

SUMMARY
Apoptosis-inducing factor (AIF) is critical for mitochondrial respiratory complex biogenesis and for mediating necroptotic parthanatos: these functions are seemingly regulated by enigmatic allosteric switching driven by NADH charge-transfer complex (CTC) formation. Here we define molecular pathways linking AIF’s active site to allosteric switching regions by characterizing dimer-permissive mutants using small-angle X-ray scattering (SAXS) and crystallography and by probing AIF-CTC communication networks using molecular dynamics simulations. Collective results identify two pathways propagating allostery from the CTC active site: 1) active site H454 links to S480 of AIF’s central β-strand to modulate a hydrophobic border at the dimerization interface and 2) an interaction network links AIF’s FAD cofactor, central β-strand, and Cβ-clasp whereby R529 reorientation initiates C-loop release during CTC formation. This knowledge of AIF allostery and its flavoswitch mechanism provides a foundation for biologically understanding and biomedically controlling its participation in mitochondrial homeostasis and cell death.
Keywords: Apoptosis-inducing factor, charge-transfer complex, flavoswitch, parthanatos, mitochondrial homeostasis, allostery, SAXS, X-ray crystallography, molecular dynamics
eTOC Blurb
Brosey et al. define the mechanism whereby mitochondrial Apoptosis-Inducing Factor (AIF) is allosterically modulated by charge-complex (CTC) formation with NADH. Two distinct molecular pathways couple AIF’s active site to dimerization and release of its regulatory C-loop insert with implications for AIF allosteric switching in cell survival and death
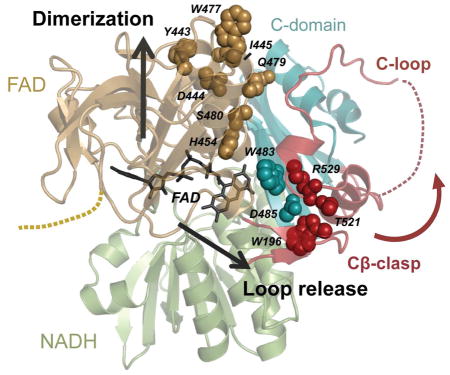
INTRODUCTION
Apoptosis-inducing factor (AIF) is a mitochondrial oxidoreductase that acts in cell survival as a supporter of respiratory complex assembly (Pospisilik et al., 2007; Vahsen et al., 2004) and in cell death as a mediator of necroptotic poly(ADP-ribose) polymerase (PARP)-1-dependent cell death (parthanatos) (Wang et al., 2009). AIF’s acts in respiratory complex biogenesis by facilitating import factor CHCHD4/MIA40 entry into mitochondria (Hangen et al., 2015; Meyer et al., 2015). In parthanatos AIF accumulates in the nucleus following release from its inner membrane tether during PARP-1 hyperactivation, where it may aid in DNA fragmentation and chromatin condensation (Moubarak et al., 2007; Yu et al., 2006). The regulatory processes surrounding AIF release from mitochondria and nuclear accumulation during parthanatos remain largely unknown.
AIF’s structure is dramatically modulated by binding NAD(P)H and forming a stable, long-lived charge-transfer complex (CTC) between its FAD cofactor and the NAD(P)H nicotinamide moiety (Churbanova and Sevrioukova, 2008). CTC formation stimulates dimerization and releases a 50-residue insert within the C-terminal domain (C-loop, residues 509–559) from contacts with the protein core and a β-hairpin insert (‘Cβ-clasp’) that bridges the FAD and NADH domains (Figure 1A, S1A–B). Interaction with NADH and CTC formation are key to AIF function in vivo. NADH binding is required for AIF’s interaction with mitochondrial import factor CHCHD4 (Hangen et al., 2015). Disease-causing mutations that hinder AIF’s ability to efficiently maintain a CTC with NADH lead to human neurodegenerative mitochondriopathies (Berger et al., 2011; Diodato et al., 2016; Ghezzi et al., 2010; Rinaldi et al., 2012; Zong et al., 2015). These interaction and disease-causing results suggest that AIF’s structural transitions are key for mitochondrial homeostasis. Structural regulation by NADH may also modulate AIF release from mitochondria following critical depletion of NAD+ levels during PARP-1-initiated cell death (Alano et al., 2010). Thus, knowing how CTC formation regulates AIF structure is vital for understanding its dual roles in cell death and survival.
Figure 1. AIF Remodeling and Dimerization in Solution Support Crystallographic Transitions.

(A) AIF domain organization with FAD (gold), NADH (green), C-terminal domains (teal), and transmembrane tether (TM). NADH binding exposes the 50-residue C-loop (dotted line) and enables dimerization. See also Figure S1.
(B) SAXS captures NADH-driven transitions in AIF78. Overlay plots of I(q) (left) and Rg-normalized Kratky transformations (I(q)/I(0)*(q*Rg)2, right) at 0 (black), 0.25, 0.50, 0.75, 1.0, 2.0, 3.5, 5.0, 10.0 molar excess NADH (rainbow). Inset shows expanded low-q region.
(C) Guinier Rg (left) and Vc-derived masses (right) for AIF78 (●) and AIF121 (■) versus molar NADH. See also Figure S2.
(D) Overlay plots and fit ratios of experimental and theoretical scattering from monomeric (left) or dimeric (right) AIF crystal structures and Bilbo-MD optimized models. See also Table 1.
(E) Superposition of theoretical SAXS envelopes with monomer and dimer crystal structures (far right) or experimental AIF121 and AIF78 SAXS envelopes with BilboMD-optimized models. C-loops and terminal residues are highlighted in blue. C-loops are modeled in both crystal structures for reference.
Yet, the structural basis of AIF allostery is unknown. Human and mouse AIF structures were analyzed at high resolution with and without NADH (Ferreira et al., 2014; Sevrioukova, 2009); dimerization surfaces of monomeric and dimeric AIF, however, are highly similar in these structures, and it is unclear how NADH binding at the active site allosterically enables AIF-CTC dimerization or how the monomer avoids spontaneously dimerizing in the absence of NADH. Prior studies showed backbone shifts in a small β-hairpin adjacent to the dimerization interface, but have not defined an allosteric pathway that links CTC formation to dimerization (Sevrioukova, 2009). Allosteric coordination between CTC formation and C-loop release is also unclear, as the C-loop remains disordered in AIF-CTC structures. AIF-CTC structures capture an ‘aromatic tunnel’ that links the active site to the C-loop insertion point and exhibits coordinated flipping of its aromatic side chains when NADH is present (Sevrioukova, 2009). It is unclear, though, whether these side chain rearrangements stimulate C-loop remodeling and release into solvent.
Here, we combined x-ray crystallography and small-angle X-ray Scattering (SAXS) to characterize dimer-permissive AIF mutants. The structure of dimer-permissive AIF W196A identifies interaction between active site residue H454 and S480 of AIF’s central β-strand as the regulatory link between the active site and dimerization surface. Disruption of H454-S480 contact in monomeric AIF, by NADH binding or by mutagenic displacement of the C-loop, exposes and organizes a hydrophobic border at the dimer interface that regulates dimerization. All-atom molecular dynamics simulations of AIF-CTC identify the Cβ-clasp as a focal point for C-loop release, uncovering a communication network linking AIF’s FAD cofactor, central β-strand, and Cβ-clasp. Complementary structure-function analyses uncover R529 reorientation within the Cβ-clasp as a stimulus for C-loop release during CTC formation. These results establish distinct molecular pathways driving allosteric switching of AIF architecture during CTC formation. Defining this novel flavoswitch mechanism provides a foundation for investigating and controlling how cellular NAD(H) regulates AIF actions in mitochondrial homeostasis and cell death.
RESULTS
AIF Remodeling and Dimerization in Solution Support Crystallographic Transitions
Existing crystal structures of monomeric AIF and dimeric AIF-CTC display similar electrostatic surfaces at the dimer interface and fail to explain how access to this surface is regulated. To test whether the crystallographic monomer and dimer accurately reveal AIF architecture in solution or reflect crystal packing, we collected SAXS data on soluble constructs with increasing NADH concentrations: AIF78 (residues 78–613, the intact mitochondrial protein save the 24-residue transmembrane insert, residues 54–77), and AIF121 (residues 121–613, excluding the disordered N-terminal linker to the membrane) (Figure 1A, S2). These constructs were biochemically validated for their ability (1) to expose the C-loop by tracking its increased proteolytic susceptibility and (2) to dimerize by observing cross-linked dimer species with SDS-PAGE in the presence of NADH (Sevrioukova, 2016) (Figure S1C).
The formation of dimeric AIF78-CTC with increasing NADH is captured in a progressive I0 rise in the scattering curve, I(q), and expansion of the Kratky maximum into a secondary shoulder (Figure 1B). Tracking radius of gyration (Rg) and mass (derived from SAXS empirical power-law relationships (Rambo and Tainer, 2013)) across the titration series suggests that dimer formation plateaus beyond a 2-fold excess of NADH ligand (Figure 1C). AIF121 exhibits similar transitions in I(q), Rg, and mass upon NADH addition, but reaches a dimeric state at equimolar NADH (Figure 1C). AIF121 may dimerize more efficiently than AIF78 in the absence of steric interference from the extended N-terminus, which flanks the dimerization surface (Figure S1B).
We compared Rg values and Porod volumes (Vp) from the SAXS experiments with theoretical scattering curves calculated from published crystal coordinates of monomeric human AIF(103–613) (density for residues 127–610, modified to include missing C-loop residues 546–558, see Table 1). Rg and Vp values derived from AIF121 SAXS experiments (Rg 26.1 Å and Vp 88,613 Å3) and crystallized monomeric AIF (Rg 24.8 Å and Vp 85,719 Å3) are similar, indicating that AIF’s monomeric architecture in solution resembles the crystal structure. Modeling missing terminal residues further improves correspondence between model and data (Figure 1D). Corresponding values derived from AIF78 (Rg 28.9 Å and Vp 101,858 Å3) are elevated relative to the crystal structure, consistent with added scattering volume from the 43-residue N-terminus. Furthermore, BilboMD modeling of the flexible N-terminus in a partially extended conformation improved the fit to the scattering data (Figure 1D–E, Table 1).
Table 1.
AIF Experimental and Modeled SAXS Parameters
| Monomer | Rg (Å)a | Vp (Å3)b | Porod Coefficientc | Dmax (Å)d |
|---|---|---|---|---|
| AIF Crystale | 24.8 | 85719 | 4.0 | 75 |
|
| ||||
| AIF121 | 26.1 | 88613 | 4.0 | 86 |
| AIF121 Model (Crystal + flexible terminif) | 25.3 | 86512 | 3.9 | 81 |
|
| ||||
| AIF78 | 28.9 | 101858 | 3.9 | 108 |
| AIF78 Model (Crystal + flexible terminig) | 29.0 | 101909 | 4.0 | 106 |
|
| ||||
| Dimer | Rg (Å)a | Vp (Å3)b | Porod Coefficientc | Dmax (Å)d |
|
| ||||
| AIF Crystalh | 34.5 | 215755 | 3.2 | 107 |
| AIF Crystal +Open C-loopi | 39.5 | 297981 | 2.9 | 128 |
|
| ||||
| AIF121/NADH | 38.3 | 238403 | 3.2 | 137 |
| AIF121 (Crystal + Open C-loopi + Flexible terminif) | 38.3 | 270576 | 3.0 | 132 |
|
| ||||
| AIF78/NADH | 40.1 | 291064 | 3.0 | 153 |
| AIF78 (Crystal + Open C-loopi + Flexible terminig) | 39.5 | 302086 | 2.9 | 131 |
Radius-of-gyration
Porod volume
Porod coefficients calculated as in (Rambo and Tainer, 2011)
Maximum dimension
PDB: 4BV6, missing C-loop residues 546–558 added.
Residues 121–126, 611–619 modeled by BilboMD.
Residues 78–126, 611–619 modeled by BilboMD.
PDB: 4BUR, chains a and c
C-loop residues 517–550 (chain a), 518–557 (chain c) modeled by BilboMD.
Rg and Vp values from both dimeric AIF121 and AIF78 bound to NADH are increased relative to parameters from theoretical scattering of the crystallized dimer (Table 1), and the scattering data is not well fit by the simulated curves (Figure 1D). To test whether an alternate dimer interface at the NADH or C-terminal domain could explain the discrepancy, we calculated and compared theoretical scattering from symmetry-related dimers taken from the crystal structure with dimeric AIF121/NADH SAXS curves (Figure S1D). Of the three possible domain interfaces, however, the reported interface at the FAD domain still provides the best fit to the data. To examine whether release and flexible extension of the C-loop (absent in the crystallized dimer), could improve correspondence with the SAXS data, we added missing C-loop residues in a bound, ‘closed’ conformation to the dimer structure and sampled flexible conformations using BilboMD (Figure 1D–E). For both AIF121 and AIF78, the conformation of best fit exhibits an ‘open’ C-loop that is displaced away from the protein core (Figure 1E). Dimer conformations with open, extended C-loops were still selected when terminal residues were added and flexibly sampled by BilboMD, indicating that C-loop extension does not arise as a compensation for absent terminal residues. (Figure 1D–E). Thus, the SAXS data support the crystallographic FAD dimer interface and the presence of flexibly extended C-loops in solution.
To further compare solution and crystal architectures, we calculated scattering envelopes from AIF78 and AIF121 SAXS data and from theoretical scattering curves of unmodified AIF crystal structures, comparing these to models including the C-loop and flexible termini (Figure 1E). SAXS envelopes from monomeric AIF78 and AIF121 roughly display AIF’s tri-domain organization with extensions that align well with the flexible N-terminus when compared to atomic models (Figure 1E, top). The SAXS envelope for dimeric AIF121/NADH exhibits the bilobal architecture observed in the theoretical envelope calculated from simulated crystal structure scattering, but includes added volume at each lobe. Superposition of the BilboMD-optimized dimer model places this added volume near the ‘open’ C-loops, suggesting that flexible sampling of the C-loop accounts for differences in the envelope shapes (Figure 1E, bottom). The SAXS envelope for dimeric AIF78/NADH also exhibits two-fold symmetry, but differs in shape relative to the AIF121/NADH and crystal envelopes. Superposition of the dimeric AIF78 model places the open C-loop and extended N-termini near regions of greatest difference. Hence, differences between theoretical scattering envelopes from crystal structures and experimental SAXS envelopes reflect the presence of flexible AIF motifs captured by the scattering data.
To complement the SAXS analysis, we biochemically validated the crystallographic dimerization surface. Targeted mutation of residues acting in AIF’s dimer interface (E413A/R422A/R430A) disrupts dimerization (Ferreira et al., 2014). To test if this interference arises from disrupting binding interactions, rather than a secondary allosteric effect, we probed AIF78 cysteine mutants at interface residues R422 and R449 with PEG-maleimide (PEG-MEM) (Figure S1E). Adding NADH and forming AIF78-CTC shielded these residues from PEG-MEM labeling, showing that R422 and R449 are at the dimer interface, rather than mediating a secondary allosteric contribution to dimerization.
AIF Dimerization is Coupled to C-Loop Remodeling
Crystal structures of monomeric AIF and dimeric AIF-CTC resemble their SAXS-derived architectures in solution. To test how NADH binding allosterically regulates dimer interface access and facilitates C-loop release, we made mutants targeting the dimerization interface or the C-loop, then assessed their ability to dimerize or to release the C-loop upon binding NADH (Figures 2, S2–3). Neutralization of the dimer interface, as seen for obligate monomer mutant E413A/R422A/R430A, greatly compromises, but not does not eliminate dimerization in the presence of NADH as assessed by BS3 cross-linking and SAXS (Figures 2A, S3A). The ability of the monomeric mutant to release its C-loop remains largely undisturbed (Figure 2A). In contrast, full removal of the C-loop (replacing residues 511–558 with a GGGSSS linker) renders AIF fully permissive to dimerization in the absence of NADH binding and CTC formation (Figures 2B, S3B). Thus, C-loop release can occur independently of dimerization, but dimerization is allosterically coupled to C-loop displacement.
Figure 2. AIF Dimerization is Coupled to C-Loop Remodeling.

(A) Limited proteolysis (left) and cross-linking (middle) of AIF78 E413A/R422A/R430A (‘Monomer’) show intact C-loop release, but impaired dimerization with increasing NADH. (Ø – no protease or BS3). The mid-q region is increased in the Kratky profile of NADH-bound ‘Monomer’, consistent with C-loop remodeling, but lacks the distinct shoulder of the wild-type dimer (right). See also Figures S2–3.
(B) Limited proteolysis and BS3 cross-linking analyses demonstrate permissive dimerization for AIF78 C-loop mutant without NADH. The mutant resembles the AIF-CTC dimer in the Kratky analysis. (C) Limited proteolysis (top) and cross-linking analyses (bottom) of C-loop salt bridge mutants and Cβ-clasp mutants without NADH. C-loop salt bridge mutants show partial C-loop destabilization, while Cβ-clasp mutants show full C-loop destabilization and permissive dimerization without ligand.
(D) SAXS Rg and mass analyses of C-loop salt bridge mutants (top) and Cβ-clasp mutants (bottom). C-loop salt bridge mutants dimerize more efficiently than wild-type AIF78, while Cβ-clasp mutants are dimerization permissive at all NADH concentrations. See also Figure S2.
To more selectively probe how C-loop perturbation affects dimerization, we assessed single-point mutants targeting C-loop salt bridges to the protein core (E531, E533, E535) or the hydrogen bond network of the Cβ-clasp (W196A, R529A, D485A) (Figures 2C–D, S2). Point mutations along the C-loop backbone promoted partial C-loop exposure without ligand and more efficient protein dimerization at lower NADH levels relative to wild-type protein (Figures 2C–D, top). Remarkably, mutation of Cβ-clasp residues resulted in nearly complete C-loop exposure and permissive dimerization without ligand, a phenotype similar to full C-loop deletion (Figures 2C–D, bottom). This direct correlation of C-loop destabilization to dimerization efficiency points to the Cβ-clasp as a trigger for mediating and initiating C-loop release during CTC formation and subsequent dimerization.
Dissection-of-Function Mutant AIF78 W196A Dimerizes without CTC Formation or Aromatic Tunnel Activation
As C-loop deletion or mutagenic displacement can induce dimerization in the absence of CTC formation, we crystallized and determined the structure of the W196A mutant to unveil allostery in AIF dimerization. We also crystallized the obligate monomer mutant (AIF78 E413A/R422A/R430A) as a monomeric state reference (Figure S4, Table S1). Comparison of the monomer mutant structure to the wild-type protein (PDB: 4BV6) shows high similarity (backbone RMSD 0.36 Å) and an absence of allosteric effects from altering dimerization interface residues (Figure S4A). This supports our biochemical observation that perturbing the dimeric interface affects dimerization but has minimal impact on C-loop remodeling.
AIF78 W196A forms the dimeric interface seen in the wild-type AIF-CTC crystal structure (PDB: 4BUR) and is missing density for the displaced C-loop insert (Figure 3A). However, several key side chain rearrangements seen in the active site of wild-type AIF-CTC are absent in the W196A mutant. Residues that stabilize the FAD+ cofactor near the NADH binding site (P173, K177, E314, W483) are moderately shifted upon CTC formation and also within the W196A mutant. However, W196A residue K177 maintains monomeric hydrogen bonding contact with the FAD+ oxidized isoalloxazine ring (Figure 3B). Similarly, many residues involved in binding and retaining NADH in the active site pocket (G308, F310, L311, E314, E336, G399) maintain monomeric positions except E453 and H454. These two residues reorient to assume positions more consistent with AIF-CTC (Figure 3B). To ensure that selective E453 and H454 reorientation did not arise from phase bias during molecular replacement, we calculated simulated annealing |Fo-Fc| omit maps and verified these residue positions (Figure S4B). H454 is the sole, nonconserved residue within the NADH binding pocket, and its hydrogen bond interaction with the nicotinamide group enables AIF to form long-lived charge-transfer complexes with NADH (Churbanova and Sevrioukova, 2008). That permissive dimerization in the W196A mutant preferentially alters this residue supports its importance in mediating CTC-driven allostery. Rearrangement of the entire active site architecture thus is not required for dimerization.
Figure 3. The Dissection-of-Function Mutant AIF78 W196A Dimerizes Without CTC Formation or Aromatic Tunnel Activation.

(A) The dimeric W196A mutant crystal structure lacks C-loop density. FAD domain (gold), NADH domain (green), C-terminal domain (teal), β-hairpin and C-loop boundaries (red). See also Figure S4, Table S1.
(B) W196A FAD isoalloxazine ring (left) and NADH binding pocket (right) (color) overlaid with structures of wild-type monomer (light gray, PDB: 4BV6) and dimer (dark gray, PDB: 4BUR).
(C) W196A C-terminal aromatic tunnel (left) and the border region of the C-loop insertion (right) (color) overlaid with structures of wild-type monomer (light gray) and dimer (dark gray).
In addition, several residues that form the aromatic tunnel connecting the active site to the opposite end of the C-terminal domain (F310, F482, Y492, W579, F308) remain largely unchanged relative to the monomeric state (Figure 3C). In wild-type AIF-CTC, the F310 aromatic side chain moves into the active site to engage the NADH nicotinamide group by π-stacking interactions. Reorientation of F310 and H454 may stimulate ring flipping of neighboring F482 and Y492 residues, propagating aromatic side chain rearrangement down the aromatic tunnel and stabilizing AIF-CTC by electron delocalization (Sevrioukova, 2009). Yet, in the W196A mutant, aromatic side chain reorientations are not seen (Figure 3C). Aromatic residue Y347, which caps the tunnel at the domain end and borders the insertion point of the displaced C-loop, exhibits heterogeneity relative to monomeric AIF (Figure 3C) that appears to arise from C-loop displacement, rather than dimerization per se. Thus, activation of the C-terminal aromatic tunnel is not essential for dimerization.
Central β-strand Pivoting in the W196A Mutant Creates a Hydrophobic Border near the Dimerization Interface
Most structural changes shared by the W196A mutant and the wild-type AIF-CTC dimer cluster to a central β-strand that bridges the FAD-binding and C-terminal domains and to the β-hairpin that caps the interface between these two domains (Figure 4A). The central β-strand runs from the dimerization surface to the Cβ-clasp and forms an interior boundary between the clasp and the NADH active site. With the C-loop displaced, the strand pivots ~11 degrees about its active site center at residue W483 (Figure 4A). Near the Cβ-clasp, this rotation draws the turn at the strand base away from the clasp toward the protein interior, while the freed β-hairpin shifts forward toward the pocket exposed by the missing C-loop (Figure 4B). Residues that packed against the Cβ-clasp in the monomeric state (W483, F181, R321, F582) align to conformations more consistent with wild-type AIF-CTC.
Figure 4. Central β-strand Pivoting in the W196A Mutant Creates a Hydrophobic Border near the Dimerization Interface.

(A) Overlay of monomeric wild-type AIF (gray, PDB: 4BV6) and AIF78 W196A (color) highlights rotation of AIF’s central β-strand and capping β-hairpin at the FAD/C-domain interface.
(B) The Cβ-clasp region highlighting displacement of the clasp β-hairpin and the central β-strand.
(C) The H454-S480 hydrogen-bonding network (dashed line) in monomeric wild-type AIF (left) that is reorganized into S480-H457-Y476 in dimeric W196A (right). Oxygen, red; nitrogen, blue.
(D) The D444-R450 hydrogen bonding network (dashed line) in monomeric wild-type AIF (left) that is reorganized to include Q479 in dimeric W196A (right).
(E) The W477/I445/Y443 border (left) in monomeric wild-type AIF (top right) aligns to form a contiguous hydrophobic surface against the polar center of the dimer interface in dimeric W196A (bottom right).
At the opposite end near the dimerization interface, the central β-strand tilts forward into the region previously occupied by the C-loop and drives reorganization of hydrogen bond networks that connect it to the active site and capping β-hairpin (Figure 4C–D). Notably, β-strand displacement disrupts S480 hydrogen bonding to key active site residue H454. In monomeric AIF structures, this interaction appears to hold H454 away from its optimal NADH binding position and to prevent central β-strand pivoting. Loss of the H454-S480 interaction in the W196A mutant causes H454 to shift toward its NADH binding position, while S480 forms new hydrogen bonds with H457 and Y476 (Figure 4C). The adjacent β-strand residue Q479 also forms new hydrogen bonds with residues D444 and R450 from the capping β-hairpin (Figure 4D). In the monomer, these new interactions position Q479’s polar amide group near the interior-facing I445 side chain of the β-hairpin (Figure 4E). In AIF-CTC and the W196A mutant, the I445 side chain reorients away from this polar neighbor toward the solvent and forms a new, contiguous hydrophobic surface with W477 and Y443 at the dimerization interface edge. This hydrophobic triplet packs against and aligns with polar residues of the dimer interface (Figure 4E). Thus, while the electrostatic core of the dimerization interface is continually exposed to solvent, exposure of the hydrophobic border is allosterically modulated by pivoting of the central β-strand, enabled by CTC formation in wild-type AIF or mutagenic displacement of the C-loop in the W196A mutant.
An Allosteric Pathway Links AIF’s Active Site to the Dimerization Surface
To test the H454-S480 interaction as an allosteric pathway linking AIF’s active site to the dimerization surface, we created a panel of point mutants targeting active site residue H454 and β-strand residue S480. We also evaluated point mutations of Y443, I445, and W477 from the hydrophobic border to assess their proposed contributions to the dimerization interface.
Consistent with other reported H454 mutations (Churbanova and Sevrioukova, 2008; Villanueva et al., 2015), substitution of the active site histidine eliminates AIF’s ability to form a charge-transfer complex with NADH, and the protein rapidly converts excess NADH to NAD+ through catalytic hydride removal (Figure S5A). Biochemical and SAXS analyses show that the H454A mutant exists as a dimer in solution in the absence of NADH and uphold H454 as the key active site residue that regulates AIF dimerization (Figure 5A–B, S2). Notably, the C-loop remains intact in this mutant (Figure S5B), indicating that C-loop remodeling can be bypassed by targeting residues with direct involvement in the dimerization pathway.
Figure 5. An Allosteric Pathway Links AIF’s Active Site to the Dimerization Surface.

(A) Cross-linking analysis of H454A and S480A demonstrate permissive dimerization in the absence of ligand, while dimerization of Y443A/I445A is hindered with addition of NADH. See also Figures S5, S6.
(B) SAXS Rg and mass analyses of H454A, S480A (top) and Y443A/I445A (bottom) demonstrate permissive or hindered dimerization, respectively, relative to wild-type AIF. See also Figure S2.
Complementary alanine substitution of S480 also results in obligately dimeric AIF, unveiling the H454-S480 interaction in restraining central β-strand reorientation. (Figure 5A–B). Interestingly, formation of the hydrogen bond network among S480, H457, and Y476, as seen in dimeric AIF structures, does not appear essential for mediating dimerization, as the alanine mutant cannot form hydrogen bonds with H457 and Y476. Thus, loss of steric restraint on the central 3-strand imposed by H454-S480 interaction seems more important for regulating dimerization than formation of a new hydrogen bond network with H457 and Y476. The C-loop also remains intact in ligand-free S480A, similar to H454A, but is allosterically responsive when NADH is added (Figure S5B).
Single point mutations of hydrophobic border residues exhibit a wild-type ability to dimerize (Figure S5). Neutralizing two (Y443A/I445A) or all three (Y443A/I445A/W477A) residues of the hydrophobic border compromises dimerization in a manner similar to the original AIF monomer mutant (E413A/R422A/R430A) (Figure 5A–B, S2, S5C). C-loop remodeling remains intact in double and triple AIF border mutants, but requires higher levels of NADH to expose the loop completely (Figure S5C). Thus, the hydrophobic border serves as a key component of the dimerization interface and an allosteric endpoint for triggering dimerization through CTC formation.
To assess conformational variation across all AIF phenotypes, we calculated pairwise SAXS volatility of ratio (VR) values for each mutant relative to pure monomer (AIF78) or dimer (AIF78/NADH) states and assembled them into SAXS similarity matrices (SSM) (Hura et al., 2013) (Figure 6). Clustering the ligand-free SSM resolves two distinct groups corresponding to wild-type monomer or perturbed dimer-permissive states (Figure 6A). Previously identified dimer-permissive mutants are well separated into the latter cluster, but exhibit a continuum of agreement with the pure dimer state. Notably, mutants with C-loops intact (H454A, S480A) tend to exhibit less agreement than mutants with open or absent C-loops. Complementary analysis for NADH-bound AIF mutants also resolves two clusters with the larger following AIF-CTC (Figure 6B). A smaller cluster captures mutants of the dimer interface (E413A/R422A/R430A and Y443A/I445A) and dimer-permissive H454A, which exhibit intermediate agreement between the two states. This divergence could be attributed to C-loop conformations that differ from wild-type (open for interface mutants and closed for H454A with excess NADH). That SSM cluster analysis can classify allostery mutants generated by our structural predictions (H454A, S480A, Y443A/I445A) relative to wild-type AIF supports the proposed mechanism for allosteric dimerization.
Figure 6. SSM Analysis Resolves AIF Allostery Mutants.

(A) VR SAXS similarity matrix (SSM) correlating ligand-free mutants with monomeric (AIF78) or dimeric (AIF78/NADH) states reveals two conformational clusters. Lower VR values represent higher correspondence.
(B) VR SAXS similarity matrix (SSM) correlating NADH-bound mutants with monomeric (AIF78) or dimeric (AIF78/NADH) states reveals major and minor conformational clusters.
Capturing a Pre-release Intermediate
Our analysis of mutants defines a pathway between the CTC active site and dimerization interface and supports central β-strand pivoting as critical for forming the dimer binding surface. How CTC formation is channeled into internal displacement of AIF’s C-loop, however, is unclear from the end states captured by W196A or AIF-CTC crystal structures. To gain insight into intermediate states that precede C-loop release, we generated two 500-ns, all-atom molecular dynamics simulations for AIF in the absence of ligand and for a monomeric AIF/FADH−/NAD+ complex designed to model the NADH charge-transfer complex prior to C-loop release and dimerization. The starting structure for monomeric AIF was prepared from published coordinates (Ferreira et al., 2014) with disordered C-loop residues added. We modeled the CTC in a post-hydride transfer state (FADH−/NAD+) based upon the work of Sorrentino and colleagues (Sorrentino et al., 2015), where photoreduced AIF (FADH-) in complex with NAD+ reconstituted biochemical and structural properties of AIF-CTC.
Initial inspection of selected snapshots from CTC trajectories reveals that active site residues H454 and F310 reorient to engage the NAD+ ligand early in the simulation period, while aromatic tunnel residues also show signs of repositioning, suggesting that activation of the C-terminal aromatic tunnel follows conformational rearrangement in the CTC active site (Figure 7A). K177, which hydrogen bonds to FAD N5 in the absence of ligand, is also partly displaced, as N5 protonation of the reduced FAD precludes this interaction (Figure S7). Further inspection of trajectory snapshots reveals increased C-loop heterogeneity in the AIF/FADH−/NAD+ complex relative to ligand-free AIF, though the loop itself does not fully release from the protein core during the simulation (Figure 7B). Backbone RMSDs along consecutive segments of the C-loop show that the greatest fluctuations fall within residues 540–559, which lack contacts to the protein core; however, elevated fluctuations are also seen within residues 520–529, which frame the Cβ-clasp (Figure 7C, S8). Residues participating in salt bridges that hold the C-loop to the protein core (E531/R201, E535/R463) exhibit a propensity to separate in the CTC trajectories (Figure 7D,E). This is seen particularly for the E535/R463 salt bridge, which fronts the central β-strand near the dimerization interface and blocks the forward strand shift seen in W196A and AIF-CTC crystal structures. The rearrangement of AIF’s active site, elevation of C-loop dynamics, and increased lability of C-loop salt bridges suggest a ‘pre-release’ state for AIF-CTC.
Figure 7. Capturing a Pre-release Intermediate.

(A) Ligand-free (left) and CTC (right) trajectories reveal reorientation of active site residues to accommodate the NADH ligand. No ligand: 60 ns (gray), 225 ns (gold), 500 ns (red). CTC: 25 ns (gray), 225 ns (gold), 500 ns (red). See also Figure S7.
(B) Overlaid frames from ligand-free (top) and CTC (bottom) trajectories highlight increased mobility of the C-loop in the FADH−/NAD+ complex.
(C) Average RMSD values for 10-residue segments along the C-loop taken from single 500-ns ligand-free and CTC trajectories (error bars represent standard deviations). See also Figure S8.
(D) Enlarged view of residues participating in C-loop salt bridges from overlays in (B).
(E) Hydrogen bond occupancies of C-loop salt bridges over the course of each trajectory.
A Communication Focal Point Involving the Cβ-clasp is Enhanced in AIF-CTC
To identify key residues involved in communication between the CTC active site and the C-loop, we employed mutual information (MI) analysis of backbone and side chain dihedrals of AIF’s active site residues to assess their correlation with the rest of the protein (Bowman and Geissler, 2012). We chose a subset of residues that directly interact with the NAD+ ligand (F310, E453, H454), as well as those that contact the FAD cofactor (K177, E314, S484). MI values were calculated in aggregate from the two 500-ns trajectories simulated for each system to provide ~1 μs of simulation time for the calculation.
Plotting total active site MI values from the ligand-free simulation along AIF’s sequence and mapping them to AIF’s structure reveals a communication pathway between the active site and residues that form AIF’s central β-strand and Cβ-clasp (Figure 8A,B). Additional C-loop residues flanking the Cβ-clasp and residues extending into the C-terminal domain also exhibit moderate correlation with the active site (Figure 8B). This coupling of the active site, central β-strand, and Cβ-clasp implicates these regions in AIF allostery. Communication within this network is greatly enhanced in AIF-CTC, as seen by a rise in associated MI values (Figure 8A,B). New regions of correlation appear in the CTC simulations for residues that interact with the NAD+ ligand outside of the CTC active site, such as G308 and E336, and neighboring regions that contact these residues – G308 (residues 305–324), E336 (residues 330–365), (Figure 8A). Inter-residue variation among mutual information values is also elevated in the CTC simulations relative to the ligand-free trajectories.
Figure 8. A Communication Focal Point Involving the Cβ-clasp is Enhanced in AIF-CTC.

(A) Sequence plot of total mutual information values correlating active site dihedral conformations to the rest of AIF. No ligand (●), CTC (○).
(B) Scaled color maps of mutual information values for ligand-free (left) and CTC (right) trajectories highlighting increased active site correlation at the Cβ-clasp and central β-strand.
(C) Table of ranked total MI values for ligand-free and CTC trajectories. Residues associated with the central β-strand or the Cβ-clasp are indicated in bold.
To identify specific residues with the greatest active site correlation in the absence and presence of NAD+ ligand, we ranked total MI values for the two systems and selected the top ten residues of each for further examination (Figure 8C). Residues from the central β-strand are present in both rankings (AIF: 481–483; AIF-CTC: 481–485). Notably, the highest ranked residues from AIF-CTC (K177, W483, R529) form an interface among the reduced FAD cofactor (K177), the central β-strand (W483), and the Cβ-clasp (R529), providing a direct path for CTC communication with the Cβ-clasp (Figure S7). Examination of this region over selected trajectory snapshots reveals that R529 rotates and moves away from its Cβ-clasp contacts toward W483. Full reorientation of R529 into an interaction with W483 should disrupt the clasp and facilitate C-loop release.
The active site correlation and reorienting motions observed for R529 in the MD trajectories explain experimental observations supporting the Cβ-clasp as a focal point for C-loop remodeling. Neutralizing clasp residues (R529A, D485A, W196A) has a greater effect on C-loop perturbation than targeting residues elsewhere in the C-loop (E531A, E533A, E535A) (Figure 2). Cβ-clasp proximity to the site of FAD reduction suggests that hydride transfer and FAD reduction during CTC formation initiates C-loop remodeling, while dimerization relies more directly upon NADH occupancy of the active site. Thus, CTC formation likely stimulates separate molecular pathways to trigger AIF allostery.
DISCUSSION
AIF’s ability to form a charge-transfer complex with NADH is key to mitochondrial homeostasis and human health. Yet to date, structural analyses have failed to define how CTC formation contributes to AIF dimerization and C-loop release. Here our collective results reveal that NADH-driven allostery is mediated through two distinct molecular pathways from the active site: via the non-conserved H454 active site residue to the dimerization surface and via the FAD cofactor interface to the Cβ-clasp (Figure 9). NADH binding and reorientation of H454 disrupts its S480 interaction and frees the central β-strand to pivot forward, exposing and aligning a hydrophobic border (residues Y477, I445, Y443) that facilitates dimerization. Computational simulations link CTC formation to increased communication of the active site to the triad of residues adjoining the reduced FAD cofactor (K177, W483, R529) and point to R529 reorientation within the Cβ-clasp as an initiating event in AIF C-loop remodeling. Such arginine switches, as captured by combined solution and crystal structures, are implicated in other ligand sensing and allosteric responses to stress, such as RAD50 ATP-hydrolysis dependent conformations and NBS1 phosphoprotein binding-dependent conformations in DNA break repair (Deshpande et al., 2014; Williams et al., 2009).
Figure 9. Allosteric Regulation by NADH.

NADH binding and CTC formation trigger allosteric remodeling of residues linking active site H454 to the hydrophobic border of the dimerization interface (gold spheres) and act through the Cβ-clasp to release the C-loop (teal and red spheres). Transmembrane tether (TM); pale spheres, electrostatic residues at the interface.
While NADH binding and hydride transfer can ‘switch’ AIF from an oxidized to a CTC state in vitro, we propose that protein partners and small molecule binders can target AIF’s two molecular pathways to hinder or capitalize upon these transitions in a cellular context. Notably, opening the Cβ-clasp exposes a pocket targeted by electron acceptor substrates in conventional oxidoreductases. Crystal structures of dimeric AIF, however, have captured NAD+ occupying this site (Ferreira et al., 2014), suggesting that secondary NAD+ binding could block reassembly of the Cβ-clasp and prolong AIF-CTC lifetimes. Binding and import of mitochondrial import factor CHCHD4 relies on AIF-CTC and may enable AIF-CTC architectures via protein interaction (Hangen et al., 2015). In contrast, cell death regulator heat-shock protein 70 (Hsp70) (Schmitt et al., 2003) and E3-ubiquitin ligase X-linked inhibitor of apoptosis protein (XIAP) (Lewis et al., 2011) target residues near AIF’s Cβ-clasp (K192, K194) and dimerization interface (K255), respectively, suggesting a requirement for the closed, monomeric architecture of oxidized AIF. The recent discovery of added mitochondrial binding partners of AIF (Lenhausen et al., 2016; Nakao et al., 2015) and a PARP-1-dependent AIF-associated nuclease (Wang et al., 2016) opens further opportunities for finding how the mechanism for architectural switching defined here regulates AIF’s mitochondrial and cell death functions in vivo.
Recent biochemical characterization of disease-causing AIF mutants points to CTC persistence as critical for homeostatic AIF stability and function (Rinaldi et al., 2012; Sevrioukova, 2016; Sorrentino et al., 2015). While affected residues are distributed throughout AIF, many occur in locations linked to CTC formation, including the NADH binding site (G308E and G338E) (Berger et al., 2011; Diodato et al., 2016), dimerization interface (R422W, R422Q, R430C, A472V, P475L) (Zong et al., 2015), C-loop salt bridges (ΔR201) (Ghezzi et al., 2010), and the cavity shielded by the Cβ-clasp (E493V) (Rinaldi et al., 2012). Among disease-related mutants studied in vitro, efficiency of CTC formation varies (enhanced, reduced, or unchanged relative to wild-type). However, CTC lifetime is substantially reduced in all mutants compared to wild-type AIF (Rinaldi et al., 2012; Sevrioukova, 2016), indicating that long-lived CTC stability is critical for respiratory complex assembly and human health. A subset of these mutants (V243L, G262S, G338E), while stably expressed and purified in vitro, result in diminished protein levels in patient cells (Sevrioukova, 2016), suggesting that CTC formation may also sustain AIF stability in a cellular context. While AIF-CTC would seem to be the physiologically relevant form for mitochondrial homeostasis, detection of endogenous dimers in mitochondrial isolates has yielded conflicting results (Churbanova and Sevrioukova, 2008; Ferreira et al., 2014); further studies in live cells may better establish physiologic populations of AIF oligomers.
AIF’s monomer-dimer equilibrium may regulate AIF participation in cell death with monomer favored for mitochondrial release (Sevrioukova, 2009). Rapid cellular decline in NAD+, resulting from acute PARP-1 hyperactivation, could favor loss of AIF-CTC and reversion to monomeric AIF. The membrane tether of monomeric AIF may, in turn, present a more efficient calpain I substrate in the absence of steric hindrance from the nearby dimerization interface. Interaction between AIF and free chains of oligo(poly(ADP-ribose)) (PAR) generated by PAR glycohydrolase (PARG) activity on PARP-1 substrates may stimulate AIF release from mitochondria during parthanatos (Wang et al., 2011; Yu et al., 2006). We do not detect binding between purified PAR chains and AIF or AIF-CTC, but found evidence of C-loop destabilization in the reported release-resistant PAR-binding mutant (R589A/K590A/R593A), which could explain the protective phenotype seen in this AIF mutant during parthanatos (Wang et al., 2011).
NADH-driven allostery links AIF’s support of mitochondrial homeostasis to cellular metabolic health. Utilizing cytoplasmic NADH pools, AIF may modulate respiratory complex assembly during short-term changes in glycolytic flux, cellular redox status, and NAD+ synthesis/consumption (Verdin, 2015). AIF switching might also aid mitochondrial regulation during sustained changes in NAD+ levels in response to diet, aging, or chronic, disease-induced PARP-1 hyperactivation, complementing NAD+-driven transcriptional regulation of mitochondrial biogenesis (Fang et al., 2014; Verdin, 2015). Molecular knowledge of AIF allosteric switching provides a framework for understanding how AIF contributes to cell survival or demise during these events through interactions with protein partners and other small molecule regulators. The ability to decouple the two molecular pathways modulating AIF structure suggests a strategy for developing small molecule compounds to selectively activate or inhibit AIF allostery. Such a small molecule toolbox could prove invaluable for investigating the physiological impact of AIF structural transitions in situ and enable restoring function to human AIF mutations that are refractory to other forms of NAD+ supplementation or treatment.
EXPERIMENTAL PROCEDURES
AIF Construct Subcloning, Expression, and Purification
The AIFM1 plasmid was obtained from the DNASU Plasmid Repository (Cormier et al., 2010; Seiler et al., 2014). All AIF constructs were subcloned into the pET24b vector with C-terminal Prescission protease cleavage site and 6X histidine affinity tag. AIF point mutants were generated by Gibson assembly. Wild-type and mutant AIF constructs were expressed and purified as described in Supplemental Experimental Procedures.
AIF Biochemical Assays
Proteinase K limited proteolysis, BS3 cross-linking reactions, PEG-MEM labeling analysis, and NADH oxidation assays were performed as described in the Supplemental Experimental Procedures.
SAXS Data Collection and Analysis
AIF SAXS samples were prepared and analyzed as described in the Supplemental Experimental Procedures. SAXS data were collected at the ALS beamline 12.3.1 LBNL Berkeley, California (Classen et al., 2013). The X-ray wavelength was λ 1.03 Å, and the sample-to-detector distances were set to 1.5 m resulting in scattering vectors, q, ranging from 0.01 Å−1 to 0.33 Å−1. The scattering vector is defined as q = 4π sinθ/λ, where 2θ is the scattering angle. All experiments were performed at 20°C (Dyer et al., 2014). SAXS data were assessed for radiation-dependent aggregation and analyzed using the Atsas Primus module (Petoukhov et al., 2012) and Scatter (v. 2.3h).
Molecular envelope calculations were performed with DAMMIF (Franke and Svergun, 2009); ten DAMMIF runs were averaged in DAMAVER to produce the final envelope (Volkov and Svergun, 2003). SAXS envelopes for AIF crystal structures were calculated using theoretical scattering curves generated by the FoXS server (Schneidman-Duhovny et al., 2013). Missing C-loop or N-terminal residues were added to AIF crystal structures in Chimera (Pettersen et al., 2004), and models were passed through the BilboMD sampling server to identify C-loop or termini conformations of best-fit to the scattering data (Pelikan et al., 2009). Application of a minimal ensemble search (MES) (described in (Pelikan et al., 2009)) showed only moderate improvement in the fit to the data when two or more models were considered. Best-fitting AIF models selected by BilboMD were evaluated for -goodness-of-fit by FoXS and manually placed into SAXS envelopes with Chimera (Pettersen et al., 2004).
SAXS similarity matrices (SSM) were generated using the SAXS similarity webapp hosted by the ALS SIBYLS beamline (http://sibyls.als.lbl.gov/saxs_similarity/) (Hura et al., 2013). SAXS data from this publication are deposited at sibyls.als.lbl.gov/saxs_similarity/ and are being submitted to the BIOSIS database (http://bioisis.net).
AIF Crystallization and Structure Determination
AIF Monomer (E413A/R422A/R430A) and W916A were crystallized, and structures were solved as described in Supplemental Experimental Procedures. X-ray diffraction data were collected at ALS beamline 4.2.2 LBNL Berkeley, California to 2.0 Å (‘Monomer’) and 2.27 Å (W196A). Diffraction and refinement statistics are recorded in Table S1. Structures were solved by molecular replacement with Phaser (Mccoy et al., 2007) using PDB: 4BV6 (Monomer) and PDB: 4BUR (W196A) as search models. Models were built and corrected using COOT (Emsley et al., 2010) with refinement in Phenix (Adams et al., 2010). The AIF78 Monomer structure was refined to an Rfactor = 15.7% and an Rfree = 19.4%, and the AIF78 W196A structure was refined to an Rfactor = 20.2% and an Rfree = 24.0%. Structures have been deposited with the PDB under accession codes 5KVI (Monomer) and 5KVH (W196A). All structural figures were prepared using PyMOL (www.pymol.org).
Molecular Dynamics Simulations and Mutual Information Analysis
Molecular dynamics simulations were run with GROMACS 4.6.7. (Pronk et al., 2013; Van der Spoel et al., 2005) at 300 K using the AMBER ff03 force field (Duan et al., 2003) with the TIP3P explicit solvent model (Jorgensen et al., 1983). Mutual Information (MI) analysis of active site residue backbone and side chain dihedral angles (K177, F310, E314, E453, H454, S484) was conducted with the CARDS framework using in-house scripts (Bowman and Geissler, 2012). Full details are given in Supplemental Experimental Procedures.
Supplementary Material
Highlights.
Disruption of AIF’s C-loop causes dimerization in the absence of NADH ligand.
Active site residue H454 is coupled to AIF’s dimerization surface.
AIF dimerization is regulated by a hydrophobic border at the dimer interface.
The Cβ-clasp is a focal point for C-loop remodeling during CTC formation.
Acknowledgments
We thank the Berkeley Lab Advanced Light Source (ALS) and staff at beam lines 4.2.2 and 12.3.1 for aiding X-ray crystallographic and X-ray scattering data collection. This work was supported by National Institutes of Health (NIH) Structural Cell Biology of DNA Repair Machines P01 grant CA92584 (GLH, JAT, TE) and chair funds from Washington University School of Medicine to T.E. C.A.B. was supported by a NIH National Research Service Award (F32-GM109591). J.A.T. is supported by a Robert A. Welch Chemistry Chair, the Cancer Prevention and Research Institute of Texas, and the University of Texas System Science and Technology Acquisition and Retention. SAXS experiments at the ALS 12.3.1 SIBYLS beamline are supported by United States Department of Energy program Integrated Diffraction Analysis Technologies (IDAT).
Footnotes
AUTHOR CONTRIBUTIONS
C.A.B. and W.L. prepared reagents and conducted biochemical experiments. C.A.B. designed SAXS experiments, crystallized and determined structures, and analyzed data. C.H. performed simulations and trajectory analysis calculations. S.S. and G.B. carried out and advised on MI analysis of the simulations. K.B. and G.H. collected SAXS data and advised on SAXS analysis. J.N. collected and processed x-ray diffraction data. C.A.B., T.E., and J.A.T interpreted the data and wrote the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallographica Section D-Biological Crystallography. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alano CC, Garnier P, Ying W, Higashi Y, Kauppinen TM, Swanson RA. NAD+ depletion is necessary and sufficient for poly(ADP-ribose) polymerase-1-mediated neuronal death. J Neurosci. 2010;30:2967–2978. doi: 10.1523/JNEUROSCI.5552-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger I, Ben-Neriah Z, Dor-Wolman T, Shaag A, Saada A, Zenvirt S, Raas-Rothschild A, Nadjari M, Kaestner KH, Elpeleg O. Early prenatal ventriculomegaly due to an AIFM1 mutation identified by linkage analysis and whole exome sequencing. Mol Genet Metab. 2011;104:517–520. doi: 10.1016/j.ymgme.2011.09.020. [DOI] [PubMed] [Google Scholar]
- Bowman GR, Geissler PL. Equilibrium fluctuations of a single folded protein reveal a multitude of potential cryptic allosteric sites. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:11681–11686. doi: 10.1073/pnas.1209309109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churbanova IY, Sevrioukova IF. Redox-dependent changes in molecular properties of mitochondrial apoptosis-inducing factor. J Biol Chem. 2008;283:5622–5631. doi: 10.1074/jbc.M709147200. [DOI] [PubMed] [Google Scholar]
- Classen S, Hura GL, Holton JM, Rambo RP, Rodic I, McGuire PJ, Dyer K, Hammel M, Meigs G, Frankel KA, et al. Implementation and performance of SIBYLS: a dual endstation small-angle X-ray scattering and macromolecular crystallography beamline at the Advanced Light Source. Journal of Applied Crystallography. 2013;46:1–13. doi: 10.1107/S0021889812048698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cormier CY, Mohr SE, Zuo D, Hu Y, Rolfs A, Kramer J, Taycher E, Kelley F, Fiacco M, Turnbull G, et al. Protein Structure Initiative Material Repository: an open shared public resource of structural genomics plasmids for the biological community. Nucleic Acids Res. 2010;38:D743–749. doi: 10.1093/nar/gkp999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande RA, Williams GJ, Limbo O, Williams RS, Kuhnlein J, Lee JH, Classen S, Guenther G, Russell P, Tainer JA, et al. ATP-driven Rad50 conformations regulate DNA tethering, end resection, and ATM checkpoint signaling. Embo Journal. 2014;33:482–500. doi: 10.1002/embj.201386100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diodato D, Tasca G, Verrigni D, D’Amico A, Rizza T, Tozzi G, Martinelli D, Verardo M, Invernizzi F, Nasca A, et al. A novel AIFM1 mutation expands the phenotype to an infantile motor neuron disease. Eur J Hum Genet. 2016;24:463–466. doi: 10.1038/ejhg.2015.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan Y, Wu C, Chowdhury S, Lee MC, Xiong GM, Zhang W, Yang R, Cieplak P, Luo R, Lee T, et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. Journal of Computational Chemistry. 2003;24:1999–2012. doi: 10.1002/jcc.10349. [DOI] [PubMed] [Google Scholar]
- Dyer KN, Hammel M, Rambo RP, Tsutakawa SE, Rodic I, Classen S, Tainer JA, Hura GL. High-throughput SAXS for the characterization of biomolecules in solution: a practical approach. Methods Mol Biol. 2014;1091:245–258. doi: 10.1007/978-1-62703-691-7_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallographica Section D-Biological Crystallography. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang EF, Scheibye-Knudsen M, Brace LE, Kassahun H, SenGupta T, Nilsen H, Mitchell JR, Croteau DL, Bohr VA. Defective mitophagy in XPA via PARP-1 hyperactivation and NAD(+)/SIRT1 reduction. Cell. 2014;157:882–896. doi: 10.1016/j.cell.2014.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira P, Villanueva R, Martinez-Julvez M, Herguedas B, Marcuello C, Fernandez-Silva P, Cabon L, Hermoso JA, Lostao A, Susin SA, et al. Structural insights into the coenzyme mediated monomer-dimer transition of the pro-apoptotic apoptosis inducing factor. Biochemistry. 2014;53:4204–4215. doi: 10.1021/bi500343r. [DOI] [PubMed] [Google Scholar]
- Franke D, Svergun DI. DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. Journal of Applied Crystallography. 2009;42:342–346. doi: 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghezzi D, Sevrioukova I, Invernizzi F, Lamperti C, Mora M, D’Adamo P, Novara F, Zuffardi O, Uziel G, Zeviani M. Severe X-linked mitochondrial encephalomyopathy associated with a mutation in apoptosis-inducing factor. Am J Hum Genet. 2010;86:639–649. doi: 10.1016/j.ajhg.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hangen E, Feraud O, Lachkar S, Mou H, Doti N, Fimia GM, Lam NV, Zhu C, Godin I, Muller K, et al. Interaction between AIF and CHCHD4 Regulates Respiratory Chain Biogenesis. Mol Cell. 2015;58:1001–1014. doi: 10.1016/j.molcel.2015.04.020. [DOI] [PubMed] [Google Scholar]
- Hura GL, Budworth H, Dyer KN, Rambo RP, Hammel M, McMurray CT, Tainer JA. Comprehensive macromolecular conformations mapped by quantitative SAXS analyses. Nat Methods. 2013;10:453–454. doi: 10.1038/nmeth.2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of Simple Potential Functions for Simulating Liquid Water. Journal of Chemical Physics. 1983;79:926–935. [Google Scholar]
- Lenhausen AM, Wilkinson AS, Lewis EM, Dailey KM, Scott AJ, Khan S, Wilkinson JC. Apoptosis Inducing Factor Binding Protein PGAM5 Triggers Mitophagic Cell Death That Is Inhibited by the Ubiquitin Ligase Activity of X-Linked Inhibitor of Apoptosis. Biochemistry. 2016;55:3285–3302. doi: 10.1021/acs.biochem.6b00306. [DOI] [PubMed] [Google Scholar]
- Lewis EM, Wilkinson AS, Davis NY, Horita DA, Wilkinson JC. Nondegradative ubiquitination of apoptosis inducing factor (AIF) by X-linked inhibitor of apoptosis at a residue critical for AIF-mediated chromatin degradation. Biochemistry. 2011;50:11084–11096. doi: 10.1021/bi201483g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mccoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. Journal of Applied Crystallography. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer K, Buettner S, Ghezzi D, Zeviani M, Bano D, Nicotera P. Loss of apoptosis-inducing factor critically affects MIA40 function. Cell Death Dis. 2015;6:e1814. doi: 10.1038/cddis.2015.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moubarak RS, Yuste VJ, Artus C, Bouharrour A, Greer PA, Murcia JMD, Susin SA. Sequential activation of poly(ADP-ribose) polymerase 1, calpains, and bax is essential in apoptosis-inducing factor-mediated programmed necrosis. Molecular and Cellular Biology. 2007;27:4844–4862. doi: 10.1128/MCB.02141-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakao LS, Everley RA, Marino SM, Lo SM, de Souza LE, Gygi SP, Gladyshev VN. Mechanism-based proteomic screening identifies targets of thioredoxin-like proteins. J Biol Chem. 2015;290:5685–5695. doi: 10.1074/jbc.M114.597245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelikan M, Hura GL, Hammel M. Structure and flexibility within proteins as identified through small angle X-ray scattering. General Physiology and Biophysics. 2009;28:174–189. doi: 10.4149/gpb_2009_02_174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petoukhov MV, Franke D, Shkumatov AV, Tria G, Kikhney AG, Gajda M, Gorba C, Mertens HDT, Konarev PV, Svergun DI. New developments in the ATSAS program package for small-angle scattering data analysis. Journal of Applied Crystallography. 2012;45:342–350. doi: 10.1107/S0021889812007662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--a visualization system for exploratory research and analysis. Journal of Computational Chemistry. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Pospisilik JA, Knauf C, Joza N, Benit P, Orthofer M, Cani PD, Ebersberger I, Nakashima T, Sarao R, Neely G, et al. Targeted deletion of AIF decreases mitochondrial oxidative phosphorylation and protects from obesity and diabetes. Cell. 2007;131:476–491. doi: 10.1016/j.cell.2007.08.047. [DOI] [PubMed] [Google Scholar]
- Pronk S, Pall S, Schulz R, Larsson P, Bjelkmar P, Apostolov R, Shirts MR, Smith JC, Kasson PM, van der Spoel D, et al. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics. 2013;29:845–854. doi: 10.1093/bioinformatics/btt055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambo RP, Tainer JA. Characterizing Flexible and Intrinsically Unstructured Biological Macromolecules by SAS Using the Porod-Debye Law. Biopolymers. 2011;95:559–571. doi: 10.1002/bip.21638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambo RP, Tainer JA. Accurate assessment of mass, models and resolution by small-angle scattering. Nature. 2013;496:477–481. doi: 10.1038/nature12070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinaldi C, Grunseich C, Sevrioukova IF, Schindler A, Horkayne-Szakaly I, Lamperti C, Landoure G, Kennerson ML, Burnett BG, Bonnemann C, et al. Cowchock syndrome is associated with a mutation in apoptosis-inducing factor. Am J Hum Genet. 2012;91:1095–1102. doi: 10.1016/j.ajhg.2012.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt E, Parcellier A, Gurbuxani S, Cande C, Hammann A, Morales MC, Hunt CR, Dix DJ, Kroemer RT, Giordanetto F, et al. Chemosensitization by a non-apoptogenic heat shock protein 70-binding apoptosis-inducing factor mutant. Cancer Res. 2003;63:8233–8240. [PubMed] [Google Scholar]
- Schneidman-Duhovny D, Hammel M, Tainer JA, Sali A. Accurate SAXS profile computation and its assessment by contrast variation experiments. Biophysical Journal. 2013;105:962–974. doi: 10.1016/j.bpj.2013.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiler CY, Park JG, Sharma A, Hunter P, Surapaneni P, Sedillo C, Field J, Algar R, Price A, Steel J, et al. DNASU plasmid and PSI:Biology-Materials repositories: resources to accelerate biological research. Nucleic Acids Res. 2014;42:D1253–1260. doi: 10.1093/nar/gkt1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevrioukova IF. Redox-linked conformational dynamics in apoptosis-inducing factor. J Mol Biol. 2009;390:924–938. doi: 10.1016/j.jmb.2009.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevrioukova IF. Structure/Function Relations in AIFM1 Variants Associated with Neurodegenerative Disorders. J Mol Biol. 2016 doi: 10.1016/j.jmb.2016.05.004. [DOI] [PubMed] [Google Scholar]
- Sorrentino L, Calogero AM, Pandini V, Vanoni MA, Sevrioukova IF, Aliverti A. Key Role of the Adenylate Moiety and Integrity of the Adenylate-Binding Site for the NAD(+)/H Binding to Mitochondrial Apoptosis-Inducing Factor. Biochemistry. 2015;54:6996–7009. doi: 10.1021/acs.biochem.5b00898. [DOI] [PubMed] [Google Scholar]
- Vahsen N, Cande C, Briere JJ, Benit P, Joza N, Larochette N, Mastroberardino PG, Pequignot MO, Casares N, Lazar V, et al. AIF deficiency compromises oxidative phosphorylation. Embo Journal. 2004;23:4679–4689. doi: 10.1038/sj.emboj.7600461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJC. GROMACS: Fast, flexible, and free. Journal of Computational Chemistry. 2005;26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- Verdin E. NAD(+) in aging, metabolism, and neurodegeneration. Science. 2015;350:1208–1213. doi: 10.1126/science.aac4854. [DOI] [PubMed] [Google Scholar]
- Villanueva R, Ferreira P, Marcuello C, Uson A, Miramar MD, Peleato ML, Lostao A, Susin SA, Medina M. Key Residues Regulating the Reductase Activity of the Human Mitochondrial Apoptosis Inducing Factor. Biochemistry. 2015;54:5175–5184. doi: 10.1021/acs.biochem.5b00696. [DOI] [PubMed] [Google Scholar]
- Volkov VV, Svergun DI. Uniqueness of ab initio shape determination in small-angle scattering. Journal of Applied Crystallography. 2003;36:860–864. doi: 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, An R, Umanah GK, Park H, Nambiar K, Eacker SM, Kim B, Bao L, Harraz MM, Chang C, Chen R, Wang JE, Kam T-I, Jeong JS, Xie Z, Neifert S, Qian J, Andrabi SA, Blackshaw S, Zhu H, Song H, Ming G, Dawson VL, Dawson TM. A nuclease that mediates cell death induced by DNA damage and poly(ADP-ribose) polymerase-1. Science. 2016;354:82–95. doi: 10.1126/science.aad6872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YF, Dawson VL, Dawson TM. Poly(ADP-ribose) signals to mitochondrial AIF: A key event in parthanatos. Experimental Neurology. 2009;218:193–202. doi: 10.1016/j.expneurol.2009.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YF, Kim NS, Haince JF, Kang HC, David KK, Andrabi SA, Poirier GG, Dawson VL, Dawson TM. Poly(ADP-Ribose) (PAR) Binding to Apoptosis-Inducing Factor Is Critical for PAR Polymerase-1-Dependent Cell Death (Parthanatos) Science Signaling. 2011:4. doi: 10.1126/scisignal.2000902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams RS, Dodson GE, Limbo O, Yamada Y, Williams JS, Guenther G, Classen S, Glover JN, Iwasaki H, Russell P, et al. Nbs1 flexibly tethers Ctp1 and Mre11-Rad50 to coordinate DNA double-strand break processing and repair. Cell. 2009;139:87–99. doi: 10.1016/j.cell.2009.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu SW, Andrabi SA, Wang H, Kim NS, Poirier GG, Dawson TM, Dawson VL. Apoptosis-inducing factor mediates poly(ADP-ribose) (PAR) polymer-induced cell death. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:18314–18319. doi: 10.1073/pnas.0606528103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong L, Guan J, Ealy M, Zhang QJ, Wang DY, Wang HY, Zhao YL, Shen ZR, Campbell CA, Wang FC, et al. Mutations in apoptosis-inducing factor cause X-linked recessive auditory neuropathy spectrum disorder. Journal of Medical Genetics. 2015;52:523–531. doi: 10.1136/jmedgenet-2014-102961. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.