informed consent approved by the cancer institution review board according to the Declaration of Helsinki. Further details are in Supplemental Materials.
Cell lysis and immunoblot
Cells were lysed in RIPA buffer. Nuclear extracts were prepared with NE-PER (Pierce). Proteins were separated by SDS-PAGE and blots probed with commercially available antibodies and detected by addition of chemiluminescent substrate (Pierce) followed by quantification by Chemi-Doc system with Quantity One software (Bio-Rad Laboratories).
Assessment of cell death
Cell death was assessed using either Annexin-V/PI staining or MTS assay as previously described15. Further details are in Supplemental Materials.
Immunofluorescent Staining
Approximately 200,000 cells were cytospinned to slide. Cells were then fixed, permeabilized, blocked, and stained for confocal microscopy as previously described16. Further details are in Supplemental Materials.
Animal studies
All experiments were carried out under protocols approved by The Ohio State University Institutional Animal Care and Use Committee. All treatments were administered by oral gavage. Overall survival was the primary endpoint.
TCL1 transplant mouse model
CD19+CD5+ cells (1×107) from the spleen of a TCL1 transgenic mouse with active CLL-like leukemia and palpable splenomegaly were engrafted by tail vein into a C57BL/6 mouse. At leukemia onset, engrafted mice were randomly assigned to treatment conditions: KPT-330 (15mg/kg), KPT-8602 (15mg/kg), ibrutinib (20mg/kg), and vehicle control (0.5% methylcellulose/1% Tween80). Disease progression was monitored weekly by CD45/CD5/CD19 flow cytometry and peripheral blood smears, spleen size, health, and weight loss. Mice were sacrificed on development of splenomegaly and presence of other disease criteria causing discomfort.
MV4-11 xenograft mouse model
Splenocytes (0.5×104) from MV4-11 transplanted NSG mice were intravenously injected into NSG mice via tail vein as previously described16. One week after tumor inoculation, mice were given vehicle control 5x/week, KPT-8602 at 15mg/kg 5x/week (daily from Monday-Friday with a break Saturday-Sunday), KPT-8602 at 15mg/kg 2x/week, or KPT-330 at 20mg/kg 2x/week. Mice were monitored daily for clinical signs of leukemia, such as weight loss and hind-limb weakness/paralysis.
Statistics
All analyses were performed by the OSU Center for Biostatistics; using previously described models15. For survival experiments, survival curve estimates were calculated using the Kaplan-Meier method and differences in curves were assessed using the log-rank test. Spleen volume was compared between groups using ANOVA, and mixed effects models were used to assess changes in %CD19+/CD5+ cells over time. Where applicable, data were log-transformed to reduce skewness. P-values were adjusted for multiple comparisons using Holm's stepdown procedure or Dunnett's test, were appropriate. All analyses were performed using SAS/STAT software, version 9.3 (SAS Institute, Inc., Cary, NC).
Results
KPT-8602 binds in the NES-binding groove of XPO1
The novel SINE, KPT-8602, forms a covalent bond with the same reactive cysteine that KPT-330 binds through a warhead with an activated Michael acceptor in the Cα that is similar to KPT-330's (Fig.1A). We have solved the 2.5 Å resolution X-ray structure of KPT-8602 bound to the Saccharomyces cerevisiae XPO1 (ScXPO1) in complex with human RanGppNHp and S. cerevisiae RanBP1 (Yrb1) (Fig.1B, Supplementary Table 1). As previously reported, Thr539 of ScXPO1 was mutated to cysteine (ScXPO1 does not contain a reactive cysteine) to allow covalent conjugation of KPT-860215, 22-24. Structure of the KPT-8602-bound ScXPO1-Ran-Yrb1 complex is very similar to other inhibitor-bound ScXPO1-Ran-Yrb1 complexes (Cα rmsd of 0.2-0.3 Å). KPT-8602 binds in the NES-binding groove of XPO1, forms a covalent bond with the reactive Cys539 (equivalent to Cys528 of human XPO1) and buries a surface area of 318.1 Å2.
Figure 1. KPT-8602 binds in the NES-binding groove of XPO1.
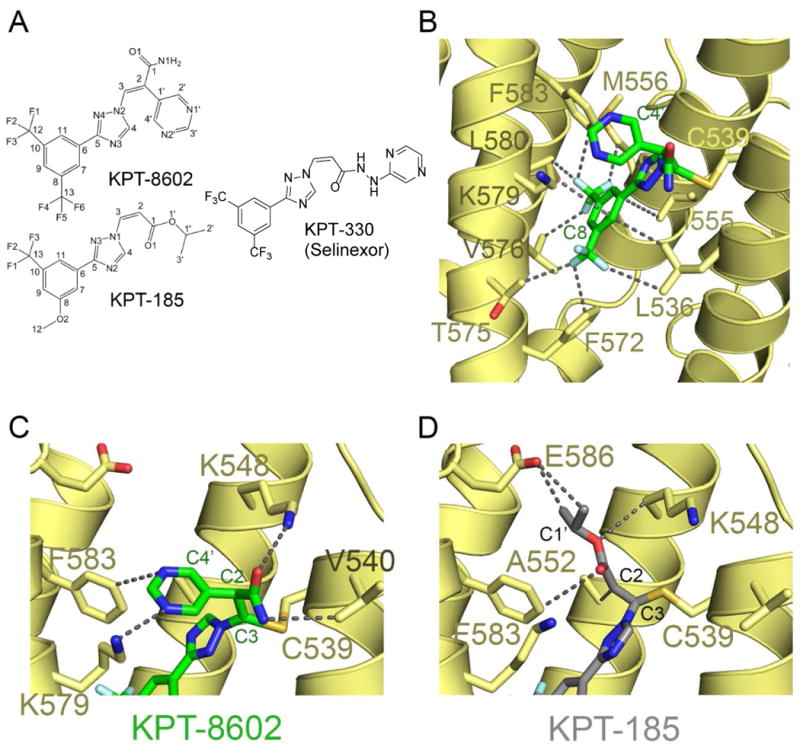
(A) Chemical structures of KPT-8602, KPT-185, and KPT-330 (selinexor).
(B) KPT-8602 (green) binds to the NES binding groove of XPO1 (yellow). Select inhibitor-XPO1 interactions (<4Å) are shown as dotted lines.
(C) Zoomed-in view of the KPT-8602 (green) Michael acceptor sidechain. Interactions with XPO1 (yellow) are show with dotted lines.
(D) Zoomed-in view of the KPT-185 (PDB ID 4GMX; grey) Michael acceptor sidechain. Interactions with XPO1 (yellow) are show with dotted lines for comparison with (C).
KPT-8602 is oriented in the groove similar to other SINE compounds15, 23. The NES groove of XPO1 remains largely the same when bound to KPT-8602, KPT-185 or Leptomycin B (Cα rmsds of 0.2-0.3 Å)15, 22. KPT-185 is a first generation SINE analog of KPT-330 that shares almost identical in vitro activities and the structure of its complex with XPO1 was previously reported. While both KPT-185 and KPT-8602 contain a similar substitution (trifluoromethyl phenyl triazole) in the Cβ, the Michael acceptor of KPT-8602 is activated with a pyrimidyl group at the Cα. The trifluoromethyl phenyl core of KPT-185 and KPT-8602 interact with the XPO1 groove in a similar fashion (Fig.1B and Supplementary Table 2), but the triazole moiety in KPT-8602 is rotated 180° relative to that of KPT-185, thus positioning the Cβ 2.4 Å away from the Cβ of KPT-185 and towards the solvent. Repositioning of the Cβ shifts the entire Michael acceptor arm such that its amide and pyrimidine form hydrogen bonds with Lys548 and Lys579 of XPO1 and make van der Waals contact with Val 540 and Phe583 of XPO1 (Fig.1C, Supplementary Fig.1). Accordingly, the Cys539 sidechain of XPO1 that forms a covalent bond with KPT-8602 adopts an alternate rotamer conformation (Fig.1C). By comparison, while the Michael acceptor arm of KPT-8602 point out of the XPO1 groove toward solvent, the equivalent portion of KPT-185 binds deep into the XPO1 NES groove (Fig.1D).
KPT-8602 inhibits XPO1-cargo interactions
We confirmed the ability of KPT-8602 to inhibit XPO1-NES interactions in a similar manner to other SINE compounds. We evaluated the ability of KPT-8602 to prevent pull-down of purified recombinant XPO1 with immobilized NESs. KPT-8602 successfully inhibited three different classical NESs/cargos (PKINES, MVM-NS2NES and full-length Snurportin) efficiently (Fig.2A) although higher concentrations are needed to completely inhibit XPO1 (10-20μM; compared to 5μM for KPT-185 or KPT-330). Unlike KPT-185, both KPT-8602 and KPT-330 required longer incubation times with XPO1 to completely inhibit XPO1 binding to PKINES (60min; compared to 5min for KPT-185; Fig.2B).
Figure 2. KPT-8602 inhibits XPO1-NES interactions.
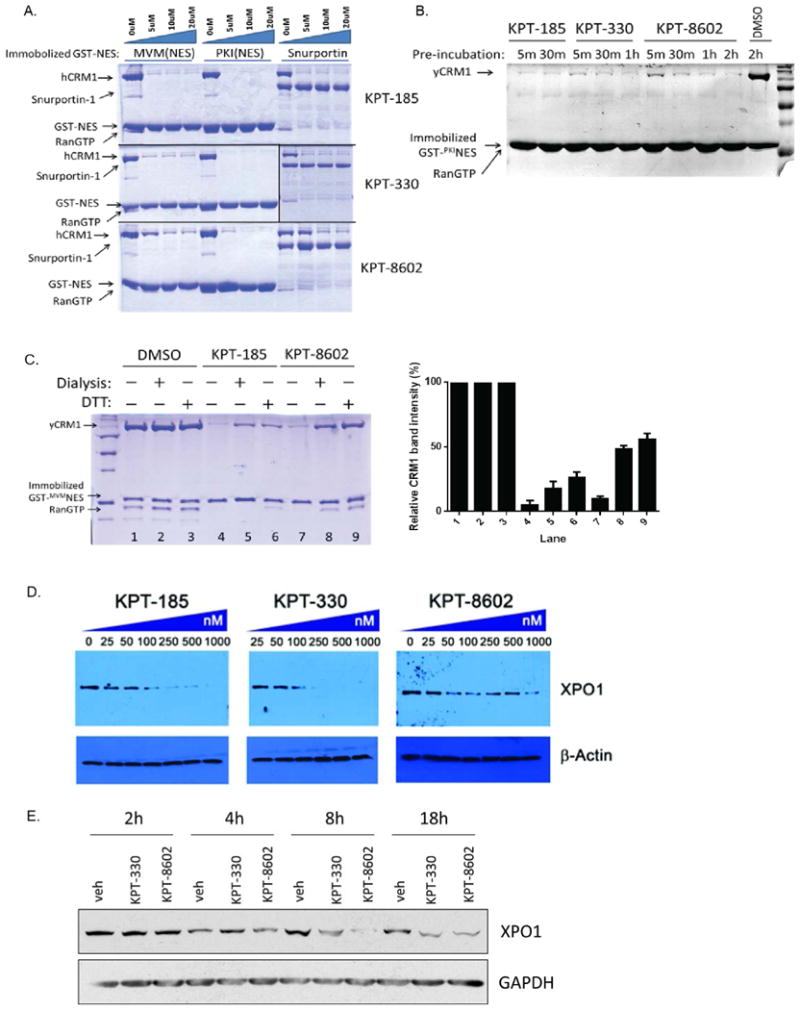
(A) KPT-8602 inhibits XPO1-cargo interactions. Pull-down binding assays using immobilized GST-MVM-NS2NES or GST-PKINES or GST-Snurportin with purified recombinant XPO1 (pre-incubated with indicated inhibitors in 1:0, 1:2, 1:4, 1:8 molar ratio) and RanGTP. Bound proteins were resolved with SDS-PAGE and Coomassie Blue staining.
(B) KPT-8602 requires slightly longer incubation time than KPT-185 to achieve complete XPO1 inhibition. Pull-down binding assays of immobilized GST-PKINES with XPO1 (pre-incubated with 10uM of indicated inhibitors for different times) and RanGTP were performed as in (A).
(C) KPT-8602 conjugation to XPO1 is reversible. Triplicate sets of XPO1 was incubated with 10uM of respective inhibitors and subjected to dialysis or DTT treatment before binding assay with RanGTP and immobilized GST-MVMNES. XPO1 band intensities were quantified and normalized to DMSO treated controls and plotted as percentages on the right panel.
(D) HT1080 cells treated with 25-1000 nM KPT-185, KPT-330 or KPT-8602 for 18 hours were harvested and cell lysates were probed with specific antibodies to observe degradation of XPO1.
(E) Freshly isolated CLL cells from a representative patient were treated with vehicle (DMSO), 0.5 μM KPT-330, or 0.5 μM KPT-8602. Total protein was extracted at 2h, 4h, 8h, 18h after treatment initiation. N=3.
To confirm that the novel structure of KPT-8602 did not result in altered binding kinetics to XPO1 we assessed the reversibility of KPT-8602 conjugation to XPO1 as previously described22. Treatment with either dialysis or DTT to remove unconjugated/deconjugated inhibitors was able to remove similar amounts of KPT-8602 and KPT-185 from XPO1 after 24 hours, suggesting that KPT-8602 binds XPO1 in a slowly reversible manner similar to first-generation SINEs (Fig.2C).
Finally, we examined inhibitor-induced degradation of XPO1 by KPT-8602. In addition to inhibition of NES binding activity, proteasome-mediated degradation of XPO1 is another phenomenon that is consistently observed in KPT-treated cells9. The mechanism of this KPT-induced degradation of XPO1 is not understood. We also examined the effect of KPT-8602 treatment on proteasome-mediated degradation of XPO1. KPT-8602 induces XPO1 degradation in HT1080 cells in a dose-dependent manner similar to other SINE compounds although higher concentrations of KPT-8602 are required to yield degradation responses similar to those with KPT-185 and KPT-330 (Fig.2D). This phenomenon was also evaluated in human primary CLL cells. In contrast to HT1080 cell lines, KPT-8602 and KPT-330 decreased XPO1 total protein as early as 4 hours post-treatment (Fig.2E) with similar efficency.
KPT-8602 induces apoptosis of primary CLL cells and significantly inhibits proliferation of diffuse large B-cell lymphoma cell lines
The ability of KPT-8602 to induce cell death in primary CLL cells was evaluated. As shown in Figure 3A and Supplementary Figure 6, treatment with KPT-8602 induced dose-dependent killing of primary CLL cells when compared to vehicle as measured by MTS analysis. We then tested the ability of selected concentrations of KPT-8602 to induce apoptosis of primary CLL cells in the presence of fetal bovine serum (FBS) or human serum (HS). As shown in Figure 3B, KPT-8602 was effective in inducing apoptosis of CLL cells in both FBS and HS.
Figure 3. KPT-8602 induces apoptosis of primary CLL cells and significantly inhibits proliferation of diffuse large B-cell lymphoma cell lines.
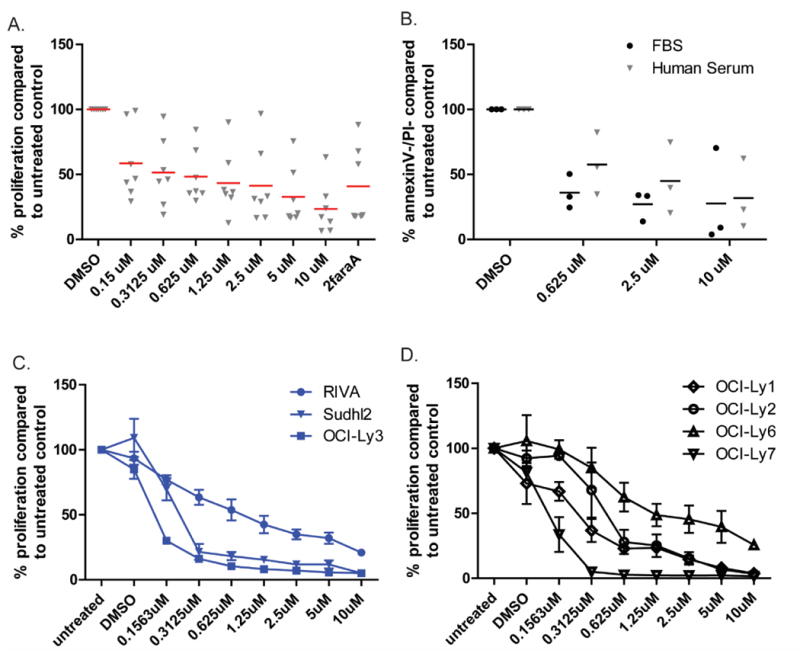
(A) KPT-8602 induces a dose-dependent cytotoxicity of CD19+ cells from CLL patients (n=7) as measured by MTS at 48h. 2faraA indicates fludarabine treatment. Red bars indicate averages.
(B) KPT-8602 induces a dose-dependent cytotoxicity of CD19+ cells from CLL patients (n=3) in the presence of 10% fetal bovine serum (FBS) or human serum. Cytotoxicity after 24h was measured by annexin-V/PI flow cytometry. Viable populations were calculated as a percent of viability of vehicle control (DMSO). Black bars indicate averages.
(C) KPT-8602 induces a dose-dependent cytotoxicity of activated B-cell (ABC) subtype of diffuse large B-cell lymphoma cell lines as measured by MTS.
(D) KPT-8602 induces a dose-dependent cytotoxicity of germinal center (GC) subtype of diffuse large B-cell lymphoma cell lines as measured by MTS.
We confirmed the effects of KPT-8602 on XPO1-dependent nuclear export by examining the cellular localization of IkB, a well-known cargo protein of XPO1, in human CLL cells after KPT-8602 treatment. Nuclear retention of IkB was observed within 8 hours of treatment of KPT-330 and KPT-8602 (Supplementary Fig.2). Next, we assessed the ability of KPT-8602 to inhibit the proliferation of a panel of cell lines representative of diffuse large B-cell lymphoma (DLBCL) both activated B-cell like (ABC-DLBCL) or germinal center subtypes (GC-DLBCL). As shown in Figure 3C KPT-8602 inhibited proliferation in ABC-DLBCL cell lines and in GC-DLBCL cell lines (Fig.3D).
KPT-8602 possesses reduced central nervous system penetration
We assessed the capacity of KPT-8602 to cross the blood brain barrier in three mammalian species (mouse, rat, and monkey) compared to that of KPT-330 (selinexor), which is currently in clinical trials. As shown in Table 1, KPT-8602 possesses a reduced central nervous system (CNS) penetration in all species compared to KPT-330. Additionally, KPT-8602 does not accumulate in plasma after repetitive dosing (data not shown).
Table 1. Brain penetration of KPT-8602 and KPT-330 across species.
| Compound | Species | Dose (po; mg/kg) | Mean Concentration at 2h post dose | Brain/Plasma Ratio | |
|---|---|---|---|---|---|
| Plasma (h.ng/mL) | Brain (h.ng/g) | ||||
| KPT-8602 | Mouse | 5 | 550 | 105 | 0.19 |
| Rat | 5 | 320 | 70 | 0.22 | |
| Monkey | 10 | 1310 | <32 | <0.02 | |
| KPT-330 (selinexor) | Mouse | 10 | 1760 | 1295 | 0.71 |
| Rat | 10 | 1670 | 1207 | 0.72 | |
| Monkey | 10 | 2545 | 1565 | 0.6 | |
KPT-8602 prolongs survival in a mouse model of CLL
The Eμ-TCL1 transgenic mouse develops a disease very similar to that observed in human CLL patients and this model has been used extensively to evaluate experimental therapeutics in CLL25, 26. We and others have shown27 that the B-cell malignancy occurring in the Eμ-TCL1 mice is amenable to adoptive cell transfer by engrafting splenic white blood cells from Eμ-TCL1 mice with active CLL-like leukemia (>10% CD45+CD5+CD19+ B-cells in peripheral blood; WBC>60) and palpable splenomegaly into background strain WT C57BL/6 mice. This produces a homogeneous population with similar pathologic findings to the Eμ-TCL1 transgenic mice and a more rapid disease acquisition (weeks instead of months), although time-to-disease and disease progression can be quite variable depending on the donor used for the engraftment.
We therefore employed the Eμ-TCL1-C57BL/6 transplant model described above to evaluate the potential of KPT-8602 to abrogate CLL progression in vivo. We hypothesized that increased reversibility combined with the lower brain penetration of KPT-8602 could reduce the development of constitutional symptoms (weight loss) and allow more frequent dosing, which would in turn impact its efficacy. C57BL/6 mice were engrafted with CD5+CD19+ splenocytes derived from a Eμ-TCL1 donor mouse with active disease. At the time of leukemia onset (defined as ≥10% CD45+CD5+CD19+ cells), mice were randomized to receive KPT-8602, KPT-330, or vehicle control. Our group previously showed that KPT-330 dosed 3x/week showed worse overall survival compared to KPT-330 dosed 2x/week in this model26. We first compared KPT-8602 to KPT-330 by employing the clinically-used schedule of administration via oral gavage 2x/week as we previously described26, 28. As expected, mice treated with either compound showed similar overall survival which was significantly improved over vehicle (p=0.001 and 0.028, respectively) (Supplementary Fig.3). KPT-8602 shows lower central nervous system penetration and mildly increased reversibility in XPO1 binding qualities that suggest an enhanced tolerability and wider therapeutic index compared to KPT-330. We therefore evaluated the therapeutic benefit of KPT-8602 dosed continuously vs 2x/week in additional C57BL/6 mice engrafted with leukemic cells from a different Eμ-TCL1 donor. At leukemia onset, mice were randomized to receive vehicle or KPT-8602 daily or 2x/week by oral gavage. Mice treated daily with KPT-8602 had significantly improved survival compared to those treated only 2x/week (p=0.001) (Fig.4A). Both daily and 2x/week KPT-8602 provided significant survival benefit compared to vehicle treatment (p<0.001). Peripheral blood disease was evaluated weekly by CD45/CD5/CD19 flow cytometry. The percentage of leukemic cells in the blood was similar among groups at enrollment, but the percentage of CD5+ B-cells in mice treated with daily KPT-8602 was significantly lower than either vehicle control or mice treated with KPT-8602 2x/week at three weeks post-treatment (p<0.001, all comparisons) (Fig.4B). Spleen dimensions were taken when study removal criteria were met. Spleens derived from mice treated daily with KPT-8602 were significantly smaller compared to all the other groups (p<0.001, all comparisons) (Fig.4C). Daily KPT-8602 treatment did not cause a significant change in body weight after 28 days of treatment (Supplementary Fig.4).
Figure 4. KPT-8602 improves survival compared to KPT-330 in a mouse model of CLL.
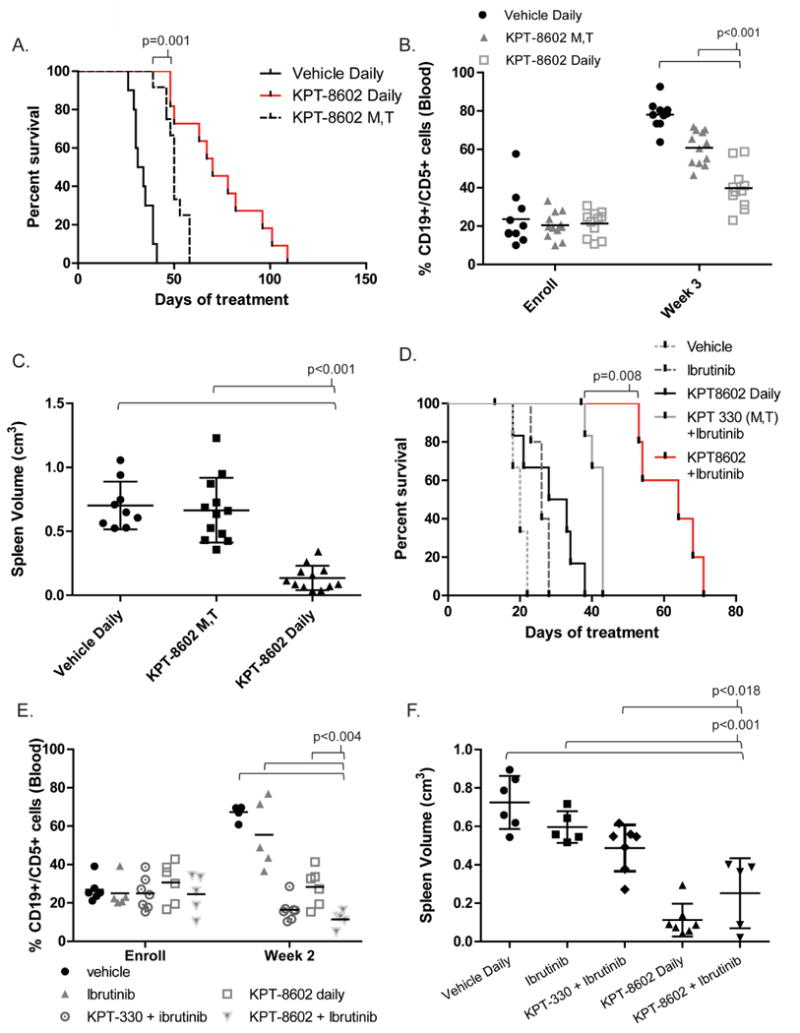
(A) Overall survival (OS) curve for C57BL/6 mice engrafted with Eμ-TCL1 leukemic splenocytes treated with KPT-8602 15 mg/kg daily (n = 14), KPT-8602 15 mg/kg 2x/week on Monday and Tuesday (KPT-8602 M,T; n = 12), or vehicle control daily (n = 12). Median OS: 70 days (KPT-8602 Daily), 50 days (KPT-8602 M,T), and 33 days (vehicle daily). Survival of mice treated daily with KPT-8602 was significantly increased compared those treated with KPT-8602 M,T (p=0.001) or vehicle daily (p<0.001). KPT-8602 M,T significantly increased survival compared to vehicle daily (p<0.001). Survival comparisons were made with the log-rank test.
(B) CD45/CD5/CD19 flow cytometry of peripheral blood was used to assess leukemic burden weekly. Percentage of leukemic cell in peripheral blood was significantly lower in mice treated with KPT-8602 daily compared to all other groups at 3 weeks post treatment initiation (p<0.001 for all comparisons). A mixed effects model was used to assess changes in %CD19+/CD5+ cells over time.
(C) Splenic volume (cm3) was measured at the time of euthanasia for all mice meeting disease progression criteria. Black bars indicate averages of each cohort. Spleen volume in the KPT-8602 daily group was significantly lower compared to all other groups (p<0.001 for all comparisons). ANOVA was used to compare spleen volume (log-transformed) in KPT-8602 vs. all other groups; p-values were adjusted using Dunnett's method.
(D) Overall survival for C57BL/6 mice engrafted with Eμ-TCL1 leukemic splenocytes treated with ibrutinib 20 mg/kg daily (n = 5), ibrutinib plus KPT-8602 15 mg/kg daily (n = 6), ibrutinib plus KPT-330 15mg/kg 2x/week (n=7), KPT-8602 15 mg/kg daily (n = 7), or vehicle control daily (n = 6). Median OS: 64 days (KPT-8602 plus ibrutinib), 43 days (KPT-330 plus ibrutinib), 26 (ibrutinib), and 20 days (vehicle). KPT-8602 plus ibrutinib increased survival compared to KPT-330 plus ibrutinib (p=0.008). Survival comparisons were made with the log-rank test and adjusted for multiple comparisons.
(E) CD45/CD5/CD19 flow cytometry of peripheral blood was used to assess leukemic burden weekly. Percentage of leukemic cell in peripheral blood was significantly lower in mice treated with KPT-8602 plus ibrutinib compared to vehicle, ibrutinib, and KPT-8602 groups at 3 weeks post treatment initiation (p<0.004). A mixed effects model was used to assess changes in %CD19+/CD5+ cells over time.
(F) Splenic volume (cm3) was measured at the time of euthanasia for all mice meeting disease progression criteria. Spleens in the combination group and KPT-8602 daily group were significantly smaller compared ibrutinib alone or vehicle (p<0.001 for all comparisons).
We have recently shown that the combination of KPT-330 and ibrutinib (bruton tyrosine kinase inhibitor) elicits a synergistic cytotoxic effect in primary CLL cells and increases overall survival compared to ibrutinib alone in a mouse model of CLL28. We therefore tested the effect of KPT-8602+ibrutinib in an additional cohort of C57BL/6 mice with engrafted leukemic cells from another Eμ-TCL1 donor. At leukemia onset, mice were randomized to receive vehicle, KPT-8602 (daily), ibrutinib (daily), or the combinations of KPT-330+ibrutinib or KPT-8602+ibrutinib. As shown in Figure 4D the combination of KPT-8602+ibrutinib was able to further improve the survival induced by KPT-330+ibrutinib. Additionally, the percentage of leukemic cells in peripheral blood was similar for all groups at enrollment, but was significantly lower in mice treated with the combination compared to mice treated with either agent alone or vehicle at week 2 (p<0.004, all comparisons) (Fig.4E). Similarly, at the last available flow cytometry analysis prior to sacrifice due to disease, peripheral blood disease was lower in mice treated with the combination compared to mice treated with ibrutinib alone or vehicle alone (p<0.001) (Supplementary Fig.6). Spleens derived from mice treated daily with KPT-8602 or the combination were significantly smaller compared to vehicle or ibrutinib alone (p<0.001 for all comparisons) (Fig.4F).
KPT-8602 significantly inhibits proliferation and induces apoptosis of AML cell lines and primary AML blasts
In order to assess the biological activity of KPT-8602 in AML, we treated a panel of AML cell lines (MV4-11, Kasumi-1, OCI/AML3) with KPT-8602 and KPT-330 and measured cell proliferation using MTS assays (Supplementary Fig.5).
Treatment with KPT-8602 at the predetermined IC50 values induced apoptosis in AML cell lines similar to KPT-330 when compared to DMSO treated controls at 48 hours (Fig.5A). We then performed proliferation assays (WST-1) using primary AML samples (n=3) using both KPT-8602 and KPT-330 and found comparable IC50 for all patients, except one in which the IC50 for KPT-8602 was higher (300nM vs. 100nM) (Fig.5B). The effect of KPT-8602 on the expression levels of two known XPO1 cargo proteins; p53 and NPM1 was assessed in AML cell lines after KPT-8602 treatment using confocal microscopy as described in methods. A significant accumulation of p53 and NPM1 in the nucleus of MV4-11 and OCI-AML3 respectively were observed after treatment with KPT-8602 (Fig.5C).
Figure 5. KPT-8602 significantly inhibits proliferation and induces apoptosis of AML cell lines and primary AML blasts and increases survival in a human leukemia xenograft model of AML compared to KPT-330.
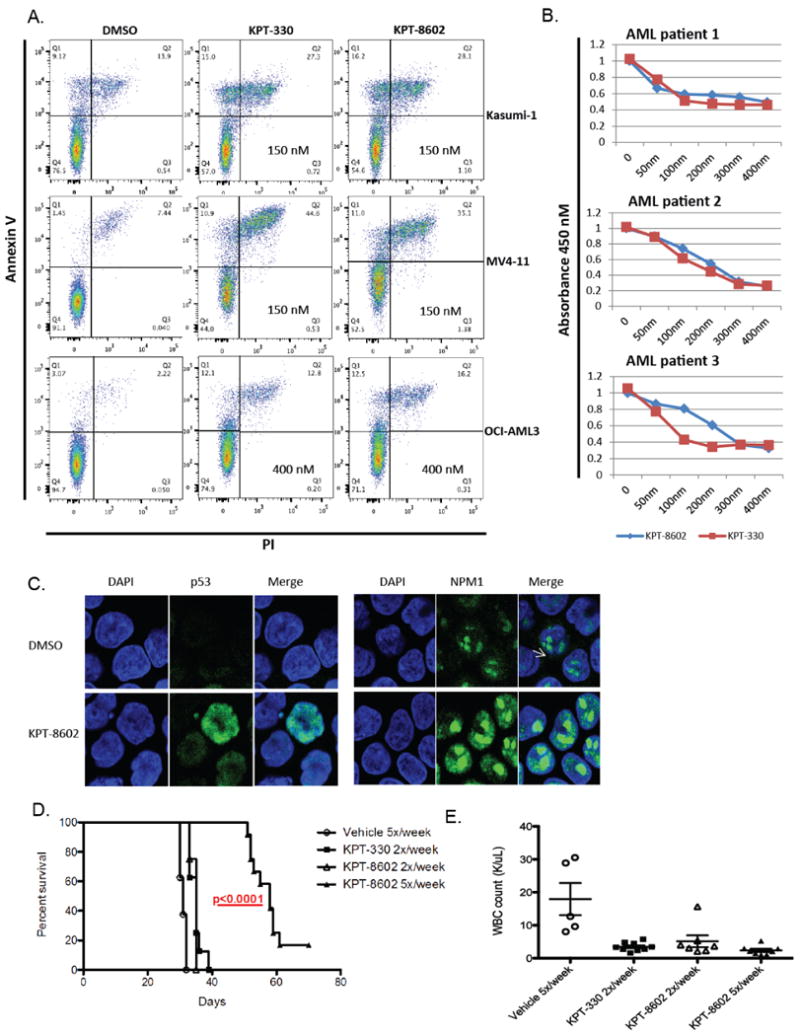
(A) Apoptosis as measured by AnnexinV/PI staining using FACS at 48 hours.
(B) WST-1 assays in 3 untreated primary AML samples.
(C) Representative confocal microscopy images of p53 and NPM1 in MV4-11 and OCI-AML3 cells. The left panel shows the DAPI staining (cell nucleus). The middle panel is p53 and NPM1 staining and the right panel is the merger of p53 and NPM1 and DAPI staining.
(D) Survival of MV4-11 xenograft mice after treatment with KPT-8602 15 mg/kg 5x/week (n = 15), KPT-8602 15mg/kg 2x/week (n = 10), KPT-330 20mg/kg 2x/week (n = 10), or vehicle control 5x/week (n = 15). Median OS: 58 days (KPT-8602 Daily), 35 days (KPT-8602 and KPT-330 M,Th), and 31 days (vehicle). Survival comparisons were made with the log-rank test.
(E) White blood cell count in KPT-8602, KPT-330 treated mice vs. vehicle control at 21 days. Comparisons were made using two-sample t-tests.
KPT-8602 prolongs survival in a human leukemia xenograft model of AML
To compare the activity of KPT-8602 and KPT-330 in AML in vivo, we used a xenograft human AML murine model (MV4-11). This model consists of NOD/SCIDγ mice that were intravenously inoculated (tail vein) with MV4-11 cells obtained from spleens of primary MV4-11 xenografts. One week post-inoculation, mice were treated with KPT-8602 at 15 mg/kg via oral gavage (5x/week or 2x/week), KPT-330 at 20 mg/kg via oral gavage (2x/week), or vehicle control (5x/week) and monitored for survival. Mice treated with KPT-330 (20mg/kg) or KPT-8602 (15mg/kg) 2x/week showed similar outcomes with improved survival when compared to vehicle-treated mice, p<0.0001.
More importantly, mice treated with KPT-8602 at 15mg/kg but given 5x/week showed strikingly better outcomes compared to KPT-8602 2x/week (15mg/kg, p<0.0001) and KPT-330 (20mg/kg, 2x/week, p<0.0001) (Fig.5D). Peripheral blood was collected from mice in each treatment group at day 21 to assess the effect of KPT-8602 on circulating leukemia burden by measuring white blood cell count (WBC). Treatment with KPT-8602 (daily and x2/week) and KPT-330 significantly reduced WBC in mice compared to those treated with vehicle (Fig.5E).
Discussion
Leptomycin B (LMB) was the first clinically developed XPO1 inhibitor29, but suspected off-target effects on molecules such as cysteine proteases and lasting toxicity in the clinic led to abandonment of its clinical development. We now believe that the long lasting toxicity of LMB is due to enhanced irreversibility of its conjugation to XPO1 as a result of XPO1-driven hydrolysis/modification of the drug22. Additionally, dose escalation studies of LMB and other synthetic nuclear export inhibitors in C57BL/6 mice suggest the low maximum tolerated dose (MTD) of LMB limits its efficacy in vivo30.
Although SINE compounds also bind covalently to XPO1, they are considered slowly-reversible inhibitors when compared to LMB22. The slowly reversible XPO1-SINE interaction is believed to contribute to the improved tolerance of SINEs over LMB as sufficient inhibitor release from XPO1 can allow essential nuclear export to resume in normal cells. This property has allowed rapid clinical development of selinexor. Despite notable clinical activity at a suboptimal dosing schedule for target inhibition based on the in vivo half-life of the compound18, 19, constitutional symptoms have been observed most likely due to the ability of selinexor to cross the blood-brain barrier. Although these side effects have been mostly manageable with supportive care, they still represent a major limitation to administering SINE compounds at a freqeuncy that provides sustained inhibition of XPO1 in cancer cells.
Our crystallographic and biochemical data show that KPT-8602 has a similar mechanism of XPO1 binding and inhibition as previous SINE compounds. We have also provided evidence that KPT-8602 inhibits XPO1 and induces similar nuclear retention of XPO1 targets similar to other SINE compounds, as expected15, 16. Additionally KPT-8602 induces comparable levels of cytotoxicity as well as inhibition of cell proliferation compared to KPT-330 in primary CLL and AML tumors and a panel of cell lines representative of AML and both molecular subtypes of DLBCL. However, in contrast to KPT-330, KPT-8602 possesses reduced blood brain barrier penetration in all analyzed species (mouse, rat, and monkey), which likely contributes to improved tolerability.
KPT-8602 was tested in the Eu-TCL1-C57BL/6 transplant model of CLL. The resulting CLL-like leukemia is uniformly fatal to all recipients that successfully engraft; however, median survival for vehicle-treated mice in each experiment depends on donor characteristics such as cell viability and leukemic burden. Additionally, oral gavage frequency may impact overall survival as demonstrated in median survival of vehicle-treated mice in Fig.4A and 4D where mice received daily and twice daily oral gavage, respectively. While further studies remain to validate nuclear accumulation of cargo proteins such as p53 and NPM1 in vivo, our results using the MV4-11 xenograft mouse model complement recent elegant studies showing that KPT-8602 is superior to KPT-330 in its ability to eliminate leukemia-initiating cells in patient-derived xenograft models of AML31. Importantly, Etchin and colleagues demonstrate that KPT-8602 spares normal CD34+ hematopoietic progenitor cells while modulating nucleocytoplasmic transport of XPO1 cargo proteins in leukemic cells in vivo.
Our data indicate that KPT-8602 allows a prolonged and frequent dosing schedule compared to KPT-330, leading to excellent therapeutic benefit and less toxicity in two murine models of hematological malignancies. These results are consistent with the lack of blood brain barrier penetration, however further studies are required to elucidate the exact mechanism driving the increase in vivo tolerability of KPT-8602. Our data suggest that the wider therapeutic window of KPT-8602 may allow increased on-target efficacy leading to even more efficacious combinations with other targeted anticancer therapies32 as exemplified by the combination of KPT-8602 with ibrutinib in a CLL mouse model.
In conclusion, we report here the preclinical therapeutic activity of the novel SINE compound, KPT-8602, an XPO1 antagonist, in CLL and AML murine models. Our results support further development of KPT-8602 as a novel therapeutic strategy for CLL and AML human patients.
Supplementary Material
Acknowledgments
We are grateful to the patients who provided blood for the above-mentioned studies. We are grateful to Dr. John B. MacMillan, PhD, for thoughtful contributions to this manuscript. Results shown in this report are derived from work performed at Argonne National Laboratory, Structural Biology Center at the Advanced Photon Source. Argonne is operated by UChicago Argonne, LLC, for the U.S. Department of Energy, Office of Biological and Environmental Research under contract DE-AC02-06CH11357. This work is funded by Cancer Prevention Research Institute of Texas (CPRIT) Grants RP120352 and RP150053 (Y.M.C.), R01 GM069909 (Y.M.C.), the University of Texas Southwestern Endowed Scholars Program (Y.M.C.), Welch Foundation Grant I-1532 (Y.M.C.), Leukemia and Lymphoma Society Scholar Award (Y.M.C.), Croucher Foundation Scholarship (H.Y.J.F.), the Leukemia and Lymphoma Society in the form of a translational grant (to JCB and RL), K12 CA133250 (to JCB), 1R01CA188269-01 (to JCB), R01CA192928 (to RL and JCB), R01CA188269 (to RG), 1F30CA196082 (to ZAH), the Department of Defense DOD W81XWH-14-01-0190 (to RL), the Leukemia and Lymphoma Society Special Fellow award (LLS 60046395 to P.R.), and the Leukemia and Lymphoma Society Scholar Award (LLS 20020030 to R.G.). This work was also supported by P30 CA016058.
Footnotes
Conflict of Interest disclosures: EB, SS, and MGK are employees of Karyopharm Therapeutics Inc. and have financial interests in this company. YMC is a consultant for Karyopharm Therapeutics Inc.
Authorship Contributions: ZAH, PR, HYJF, JCB, YMC, RG, and RL designed the experiments, analyzed the data, wrote the paper and reviewed and approved the final version. EB designed KPT-8602. EB, SM, DE, KW, JAW, JS, XY, VMG, XM, QS, TC, DML, SS, MGK, and AML planned and contributed to components of the experimental work presented (chemistry, biology, clinical, or animal studies, or statistical analysis of data), reviewed and modified versions of the paper, and approved the final version.
References
- 1.Xu D, Grishin NV, Chook YM. NESdb: a database of NES-containing CRM1 cargoes. Molecular Biology of the Cell 2012 September 15. 2012;23(18):3673–3676. doi: 10.1091/mbc.E12-01-0045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brennan CM, Gallouzi I-E, Steitz JA. Protein ligands to HuR modulate its interaction with target mRNAs in vivo. J Cell Biol. 2000;151(1):1–14. doi: 10.1083/jcb.151.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wen W, Meinkoth JL, Tsien RY, Taylor SS. Identification of a signal for rapid export of proteins from the nucleus. Cell. 1995;82:463–473. doi: 10.1016/0092-8674(95)90435-2. [DOI] [PubMed] [Google Scholar]
- 4.Fornerod M, Ohno M, Yoshida M, Mattaj IW. CRM1 is an export receptor for leucine-rich nuclear export signals. Cell. 1997;90:1051–1060. doi: 10.1016/s0092-8674(00)80371-2. [DOI] [PubMed] [Google Scholar]
- 5.Dong X, Biswas A, Suel KE, Jackson LK, Martinez R, Gu H, et al. Structural basis for leucine-rich nuclear export signal recognition by CRM1. Nature. 2009 Apr 30;458(7242):1136–1141. doi: 10.1038/nature07975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Monecke T, Guttler T, Neumann P, Dickmanns A, Gorlich D, Ficner R. Crystal structure of the nuclear export receptor CRM1 in complex with Snurportin1 and RanGTP. Science. 2009 May 22;324(5930):1087–1091. doi: 10.1126/science.1173388. [DOI] [PubMed] [Google Scholar]
- 7.Dong X, Biswas A, Suel KE, Jackson LK, Martinez R, Gu H, et al. Structural basis for leucine-rich nuclear export signal recognition by CRM1. Nature. 2009;458:1136–1141. doi: 10.1038/nature07975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fung HYJ, Fu S-c, Brautigam CA, Chook YM. Structural determinants of nuclear export signal orientation in binding CRM1. eLife. 2015 Sep 08; doi: 10.7554/eLife.10034. 12:08:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tai Y-T, Landesman Y, Acharya C, Calle Y, Zhong MY, Cea M, et al. CRM1 inhibition induces tumor cell cytotoxicity and impairs osteoclastogenesis in multiple myeloma: molecular mechanisms and therapeutic implications. Leukemia. 2014;28:155–165. doi: 10.1038/leu.2013.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Van Der Watt PJ, Maske CP, Hendricks DT, Parker MI, Denny L, Govender D, et al. The karyopherin proteins, Crm1 and Karyopherin Beta1, are overexpressed in cervical cancer and are critical for cancer cell survival and proliferation. International Journal of Cancer. 2009;124:1829–1840. doi: 10.1002/ijc.24146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tan DSP, Bedard PL, Kuruvilla J, Siu LL, Razak ARA. Promising SINEs for Embargoing Nuclear-Cytoplasmic Export as an Anticancer Strategy. Cancer discovery. 2014 doi: 10.1158/2159-8290.CD-13-1005. [DOI] [PubMed] [Google Scholar]
- 12.Falini B, Mecucci C, Tiacci E, Alcalay M, Rosati R, Pasqualucci L, et al. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. The New England journal of medicine. 2005;352:254–266. doi: 10.1056/NEJMoa041974. [DOI] [PubMed] [Google Scholar]
- 13.Yoshimura M, Ishizawa J, Ruvolo V, Dilip A, Quintas-Cardama A, McDonnell TJ, et al. Induction of p53-mediated transcription and apoptosis by exportin-1 (XPO1) inhibition in mantle cell lymphoma. Cancer science. 2014 Jul;105(7):795–801. doi: 10.1111/cas.12430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Puente XS, Pinyol M, Quesada V, Conde L, Ordonez GR, Villamor N, et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature. 2011 Jul 7;475(7354):101–105. doi: 10.1038/nature10113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lapalombella R, Sun Q, Williams K, Tangeman L, Jha S, Zhong Y, et al. Selective inhibitors of nuclear export show that CRM1/XPO1 is a target in chronic lymphocytic leukemia. Blood. 2012 Nov 29;120(23):4621–4634. doi: 10.1182/blood-2012-05-429506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ranganathan P, Yu X, Na C, Santhanam R, Shacham S, Kauffman M, et al. Preclinical activity of a novel CRM1 inhibitor in acute myeloid leukemia. Blood. 2012;120:1765–1773. doi: 10.1182/blood-2012-04-423160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ranganathan P, Yu X, Santhanam R, Hofstetter J, Walker A, Walsh K, et al. Decitabine priming enhances the anti-leukemic effects of exportin 1 (XPO1) selective inhibitor selinexor in acute myeloid leukemia. Blood. 2015 Feb 25; doi: 10.1182/blood-2014-10-607648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kuruvilla J, Gutierrez M, Shah BD, Gabrail NY, de Nully Brown P, Stone RM, et al. Preliminary Evidence Of Anti Tumor Activity Of Selinexor (KPT-330) In a Phase I Trial Ofa First-In-Class Oral Selective Inhibitor Of Nuclear Export (SINE) In Patients (pts) With Relapsed / Refractory Non Hodgkin's Lymphoma (NHL) and Chronic Lymphocytic L…. 2013;122:90–90. [Google Scholar]
- 19.Chen CI, Gutierrez M, de Nully Brown P, Gabrail N, Baz R, Reece DE, et al. Anti Tumor Activity Of Selinexor (KPT-330), A First-In-Class Oral Selective Inhibitor Of Nuclear Export (SINE) XPO1/CRM1 Antagonist In Patients (pts) With Relapsed/Refractory Multiple Myeloma (MM) Or Waldenstrom's Macroglobulinemia (WM) Blood. 2013;122(21):1942–1942. [Google Scholar]
- 20.Savona M, Garzon R, de Nully Brown P, Yee K, Lancet JE, Gutierrez M, et al. Phase I trial of selinexor (KPT-330), a first-in-class oral selective inhibitor of nuclear export (SINE) in patients (pts) with advanced acute myelogenous leukemia (AML) Blood. 2013;122(21):1440–1440. [Google Scholar]
- 21.Chook YM, Blobel G. Structure of the nuclear transport complex karyopherin-beta2-Ran x GppNHp. Nature. 1999 May 20;399(6733):230–237. doi: 10.1038/20375. [DOI] [PubMed] [Google Scholar]
- 22.Sun Q, Carrasco YP, Hu Y, Guo X, Mirzaei H, Macmillan J, et al. Nuclear export inhibition through covalent conjugation and hydrolysis of Leptomycin B by CRM1. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:1303–1308. doi: 10.1073/pnas.1217203110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Etchin J, Sun Q, Kentsis A, Farmer A, Zhang ZC, Sanda T, et al. Antileukemic activity of nuclear export inhibitors that spare normal hematopoietic cells. Leukemia. 2013 Jan;27(1):66–74. doi: 10.1038/leu.2012.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haines JD, Herbin O, de la Hera B, Vidaurre OG, Moy GA, Sun Q, et al. Nuclear export inhibitors avert progression in preclinical models of inflammatory demyelination. Nature neuroscience. 2015 Feb 23; doi: 10.1038/nn.3953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bichi R, Shinton SA, Martin ES, Koval A, Calin GA, Cesari R, et al. Human chronic lymphocytic leukemia modeled in mouse by targeted TCL1 expression. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(10):6955–6960. doi: 10.1073/pnas.102181599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhong Y, El-Gamal D, Dubovsky JA, Beckwith KA, Harrington BK, Williams KE, et al. Selinexor suppresses downstream effectors of B-cell activation, proliferation and migration in chronic lymphocytic leukemia cells. Leukemia. 2014 May;28(5):1158–1163. doi: 10.1038/leu.2014.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johnson AJ, Lucas DM, Muthusamy N, Smith LL, Edwards RB, De Lay MD, et al. Characterization of the TCL-1 transgenic mouse as a preclinical drug development tool for human chronic lymphocytic leukemia. Blood. 2006 Aug 15;108(4):1334–1338. doi: 10.1182/blood-2005-12-011213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hing ZA, Mantel R, Beckwith KA, Guinn D, Williams E, Smith LL, et al. Selinexor is effective in acquired resistance to ibrutinib and synergizes with ibrutinib in chronic lymphocytic leukemia. Blood. 2015 Apr 02; doi: 10.1182/blood-2015-01-621391. 18:00:08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Newlands ES, Rustin GJ, Brampton MH. Phase I trial of elactocin. British Journal of Cancer. 1996;74(4):648–649. doi: 10.1038/bjc.1996.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mutka SC, Yang WQ, Dong SD, Ward SL, Craig Da, Timmermans PBMWM, et al. Identification of nuclear export inhibitors with potent anticancer activity in vivo. Cancer Research. 2009;69:510–517. doi: 10.1158/0008-5472.CAN-08-0858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Etchin J, Berezovskaya A, Conway AS, Chen WC, Baloglu E, Landesman Y, et al. Nuclear Export Inhibitor KPT-8602 Is Highly Active Against Leukemic Blasts and Leukemia-Initiating Cells in Patient-Derived Xenograft Models of AML. Leukemia. in submission. [Google Scholar]
- 32.Turner JG, Dawson J, Sullivan DM. Nuclear export of proteins and drug resistance in cancer. Biochemical pharmacology. 2012;83:1021–1032. doi: 10.1016/j.bcp.2011.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.