

Abstract
Background
Dexamethasone is reported to induce both tumor-suppressive and tumor-promoting effects. The purpose of this study was to identify the genomic impact of dexamethasone in glioblastoma stem cell (GSC) lines and its prognostic value; furthermore, to identify drugs that can counter these side effects of dexamethasone exposure.
Methods
We utilized three independent GSC lines with tumorigenic potential for this study. Whole-genome expression profiling and pathway analyses were done with dexamethasone-exposed and control cells. GSCs were also co-exposed to dexamethasone and temozolomide. Risk scores were calculated for most affected genes, and their associations with survival in TCGA and REMBRANDT databases. In silico connectivity Map analysis identified camptothecin as antagonist to dexamethasone induced negative effects.
Results
Pathway analyses predicted an activation of dexamethasone network (z-score:2.908). Top activated canonical pathways included ‘role of BRCA1 in DNA damage response’ (p=1.07E-04). GSCs were protected against temozolomide-induced apoptosis when co-incubated with dexamethasone. Altered cellular functions included cell-movement, cell-survival, and apoptosis with z-scores of 2.815, 5.137, and −3.122 respectively. CEBPB was activated in a dose dependent manner specifically in slow-dividing ‘stem-like’ cells. CEBPB was activated in dexamethasone-treated orthotopic tumors. Patients with high risk score had significantly shorter survival. Camptothecin was validated as potential partial neutralizer of dexamethasone effects.
Conclusions
Dexamethasone exposure induces a genetic program and CEBPB expression in GSCs that adversely affects key cellular functions and response to therapeutics. High risk scores associated with these genes have negative prognostic value. Our findings further suggest camptothecin as a potential neutralizer of adverse dexamethasone-mediated effects.
Keywords: Dexamethasone, Glioblastoma, Glioblastoma Stem Cells, TCGA, Kaplan Meier Analysis
Introduction
Glioblastoma is the most common malignant primary brain tumor in adults,1 with a median survival of 15.3–21.7 months.2 Neural, proneural, classical and mesenchymal subtypes have been described, whereof mesenchymal glioblastoma are described as the most aggressive.3 The transcription factor C/EBPbeta (CEBPB) is proposed to be a master regulator of mesenchymal programming in glioblastoma.4 Capillaries in glioblastoma do not evince the physiological blood-brain barrier and thus exhibit increased permeability leading to edema.5 Dexamethasone is the clinical standard for treating vasogenic edema–induced local mass effects and elevated intracranial pressure in glioblastoma.6
While dexamethasone is effective in managing edema, its molecular impact remains controversial: it has been reported to induce proliferation in glioblastoma cell lines,7 while other studies demonstrated that it has inhibitory effects on proliferation of human glioblastoma cell lines in vitro.8 A rat model study generated the proposal that dexamethasone reduces both the expression and the response of vascular endothelial growth factor (VEGF), thus reducing edema.9 Dexamethasone-mediated induction of apoptosis in human glioblastoma cell lines as well as its antitumor activity in xenografted nude rats have been described,10 and it has also been suggested that dexamethasone reduces invasiveness of human glioblastoma cells in vitro and blood vessel invasion in vivo through a MAPK phosphatase-1–dependent mechanism.11 Furthermore, glioma growth was reduced in mice following treatment with dexamethasone or anti-angiopoietin 2.12 Recently, however, Wong et al. showed that glioblastoma patients who received a dexamethasone dose >4.1 mg per day had significantly shorter overall survival (OS) than those who received ≤4.1 mg per day. The study attributed this finding to immunologic interference in treatment efficacy.13 Thus, the drug’s impact on biology is controversial.
Singh et al. strongly supported the key role of the cancer stem cell hypothesis14 in glioblastoma, whereby glioblastoma stem cells (GSCs) appear to retain multiple salient features of the original tumor and contribute to therapeutic resistance and tumor recurrence.15 Since the publication of that report, GSCs derived from human tumors have become a reliable tool for investigating glioblastoma biology in the laboratory. So far there are no data on the impact of dexamethasone exposure on GSCs. Therefore, we aimed to identify key genes and molecular mechanisms regulated by dexamethasone in patient-derived GSC lines. We further aimed to assess the prognostic significance of a risk score based on these altered genes in glioblastoma patients and to identify clinically relevant antagonists.
Materials and Methods
Ethics approvals
GSC lines were derived from MD Anderson Cancer Center patients who gave written informed consent in advance and in accordance with an MD Anderson institutional review board–approved protocol (LAB04-0001).
All animal use complied with institutional and governmental laws and regulations and was approved by the MD Anderson institutional animal care and use committee (protocol 00001100-RN00) in accordance with the guidelines of the American Association for Laboratory Animal Science.
Proliferation of patient-derived glioblastoma stem cells
To obtain a robust representation of GBM, three independent MD Anderson patient-derived GSC lines (GSC1, GSC3, and GSC6) were isolated and cultured from surgical specimens as described elsewhere.15, 16 In brief, cells were grown in GSC medium consisting of Dulbecco modified Eagle medium (DMEM)/F12 (Corning, Corning, NY) including L-glutamine (Sigma, St Louis, MO), 1× penicillin/streptomycin (Corning), 1× B27 without vitamin A (Life Technologies, Carlsbad, CA), 20 ng/mL basic fibroblast growth factor (VWR, Radnor, PA), and 20 ng/mL epidermal growth factor (EMD Millipore, Billerica, MA) at 37°C in presence of 5% CO2 as defined previously by Singh et al.15 Cells were passaged according to standard protocols with 3 minutes exposure to Accutase cell detachment solution (EMD Millipore) at 37°C.
Differentiation assays
GSCs were differentiated in tissue culture flasks and in chamber slides (Labtek, Scotts Valley, CA) and pretreated with poly-L-lysine (Sigma) overnight at 37°C. The three GSC lines were allowed to attach to poly-L-lysine (PLL) coated plates. After 12 hours, the medium was changed to differentiation medium containing DMEM, 1× penicillin/streptomycin, and 10% fetal bovine serum (Denville Scientific, Holliston, MA). The medium was exchanged every 48 hours. Cells differentiated in flasks were harvested on day 7 with 3 minutes exposure to 0.25% Trypsin solution (Corning), whereas differentiated cells in chamber slides were fixed with 4% paraformaldehyde for 10 minutes at room temperature before proceeding to immunofluorescence analysis.
Flow cytometry
GSC lines were cultured in GSC medium for proliferation assays. After Accutase treatment, single cells were fixed with 4% paraformaldehyde for 10 minutes at room temperature and were washed twice with phosphate-buffered saline solution. Cells were permeabilized with permeabilization buffer containing 1% bovine serum albumin, 0.1% Triton X-100, and 0.09% sodium azide for 15 minutes at room temperature. Permeabilized cells were then incubated with mouse anti-Sox2, a stem cell marker (R&D Systems, Minneapolis, MN); another stem cell marker, rabbit anti-nestin (Thermo Scientific Pierce, Waltham, MA); and cell proliferation marker rabbit anti-Ki67 (Thermo Scientific Pierce) in permeabilization buffer for 30 minutes at room temperature. After three washes with permeabilization buffer, species-specific APC-conjugated secondary antibodies anti-mouse (BD Biosciences, San Jose, CA) and anti-rabbit (Thermo Scientific Novex, Grand Island, NY) were used for indirect labeling according to the manufacturer’s protocol. Fluorescence-activated cell sorting (FACS) analysis was performed with a Gallios flow cytometer (Beckman Coulter, Brea, CA: Flow Cytometry and Cellular Imaging core facility, MD Anderson Cancer Center). The gates for sorting were based on isotype control samples for each GSC line. The FACS data were analyzed with FlowJo software (version 10).
Western blot analysis
Total proteins were extracted from the three independent GSC lines in the presence of protease and phosphatase inhibitors (Thermo Scientific Pierce) and analyzed with Western blotting for specific proteins. Briefly, each whole-cell extract (50–100 µg) was resolved on a 10–12.5% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE, Fisher BioReagents, Pittsburgh, PA) and transferred to a nitrocellulose membrane. The membranes were probed with mouse anti-Sox2 (R&D Systems), rabbit anti-nestin (Thermo Scientific Pierce), rabbit anti-Ki67 (Thermo Scientific Pierce), and mouse anti-GAPDH (Abcam, Cambridge, MA) primary antibodies overnight at 4°C. Rabbit anti-cleaved PARP (Cell Signaling), was used as apoptosis marker. The membranes were incubated with species-specific horseradish peroxidase–conjugated secondary antibodies for 1 hour at room temperature. The membranes were exposed to Clarity Western ECL Substrate (BioRad, Hercules, CA) and scanned with a c600 imaging system (Azure Biosystems, Dublin, CA). Scanned images were analyzed with cSeries Capture Software. Specific protein bands in western blots were quantified with ImageJ software.
Immunofluorescence analysis
Cells were differentiated for 7 days on PLL coated Lab-Tek II Chamber slides (50,000 cells per chamber), fixed as described above, and stained with mouse anti-GFAP (glial fibrillary acidic protein) antibody and goat anti-mouse rhodamine–conjugated secondary antibody (both, Thermo Scientific Pierce). Nuclei were counterstained with 4',6-diamidino-2-phenylindole. Images were taken with a DFC3000 G microscope (Leica, Buffalo Grove, IL) using a 20× objective and analyzed with LAS-X software.
Orthotopic tumor formation
9 (3×3) male nude mice (strain nu/nu, 5 weeks old; obtained from the Department of Veterinary Medicine and Surgery at MD Anderson Cancer Center) were implanted with a nylon bolt in an orthotopic position 2 mm posterior and 1.5 mm lateral to the bregma in the right cerebral hemisphere. The animals were anesthetized through this procedure, receiving ketamine intraperitoneally as described elsewhere.16 One week after bolting, each animal was injected with GSC1, GSC3, or GSC6 (1×105) in DMEM (5 µL) over 5 minutes through the bolt, again under anesthesia and with intraperitoneal ketamine.16 Mice were killed with hypercarbia and decapitation at the earliest onset of neurological symptoms and their brains were collected in formalin for paraffin embedding. Embedded tumor-bearing brains were cut into 5-µm slices and deparaffinized in xylene, rehydrated in an alcohol series (100–70%) and double distilled water, and stained with hematoxylin and eosin (H&E), all according to standard protocols. H&E-stained slides were imaged with an Olympus BX51 microscope (Olympus USA, Center Valley, PA) using a 10× objective, and the images analyzed with QCapture software.
Dexamethasone, temozolomide, and camptothecin treatment
A broad range of dexamethasone concentrations has been used for in vitro studies,17 and the concentration we used (50 µM) falls within this range.18 Single cell suspension of GSCs after 3 minutes exposure to Accutase cell detachment solution (EMD Millipore) were exposed for 6 days to GSC medium containing dexamethasone 50 µM (Sigma) or GSC medium alone as control. Medium was exchanged daily. To validate the in silico prediction in vitro, GSC 3 and 6 were exposed to 0, 25, 50 and 100 µM dexamethasone over three days under stress of concomitant treatment with 500 µM of the alkylating chemotherapeutic agent temozolomide (Sigma, St Louis, MO). Medium was exchanged daily. Total proteins were extracted and analyzed with Western blotting as described above. GSCs exposed to dexamethasone for five days were co-exposed to 10 µM camtothecin19 for 24 hours and cells were harvested for total RNA isolation followed by reverse transcriptase and quantitative PCR.
Label retention analysis
GSC1 and 3 were exposed to 5 µM of CellTrace™ CFSE Cell Proliferation substrate (Thermo Scientific Novex, Grand Island, NY) for 20 minutes at room temperature followed by removal of free dye from the solution as per the manufacturer’s protocol and treated with 50 µM dexamethasone (daily exchange) for 8–10 days. Cells underwent FACS sorting for high versus low CFSE dye retaining cells. Gates for FACS were set with negative and positive samples (CFSE stained on the day of sorting).
RNA isolation, whole-genome expression profiling, reverse transcriptase and real time PCR
RNA was isolated with Purezol RNA Isolation Reagent (BioRad) according to the manufacturer’s protocol. After quality control, RNA was analyzed with the Human Transcriptome 2.0 Microarray (Affymetrix, Santa Clara, CA) according to the producer’s standards (Microarray core facility, Department of Genetics, MD Anderson Cancer Center). Probe cell intensity data (.cel format) were uploaded and normalized across all three GSC lines, either treated with dexamethasone or untreated (controls), using the Expression Console software (Affymetrix) with gene level analysis. All gene confidence levels (core, extended, and full) were included in the analysis, as recommended by the manufacturer. Expression profiles were analyzed with the Transcriptome Analysis Console (Affymetrix), employing a cutoff of ±2.0 fold-change and a normalization z-score of ≥2.0 as described in a previous publication.20, 21 In brief, the z-score adjusts data within a single probe. Values for individual genes are expressed as the standard deviation from the normalized mean of zero.22
The selected genes were assessed with the Ingenuity Pathway Analysis platform (IPA; Qiagen, Valencia, CA) to predict dexamethasone-induced cellular functions, their networks of upstream regulators and canonical pathways. A heatmap of predicted upstream regulators and differentially expressed genes was generated with the Gene E program (http://www.broadinstitute.org/).
Approximately 100–200 ng of total RNA were used as template in reverse transcriptase reaction with iScript cDNA synthesis kit (BioRad, Hercules, CA) as per manufacturer’s protocol. The cDNAs were then amplified with SYBR® Green based polymerase chain reaction (PCR) (BioRad, Hercules, CA) using CFX real-time system. Data were analyzed with BioRad CFX manager and -2[ΔΔCt] values were plotted after normalization with GAPDH (housekeeping gene), p-value were calculated by 2 tailed TTEST between samples. Gene specific primers used were: GAPDH-Forward: 5’AAGGTGAAGGTCGGAGTCAA3’; Reverse: 5’AATGAAGGGGTCATTGATGG3’. CEBPB- Forward: 5’ CGTGTACACACGCGTTCAG3’; Reverse: 5’CTCTCTGCTTCTCCCTCTGC3’. Sox2- Forward: 5’ GCTTAGCCTCGTCGATGAAC3’; Reverse: 5’AACCCCAAGATGCACAACTC3’. FKBP5- Forward: 5’ GCGGAGAGTGACGGAGTC3’; Reverse: 5’TGGGGCTTTCTTCATTGTTC3’. MT1X- Forward: 5’ GCAAATGCAAAGAGTGCAAA3’; Reverse: 5’CTTTGCAGATGCAGCCCT3’. TIMP4- Forward: 5’ ACGCCTTTTGACTCTTCCCT3’; Reverse: 5’TTTCCATCACTGAGGACCTG3’.
In vivo model
After tumour forming capacity of GSC3 was confirmed, we injected 10 mice with 1 × 105 human GSC3 as described above. Upon the day of the orthotopic injection, 5 mice in each group were treated daily with 2 mg/kg (50 µg in 100 µl H2O) dexamethasone (Sigma Aldrich) intraperitoneally, as described in previous publications.23 The 5 control animals received 100 µl H2O intraperitoneally daily. Animals mice were killed with hypercarbia and decapitation at the earliest onset of neurological symptoms, in accordance with MD Anderson’s institutional animal research protocol. Some of the brains were immediately collected in formalin for paraffin embedding and later H&E and immunohistochemical staining. Others were placed in collection solution and then processed with a NeuroCult enzymatic dissociation kit (Stemcell Technologies, Vancouver, BC, Canada) to obtain orthotopic tumor cells for ex vivo expression analysis.
Histopathological staining
Paraffin-embedded brains from the dexamethasone-treated and control mice were cut into 5-µm slices, mounted on slides, deparaffinised in xylene, rehydrated in a descending alcohol series, and every 10th slide stained with H&E according to standard protocols. To assess CEBPB protein expression in the orthotopically implanted tumours, we stained tumour samples with a CEBPB specific antibody (Abcam, San Francisco, CA, USA), and incubated overnight at 4°C.
The slides were then incubated with polyclonal secondary antibodies (Abcam) and exposed to peroxidase (Abcam) and DAB substrate (Vector Laboratories, Burlingame, CA, USA) according to the manufacturers’ protocols. Counterstaining with haematoxylin was performed with standard procedures; finally, probes were pictured with an Olympus BX51 microscope (Olympus USA, Center Valley, PA, USA) and images were acquired using QCapture software.
Risk score calculation and clinical outcome analyses
Genes with ≥3.5 fold-change in expression upon dexamethasone exposure were analyzed for survival in the patient databases of The Cancer Genome Atlas (TCGA) and the Repository of Molecular brain neoplasia data (REMBRANDT). Small nucleolar RNAs were excluded, as they are not present on the U133A microarray platform. Cox regression coefficient of each gene was calculated by using a Cox hazard model. A risk score (RS) was calculated for each patient by using the following equation:
where βi and xi are the Cox regression coefficient and expression levels of the ith gene of the signature.24 Using median risk score as a cutoff, patients were dichotomized into high- and low-risk groups. Difference in OS for the two groups was analyzed by the Kaplan-Meier method with a two-sided log rank test. OS was defined as the time between the date of pathologic diagnosis and the date of death or the date of last clinical follow-up visit. To evaluate the effect of age and functional impairment on the prognostic effects of the gene signature, we calculated the age- and Karnofsky Performance Score (KPS)-adjusted hazard ratio in a multivariable Cox regression model in the TCGA cohort. A P-value ≤0.05 was considered statistically significant. Statistical analyses were done by using R (version 3.2.2) and SPSS (version 22) software. Expression z-scores of individual genes (the gene expressions have been z-scored for each sample over the entire gene platform panel)25 in specific molecular sub-groups of GBM i.e. mesenchymal and proneural were obtained from GBM-BioDP software (http://gbm-biodp.nci.nih.gov). For single gene clinical outcome, patients were dichotomized based on median expression z-score25 and KM curve analysis were performed using GraphPad Prism 6 software. P-value is calculation is based on Gehan-Breslow-Wilcoxon test.
Connectivity Map analysis
The genes identified as being regulated by dexamethasone in GSCs were then analyzed by the pattern-matching Connectivity Map platform to identify therapeutic agents that could potentially counteract the predicted dexamethasone induced oncogenic effects. The upregulated and downregulated genes (mRNA, fold-change ≤2.0 or ≥2.0 in microarray) in their rank order were uploaded to Connectivity Map (www.broadinstitute.org), where a collection of genome-wide expression profiles from human cell lines treated with bioactive agents is archived.26 The details of Connectivity Map analysis are described elsewhere.27, 28 In brief, the platform creates a “connectivity score” of collected and uploaded data where upregulation yields a positive score and downregulation a negative score (0 if no change). Respecting means of observed results, number of available cell lines, P-values, specificity, percentage of non-null results, and enrichment scores, the program ranks the results. The enrichment score displays the degree to which a set is overrepresented at the extremes within a ranked list.29
Results
Stem cell properties and orthotopic tumor formation are confirmed in human glioblastoma-derived cells
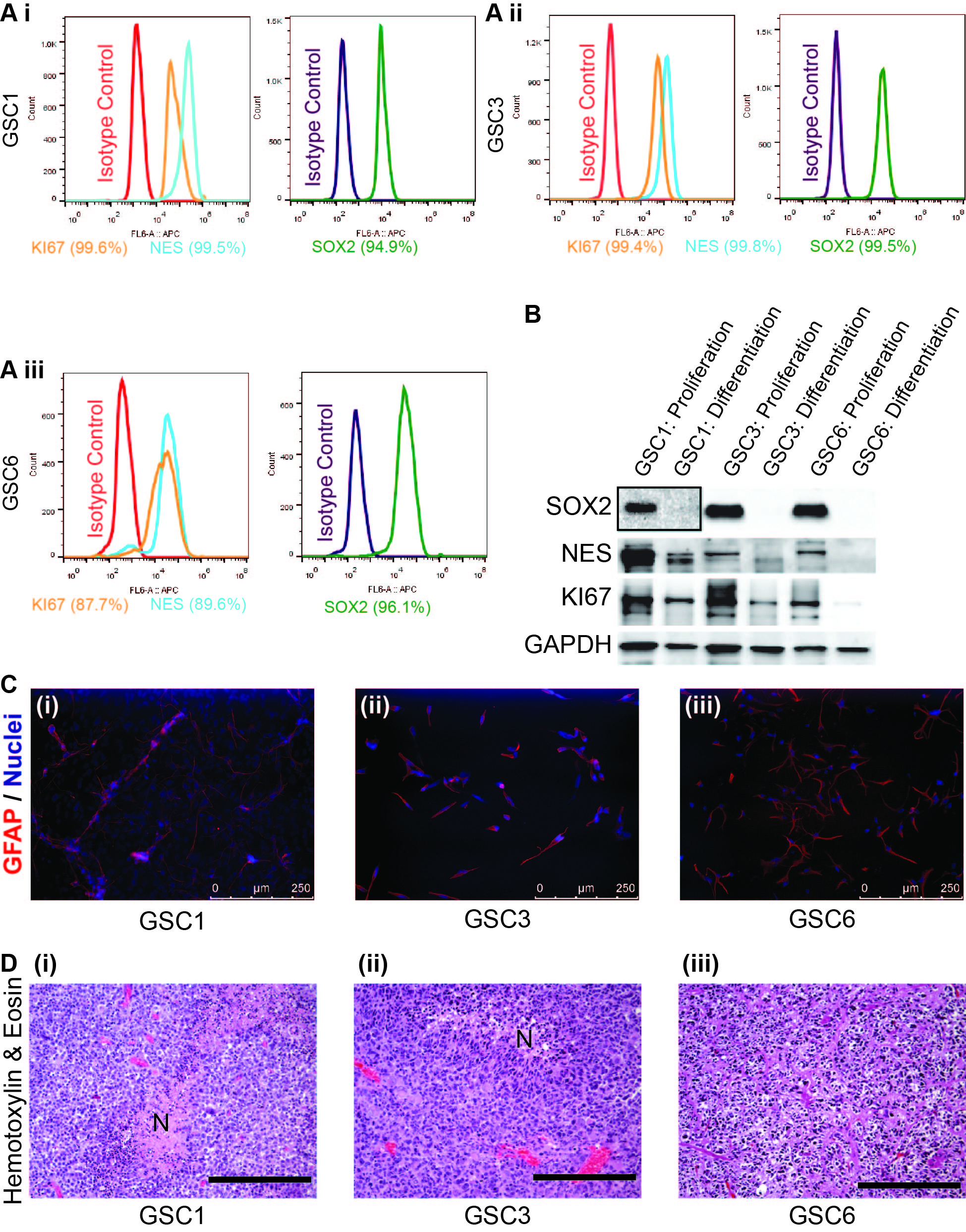
First, we sought to establish that our patient-derived cell lines GSC1, GSC3, and GSC6 had stem cell features and the potential to differentiate. FACS results show that a very high percentage of cells from all three GSC lines expressed the stem cell markers Sox2 (sex determining region Y-box 2”) and nestin (NES) and the proliferation marker Ki-67 (KI67) when cultured in GSC medium (Supplemental Figure 1A i–iii). We then exposed GSCs maintained in GSC medium to differentiation conditions. Western blot results show significantly lower protein levels of stem cell and proliferation markers under differentiation conditions than under proliferation conditions (Supplemental Figure 1B). Immunofluorescence analysis using antibody against differentiation marker GFAP show expression of the marker in all three GSC lines cultured in differentiation medium (Supplemental Figure 1C i–iii). All three GSC lines, when implanted at orthotopic locations in mouse brains, gave rise to xenograft tumors which bore hallmarks of glioblastoma, such as high cellularity, areas of necrosis, and hypervascularization (Supplemental Figure 1D i–iii).
Genes associated with cell proliferation and movement are most significantly affected by dexamethasone exposure in GSCs
Having confirmed the identity and tumorigenic potential of these GSC lines, we proceeded to assess the global impact of dexamethasone at the molecular level by microarray profiling of dexamethasone-treated GSCs (n=3) and their respective controls (n=3). This analysis of the expression profile data identified 333 altered genes, 290 upregulated and 43 downregulated (Affymetrix Human Transcriptome Array 2.0 Probeset IDs of dexamethasone-regulated genes in GSC1, GSC3, and GSC6 with cutoff >2.0 fold-change in Supplemental Table S1). Pathway analysis of the altered genes in our dataset predicted that the dexamethasone-regulated network was activated (activation z-score 2.908, P = 4.23E-08; Figures 1A and B), providing an unbiased validation of our expression profile data. Furthermore, genes associated with this dexamethasone network alone had significant activating impact on cellular functions such as proliferation (based on 30 genes; P = 6.14E-11), cell movement (based on 20 genes; P = 3.35E-09), and inhibition of cell death (based on 29 genes; P = 3.03E-1; Figure 1B).
Figure 1.
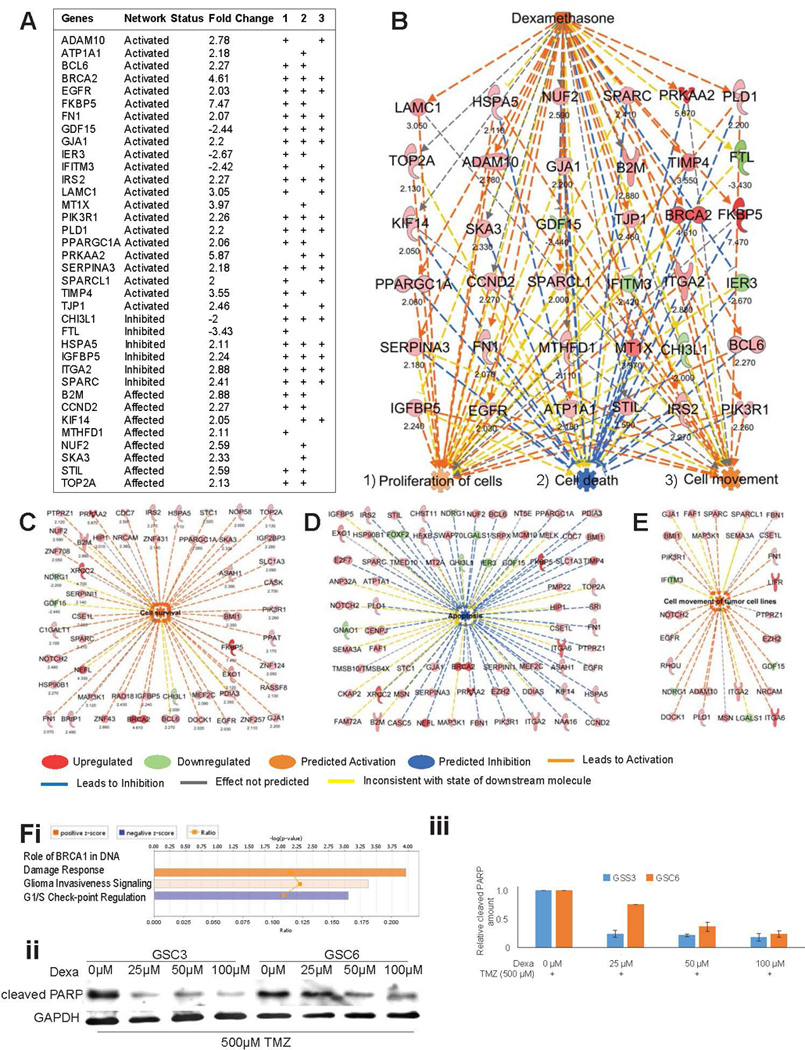
Apoptosis is inhibited and survival is activated upon dexamethasone exposure. (A) Fold-changes of gene expression in the dexamethasone network predicted its activation status. Their respective associations (+) with the cellular functions (1) proliferation, (2) cell death, and (3) cell movement (3) are shown. (B) This schematic representation shows the dexamethasone network, the genes involved in its activation, and its predicted impact on cellular functions. Numbers under the gene names indicate the fold-change in expression. (C–E) Individual genes contributing to the activated state of cell survival (C), the inhibited state of apoptosis (D), and the activated state of cell movement (E) in GSCs in response to dexamethasone are represented schematically. Numbers under the gene names indicate the fold-change in expression. Genes and cellular functions are labeled and color coded. A color index is shown at the bottom. (F) Network analysis for canonical pathway affected by dexamethasone shows “Role of BRCA1 in DNA Damage Response” with z-score 1.633 as top result (i). Scans of Western blots of whole-cell extracts of GSC3 and GSC6 treated with different concentrations of dexamethasone over three days under DNA damage stress of concomitant treatment with 500 µM Temozolomide and probed with an antibody against cleaved PARP as marker of apoptosis (GAPDH is loading control) (ii). Quantification of relative cleaved PARP amounts in western blots (shown in ii) normalized with GAPDH (iii).
Network analysis of the entire dataset of dexamethasone-induced genes showed significant activation of cell survival (z-score 5.137, P = 1.12E-08; Figure 1C), decreased apoptosis (z-score −3.122, P = 1.31E-06; Figure 1D), and increased movement (z-score 2.815, P = 1.61E-04; Figure 1E). Top affected canonical pathway was “Role of BRCA1 DNA Damage Response” (z-score 1.633, P = 1.07E-04). The in vitro validation of in silico prediction showed a decrease in cleaved PAPR with increasing concentrations of dexamethasone indicating a protective effect against DNA damage upon concomitant treatment with 500 µM Temozolomide in GSC3 and 6 (Figures 1F).
As an additional filter to predict cellular functions most affected by dexamethasone exposure, we collated a list of regulators (z-score >±2.0) whose functional status was determined by downstream gene levels in our expression data in dexamethasone-treated vs control cells (Figure 2A, Supplemental Tables S1 and S2). The cumulative effect of these upstream regulators and their respective downstream genes were then used to predict cellular functions by employing an activation z-score of ±3.0 or more as cutoff. In this analysis, hepatocyte growth factor (HGF; activation z-score 3.572, P = 1.22E-04; Figure 2B), VEGF (activation z-score 3.335, P = 8.07E-07) and VEGFA (activation z-score 3.13, P = 6.82E-06; Figure 2C), and estrogen receptor 1 (ESR1; activation z-score 3.187, P = 3.39E-05; Figure 2D) were the top activated regulatory networks, and nuclear protein transcriptional regulator1 (NUPR1; activation z-score 3.13, P = 6.82E-06; Figure 2E) was the top inhibited network. All activated upstream regulators, together with their downstream target genes, were predicted to induce activation of cell movement and survival (Figures 2B–D). The top inhibited upstream regulator (NUPR1) and its downstream targets (Figure 2E), also predicted to induce activation of cell movement and survival. Interestingly CEBPB was predicted to be upregulated by dexamethasone exposure (activation z-score 1.69, P = 7.33E-043; Supplemental Figure 2).
Figure 2.
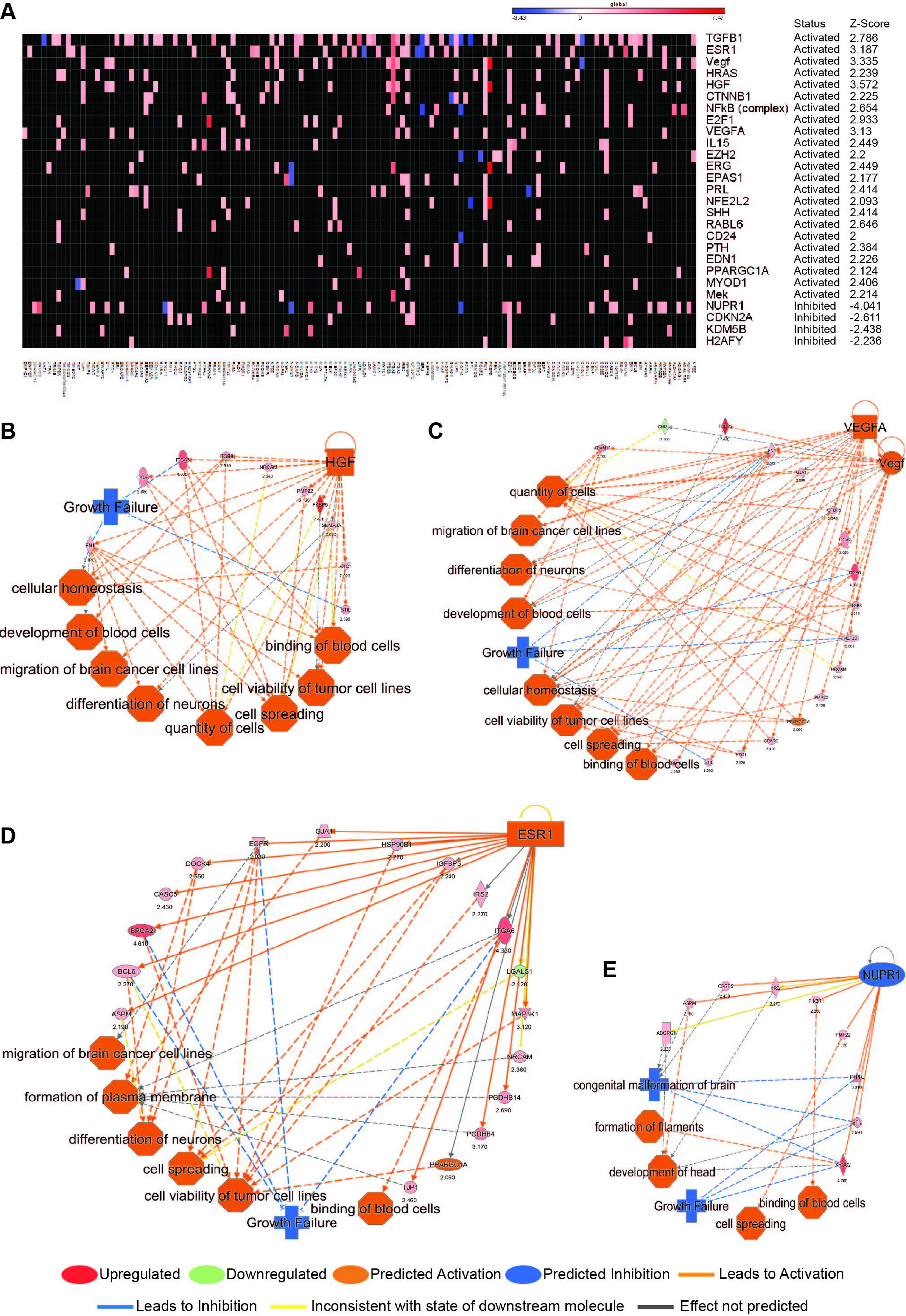
Oncogenic networks are activated by dexamethasone. (A) Heatmap shows genes and their association with upstream regulators with activation status z-scores ≥2.0. The color intensity bar above the heatmap represents the range of fold-change of gene expression: red as upregulation and blue as downregulation. (B) Schematic of the HGF network (activation z-score 3.572) with the genes associated with this top activated upstream regulator and their biologic implications. (C) Schematic of the VEGF (activation z-score 3.335) and VEGFA (activation z-score 3.31) networks with the genes associated with this top activated upstream regulator and their biologic implications. (D) Schematic of the ESR1 network (activation z-score 3.187) with the genes associated with this top activated upstream regulator and their biologic implications. (E) Schematic of the NUPR1 network (activation z-score −4.041) with the genes associated with this top activated upstream regulator and their biologic implications. Genes and cellular functions are labeled and color coded. A color index is shown at the bottom.
CEBPB, a master regulator of mesenchymal programming in glioblastoma is activated by dexamethasone
To validate the predicted upregulation of CEBPB upon exposure to dexamethasone, we exposed GSC1 and GSC3 cells to varying doses of dexamethasone for 6 days. Our results from quantitative real time PCR show a dose dependent increase in CEBPB levels with R2 values of 0.96 and 0.91 in GSC1 and GSC3 respectively (Figures 3A and 3B). To further elucidate impact of dexamethasone on CEBPB levels in specific cell populations within GSC cultures, namely CFSE high (slow dividing-‘stem-like’ cells, express high levels of Sox2 (data not shown)) and label low (fast dividing ‘non-stem-like’ cells), GSC1 and GSC3 cells were labeled with CFSE dye and treated with 50 µM dexamethasone over 8–10 days and sorted for low (fast dividing) and high (slow dividing) subpopulations (Figure 3C). Real time PCR data show endogenous levels of CEBPB is higher in CFSE high cells, a trend which was maintained upon dexamethasone exposure (Figures 3D and E). CEBPB expression levels in ex vivo isolated tumor cells showed a significant upregulation upon treatment of mice with 2 mg/kg dexamethasone over 4 weeks (Figure 3F). IHC staining against CEBPB protein confirmed this increase in tumor bearing sections of dexamethasone treated mice brains (Figure 3G).
Figure 3.

The transcription factor C/EBPbeta (CEBPB), a master regulator of the most malign subtype of glioblastoma is activated upon dexamethasone exposure in vitro, ex vivo and in vivo and dexamethasone treated thus CEBPB expressing cells slower division rate as indicator for cell “stemness”. (A) Relative expression levels of CEBPB in GSC1 upon exposure to 50, 100 and 200 µM dexamethasone for 6 days. (B) Relative expression levels of CEBPB in GSC3 upon exposure to 50, 100 and 200 µM dexamethasone for 6 days. (C) Schematic of the experiment with CFSE labeling to determine cell division rates as a readout of stemness properties of GSCs upon 8–10days exposure to 50 µM dexamethasone. (D) CFSE labeled GSC 1 cells treated with 50 µM dexamethasone over 8–10 days selected for fast and slow dividing subpopulations show a significant upregulation of CEBPB in slow dividing cells upon exposure to 50 µM dexamethasone over 8–10 days. (E) CFSE labeled GSC1 cells treated with 50 µM dexamethasone over 8–10 days selected for fast and slow dividing subpopulations show a significant upregulation of CEBPB in slow dividing cells upon exposure to 50 µM dexamethasone over 8–10 days. (F) Expression level analysis of GSC3 ex vivo derived orthotopic tumor cells shows a significant increase of CEBPB expression levels upon exposure of tumor bearing mice to 2 mg/kg dexamethasone daily for 4 weeks. (G) IHC staining with specific antibodies against CEBPB confirms the increase of CEBPB expression upon exposure of tumor bearing mice to 2 mg/kg dexamethasone. Brown nuclei are positive for CEBPB. P-values are displayed above the compared samples in each graph.
Patients with high risk scores for dexamethasone-induced genes have poorer prognosis and dexamethasone-induced upregulation of genes included in the score confirmed
Applying a most stringent cutoff of >3.5 fold-change identified 15 genes upregulated by dexamethasone in GSCs (Table 1). To assess the clinical translatability of these genes, we calculated the risk scores for each gene as described in Methods. These scores were then applied to discovery (TCGA) and validation (REMBRANDT) datasets to identify each of these glioblastoma patients as either high or low risk. Strikingly, Kaplan-Meier analyses showed a significant survival difference between high- and low-risk groups (Figure 4). Median survival durations for patients with high vs low risk score were 12.8 vs. 16.7 months in the TCGA cohort (N=515, P <0.001) and 13.1 months vs. 17.0 months in the REMBRANDT cohort (N=178, P = 0.015; Figures 4A and B). The multivariate Cox regression analysis of the TCGA cohort (hazard ratio 1.92, 95% CI 1.26 – 2.93; P = 0.002) showed that our risk score is a valid predictor of survival independent of age (hazard ratio 1.02, 95% CI 1.01–1.03: P <0.001) and KPS score (hazard ratio 0.98, 95% CI 0.97–0.99: P <0.001). The dexamethasone dependence of FKBP5, MT1X and TIMP4, three representative genes predicted in the two databases, are validated by real time PCR in vitro in GSC1 (Figure 4C) and GSC3 (Figure 4D).
Table 1.
Genes with > 3.5 fold-change in TCGA and REMBRANDT GBM patient cohorts
| Gene | Fold-change | Beta-coefficient score TCGA | Beta-coefficient score REMBRANDT |
|---|---|---|---|
| FKBP5 | 7.47 | 0.097 | 0.038 |
| PRKAA2 | 5.87 | −0.086 | −0.395 |
| PCDHB8 | 5.86 | 0.001 | 0.247 |
| ABCA5 | 4.84 | 0.218 | 0.188 |
| XRCC2 | 4.70 | 0.185 | −0.043 |
| BRCA2 | 4.61 | 0.056 | 0.044 |
| LIFR | 4.57 | −0.167 | −0.198 |
| LRRTM2 | 4.44 | 0.137 | 0.285 |
| ITGA6 | 4.33 | 0.098 | 0.208 |
| NEFL | 4.33 | 0.021 | −0.085 |
| MT1X | 3.97 | 0.041 | 0.311 |
| PCDHB12 | 3.61 | 0.03 | 0.066 |
| ALDH7A1 | 3.59 | −0.056 | 0.1 |
| TIMP4 | 3.55 | −0.007 | −0.129 |
| CHD7 | 3.52 | −0.204 | −0.13 |
Figure 4.

Glioblastoma patients with high risk score for dexamethasone-induced genes have poorer prognosis. (A) Kaplan-Meier curve analysis of the TCGA glioblastoma cohort (n=515). Median survival of glioblastoma patients with a high risk score (red) was 12.8 months, whereas that of patients with a low risk score (green) was 16.7 months (P <0.001). (B) Kaplan-Meier curve analysis of the REMBRANDT glioblastoma cohort (n=178). Median survival of glioblastoma patients with a high risk score (red) was 13.1 months, whereas that of patients with a low risk score (green) was 17.0 months (P = 0.015). (C and D) Real time PCR for FKBP5, Mt1X, and TIMP4, members of dexamethasone induced gene signature, in GSC1 and GSC3 with and without dexamethasone exposure. P-values are displayed above the compared samples in each graph.
Identification and validation of camptothecin as neutralizer of dexamethasone-induced genes in vitro
The dexamethasone-regulated genes with expression fold-change ≤−2.0 or ≥2.0 were assessed with Connectivity Map, an archive of genome-wide expression profiles from human cell lines treated with bioactive agents; this archive can be used to identify interactions between these agents and genes. This analysis was undertaken to identify therapeutics that could potentially counteract adverse gene expression changes induced by dexamethasone. We identified therapeutics with enrichment scores up to −0.992; the top-ranked compound was camptothecin (Figure 5A, Supplemental Table S3). The enrichment score reflects the degree to which a set is overrepresented at the extremes within a ranked list. To test potential antagonistic properties of camptothecin to dexamethasone induce alterations, we exposed GSC1 and GSC3 to camptothecin for 24 hours (a time point at which we did not detect significant amount of cell death. Supplemental Figure 3) after these cells were pre-exposed to dexamethasone for 5 days. Real time PCR data show a significant decrease in FKBP5 and MT1X levels in cells co-exposed to camptothecin and dexamethasone as compared to cells treated with dexamethasone alone (Figures 5B and 5C). Further analysis of TCGA expression data show that FKBP5 and MT1X genes are highly expressed in the mesenchymal subgroup (Figures 5 D and F). When the proneural subgroup of patients were further sub-classified into high and low groups, the high group of patients has significantly poorer clinical outcome (Figures 5 E and G).
Figure 5.

Identification and validation of camptothecin as neutralizer of dexamethasone induced effects in GSC. (A) A list of top ten therapeutics identified by connectivity map analysis based on dexamethasone induced genomic profiles. The ranking was automatically calculated, respecting mean observed values, number of cell lines available in the database, enrichment score, P-value, specificity, and percentage of non-null results. (B and C) In vitro exposure of GSCs with 10 µM Camptothecin and 50 µM dexamethasone (alone and in combinations), relative expression levels of FKBP5 and MT1X, two representative genes of the dexamethasone-induced oncogenic gene network in GSC1 and 3. P-values are displayed above the compared samples in each graph. (D and F) Expression of FKBP5 and MT1X in mesenchymal and proneural sub-groups of GBM patients in TCGA database. (E and G) Kaplan-Meier curve analysis of the proneural TCGA glioblastoma cohort (n=113). (E) Median survival of proneural glioblastoma patients with a high FKBP5 expression is 7.33 months, whereas that of patients with a low FKBP5 expression is 16.75 months (P = 0.0065). (G) Median survival of proneural glioblastoma patients with a high MT1X expression is 6.93 months, whereas that of patients with a low MT1X expression is 13.47 months (P = 0.045).
Discussion
This preclinical study demonstrated that dexamethasone altered genes predicted to promote oncogenic pathways in GSCs. High risk scores associated with genes most responsive to dexamethasone exposure were then found to be prognostic for poor outcome in two independent clinico-genomic glioblastoma cohorts. This is potentially of clinical importance, since dexamethasone is widely used in glioblastoma patients to treat edema and associated increased intracranial pressure during the perioperative period. Further, analysis of altered genes with Connectivity Map identified United States Food and Drug Administration (FDA)-approved drugs that can counter these adverse dexamethasone-induced effects while potentially preserving the beneficial anti-edema effects.
Upstream regulatory networks identified by this work are well known in the pathogenesis of glioblastoma. HGF has been shown to be involved in epithelial-to-mesenchymal transition through binding to the receptor tyrosine kinase c-MET, not only in embryogenesis but also in invasiveness of glioblastoma, because of its potent induction of mesenchymal transition in epithelium-driven cells.30 A recent clinical trial (phase II) investigated the antitumor activity of an anti-HGF antibody: though no significant advantage could be shown in patients with recurrent glioblastoma31, HGF’s contribution to glioblastoma malignancy is unquestioned. As a response to tissue hypoxia, the hypoxia-inducible factor 1 (HIF-1) acts as a transcription factor, controlling the expression of VEGF.32, 33 Both VEGF and HIF-1α have been shown to significantly promote both angiogenesis and invasion in glioblastoma.34 VEGFA, one of five VEGF subtypes, has been shown to be a prognostic indicator for brain tumor patients.35 Given that vascular proliferation is a key component of glioblastoma biology and that VEGF expression levels directly correlate with tumor malignancy and hence inversely with prognosis, the anti-VEGF antibody bevacizumab has been approved by the FDA for treatment of recurrent glioblastoma.36, 37 The role of estrogen receptors in glioblastoma remains controversial: some evidence shows that estrogen receptors are decreased in high-grade gliomas, while estrogen receptor expression levels correlated negatively with tumor differentiation.38 The estrogen receptor antagonist tamoxifen was shown to induce cell death in glioma cells39 and has been proposed as a potential chemotherapeutic agent in temozolomide-resistant glioblastoma.40 Our results provide a potential molecular basis for this indication of tamoxifen, especially given that most patients with recurrent glioblastoma are further exposed to dexamethasone. NUPR1, the most inhibited dexamethasone-dependent upstream regulator we found, is involved in a variety of stress-related functions in non-cancer conditions41 and as a tumor suppressor in prostate cancer.42 Our discovery of its potential involvement in glioblastoma is novel. The dose dependent increase in CEBPB (both in vitro and in vivo) upon dexamethasone exposure, a known regulator of mesenchymal program in GBM, is indicative of a potential mesenchymal shift in tumor cells (specifically in stem-like cells), which may contribute to poorer outcome of patients.
In our screen, the cellular functions most affected by dexamethasone were activation of cellular survival and migration as well as inhibition of cellular apoptosis, all functions well known to drive glioblastoma pathogenesis.43, 44 DNA repair associated genes such as the tumor suppressor Breast Cancer 1 (BRCA1) are known to be increased upon treatment with temozolomide.45 Interestingly, the activation of BRCA1 DNA damage response was the top affected canonical pathway in our screen. The functional readout of dexamethasone and temozolomide co-treatment, however, showed a decrease of apoptosis with increasing concentrations of dexamethasone. The upstream regulatory networks contributing to these changes are known to contribute to glioblastoma malignancy. These preclinical findings are of importance since they offer a possible explanation for recent findings by Wong et al. that glioblastoma patients who received higher dexamethasone doses had significantly shorter OS than those who received lower doses.13 Kaplan-Meier analysis of the risk scores associated with the most dexamethasone-responsive genes in our work supports a potentially dexamethasone-dependent significant survival difference in the two most exhaustive clinical databases, TCGA and REMBRANDT. That this prognostic association remains significant after multiple corrections indicates the robustness of our screen and the possible distinct biologic mechanism induced by dexamethasone that mediates a survival disadvantage, given that the vast majority of patients might have been under dexamethasone treatment when samples for the two databases were obtained. However, it appears likely that patients to whom larger doses of dexamethasone were administered had larger tumor and edema burden.
It remains unclear whether the described adverse effects of dexamethasone are due to its glucocorticoid or drug specific effect. Comparison with another corticosteroid would shed light on what is leading to the changes in gene transcription. However, the fact that cerebral edema is currently mainly treated with dexamethasone, an ethical dilemma might occur to test other steroids’ effects. Dexamethasone is essential not only for acute treatment of edema in glioblastoma but also as a long-term adjuvant for chemotherapy and radiation therapy.46 Identification of the FDA-approved camptothecin by our enrichment analysis suggests a potential route for future investigations to identify the drug’s potential to reduce unwanted dexamethasone effects. Our in vitro results suggest that, as predicted in silico, short term exposure of camptothecin is able to counter dexamethasone effects at least partially. However, the presented evidence that dexamethasone increases tumor cell survival is based on gene analysis, cell survival and proliferation in vitro. To provide strong evidence that dexamethasone increases tumor cell survival, both, the size of orthotopic tumors and orthotopic tumor bearing animals’ survival upon dexamethasone treatment remain to be evaluated. In brief, these findings resulting from an in vitro screen warrant further investigation in an in vivo model to uncover a definitive mechanism.
In summary, our GSC data shine light on the intriguing controversy of dexamethasone-induced effects in cancer biology and provide novel insight into the potential molecular mechanisms of these effects. We demonstrate that dexamethasone induces a gene signature that is a significant predictor of poor prognosis in glioblastoma patients. It is conceivable that neutralizing dexamethasone-induced oncogenic effects e.g. with camptothecin, may become an integral part of glioblastoma therapy. The data presented here may represent a stepping stone to improving glioblastoma treatment and prolonging patient survival.
Supplementary Material
Glioblastoma stem cells harbor characteristics of cancer stem cells. (A) Histograms show FACS data for GSC1 (i), GSC3 (ii), and GSC6 (iii) labeled with antibodies against the stemness markers SOX2 and NES and the proliferation marker KI67. Percentages of positive cells for specific markers are shown under the plots in brackets. Isotype control peaks are shown as secondary antibody control. (B) Scans of Western blots of whole-cell extracts of GSC1, GSC3, and GSC6 that were cultured under proliferative or differentiation conditions and probed with antibodies against SOX2, NES, and KI67. GAPDH is loading control. (C) Micrographs of immunofluorescence staining for differentiation marker GFAP (red) in GSC1 (i), GSC3 (ii), and GSC6 (iii) after differentiation. Nuclei are shown in blue. Scale bar = 250 µm. (D) Micrographs of H & E–stained mouse brains bearing orthotopic xenograft tumors arising from implantation of GSC1 (i), GSC3 (ii), or GSC6 (iii). Histologic examination of the tumors revealed high cellularity (purple), hypervascularity (endothelial proliferation: dark red), and necrosis (N). Scale bar = 200 µm.
{kind=link}
The transcription factor C/EBPbeta (CEBPB), a master regulator of mesenchymal transformation in glioblastoma is activated upon dexamethasone exposure (activation z-score 1.69, P = 7.33E-043). The schematic representation shows a network analysis for CEBPB as upstream regulator affected by dexamethasone upon in vitro exposure of 3 GSC lines with 50 µM dexamethasone for 6 days.
Viability percentages of camptothecin exposed GSCs for 24 and 48 hours. At 24-hour time point the difference in viability of GSC1 and GSC3 were insignificant (p=0.058), whereas that at 48 hour time point was significantly different (p=1.0314e-006).
Acknowledgments
Acknowledgements / funding
Alfred & Anneliese Sutter-Stoettner Foundation, Switzerland (MML): No role in design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
JS Dunn Sr Foundation (RRC): No role in design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Mach-Gaennslen Foundation, Switzeland (MML): No role in design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
NIH (FFL, EPS):No role in design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
University of Texas MD Anderson Cancer Center Startup Grant (RRC): No role in design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Funding: Mach-Gaensslen Foundation, Unteraegeri, Switzerland Alfred & Anneliese Sutter-Stoettner Foundation, Muenchwilen, Switzerland National Institue of Health (NIH), grant 1P50CA12CA127001, Bethesda, MD, USA John S. Dunn Sr. Distinguished Chair in Diagnostic Imaging, Houston TX, USA The University of Texas MD Anderson Cancer Center Start-up Fund, Houston TX, USA
List of abbreviations
- BRCA1
Breast cancer 1
- CEBPB
CCAAT/Enhancer Binding Protein
- DMEM
Dulbecco modified eagle medium
- ESR1
Estrogen receptor 1
- FACS
Fluorescence-activated cell sorting
- GAPDH
Glyceraldehyde-3-Phosphate Dehydrogenase
- GFAP
Glial fibrillary acidic protein
- GSC
Glioblastoma stem cell
- H & E
Hematoxylin and eosin
- HGF
Hepatocyte growth factor
- HIF-1
Hypoxia-inducible factor 1
- IACUC
Institutional animal care and use committee
- IPA
Ingenuity pathway analysis
- KPS
Karnofsky Performance Score
- NUPR1
Nuclear protein transcriptional regulator1
- OS
Overall survival
- PLL
Poly-L-lysine
- REMBRANDT
Repository of molecular brain neoplasia data
- RS
Risk score
- SOX2
SRY (Sex Determining Region Y)-Box 2
- TCGA
The cancer genome atlas
- VEGF
Vascular endothelial growth factor
- VEGFA
Vascular endothelial growth factor A
Footnotes
IRB human cells: Professor Colleen Gallagher, chief and executive director clinical ethics, The University of Texas MD Anderson Cancer Center, Houston TX, USA Phone: ++1 713 792 3204, CMGallagher@mdanderson.org …
IRB mouse work: Professor David Johnson, chair of the Institutional Animal Care and Use Committee (IACUC), The University of Texas MD Anderson Cancer Center, Houston TX, USA Phone: ++1 512 237 9511, djohnson@mdanderson.org
Ethics approvals and consent
Human cell lines: GSC lines were derived from MD Anderson Cancer Center patients who gave written informed consent in advance and in accordance with an MD Anderson institutional review board–approved protocol (LAB04-0001). Address: Professor Colleen Gallagher, chief and executive director clinical ethics, The University of Texas MD Anderson Cancer Center, Houston TX, USA Phone: ++1 713 792 3204, CMGallaghermdanderson.org
Animal work: All animal use complied with institutional and governmental laws and regulations and was approved by the MD Anderson institutional animal care and use committee (protocol 00001100-RN00) in accordance with the guidelines of the American Association for Laboratory Animal Science. Address: Professor David Johnson, chair of the Institutional Animal Care and Use Committee (IACUC), The University of Texas MD Anderson Cancer Center, Houston TX, USA Phone: ++1 512 237 9511, djohnsonmdanderson.org
Declarations
| Markus M. Luedi: | Reported no conflicts of interest |
| Sanjay K. Singh: | Reported no conflicts of interest |
| Jennifer C. Mosley | Reported no conflicts of interest |
| Masumeh Hatami: | Reported no conflicts of interest |
| Joy Gumin: | Reported no conflicts of interest |
| Erik P. Sulman: | Reported no conflicts of interest |
| Fred F. Land: | Reported no conflicts of interest |
| Frank Stueber: | Reported no conflicts of interest |
| Pascal O. Zinn: | Reported no conflicts of interest |
| Rivka R. Colen: | Reported no conflicts of interest |
| Markus M. Luedi: | Helped design the study, conduct the study, analyze the data, and write the manuscript. |
| Sanjay K. Singh: | Helped design the study, conduct the study, analyze the data, and write the manuscript. |
| Jennifer C. Mosley: | Helped design the study, conduct the study, analyze the data, and write the manuscript. |
| Masumeh Hatami: | Helped design the study, conduct the study, analyze the data, and write the manuscript. |
| Joy Gumin: | Helped design the study and conduct the study. |
| Erik P. Sulman: | Helped design the study and conduct the study. |
| Frederick F. Lang | Helped design the study and conduct the study. |
| Frank Stueber: | Helped design the study, analyze the data, and write the manuscript. |
| Pascal O. Zinn: | Helped design the study, conduct the study, analyze the data, and write the manuscript. |
| Rivka R. Colen | Helped design the study, conduct the study, analyze the data, and write the manuscript. |
Contributor Information
Dr. Markus M. Luedi, Email: mmluedi@mdanderson.org, Department of Anesthesiology, Bern University Hospital Inselspital, Bern, Switzerland and Department of Cancer Systems Imaging, The University of Texas MD Anderson Cancer Center, Houston TX, USA.
Dr. Sanjay K. Singh, Email: sksingh@mdanderson.org, Department of Diagnostic Imaging, The University of Texas MD Anderson Cancer Center, Houston TX, USA.
Jennifer C. Mosley, Email: JCMosley@mdanderson.org, Department of Cancer Systems Imaging, The University of Texas MD Anderson Cancer Center, Houston, TX, USA.
Dr. Masumeh Hatami, Email: mhatami@mdanderson.org, Department of Cancer Systems Imaging, The University of Texas MD Anderson Cancer Center, Houston, TX, USA.
Joy Gumin, Email: jgumin@mdanderson.org, Department of Neurosurgery and Brain Tumor Center, The University of Texas MD Anderson Cancer Center, Houston, TX, USA.
Prof. Erik P. Sulman, Email: epsulman@mdanderson.org, Department of Radiation Oncology, Division of Radiation Oncology, The University of Texas MD Anderson Cancer Center, Houston, TX, USA.
Prof. Frederick F. Lang, Email: flang@mdanderson.org, Department of Neurosurgery and Brain Tumor Center, The University of Texas MD Anderson Cancer Center, Houston, TX, USA.
Prof. Frank Stueber, Email: frank.stueber@mdanderson.org, Department of Anesthesiology, Bern University Hospital Inselspital, Bern, Switzerland.
Dr. Pascal O. Zinn, Email: zinn@bcm.edu, Department of Neurosurgery, Baylor College of Medicine and Departments of Neurosurgery, Cancer Systems Imaging, and Cancer Biology, The University of Texas MD Anderson Cancer Center, Houston TX, USA.
Prof. Rivka R. Colen, Email: rcolen@mdanderson.org, Departments of Cancer Systems Imaging, and Diagnostic Imaging, The University of Texas MD Anderson Cancer Center, Houston TX, USA.
References
- 1.Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med. 2008;359:492–507. doi: 10.1056/NEJMra0708126. [DOI] [PubMed] [Google Scholar]
- 2.Hegi ME, Diserens AC, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352:997–1003. doi: 10.1056/NEJMoa043331. [DOI] [PubMed] [Google Scholar]
- 3.Verhaak RG, Hoadley KA, Purdom E, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carro MS, Lim WK, Alvarez MJ, et al. The transcriptional network for mesenchymal transformation of brain tumours. Nature. 2010;463:318–325. doi: 10.1038/nature08712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Long DM. Capillary ultrastructure and the blood-brain barrier in human malignant brain tumors. J Neurosurg. 1970;32:127–144. doi: 10.3171/jns.1970.32.2.0127. [DOI] [PubMed] [Google Scholar]
- 6.McClelland S, 3rd, Long DM. Genesis of the use of corticosteroids in the treatment and prevention of brain edema. Neurosurgery. 2008;62:965–967. doi: 10.1227/01.neu.0000318183.25783.77. discussion 967-8. [DOI] [PubMed] [Google Scholar]
- 7.Zibera C, Gibelli N, Butti G, et al. Proliferative effect of dexamethasone on a human glioblastoma cell line (HU 197) is mediated by glucocorticoid receptors. Anticancer Res. 1992;12:1571–1574. [PubMed] [Google Scholar]
- 8.Kaup B, Schindler I, Knupfer H, et al. Time-dependent inhibition of glioblastoma cell proliferation by dexamethasone. J Neurooncol. 2001;51:105–110. doi: 10.1023/a:1010684921099. [DOI] [PubMed] [Google Scholar]
- 9.Heiss JD, Papavassiliou E, Merrill MJ, et al. Mechanism of dexamethasone suppression of brain tumor-associated vascular permeability in rats. Involvement of the glucocorticoid receptor and vascular permeability factor. J Clin Invest. 1996;98:1400–1408. doi: 10.1172/JCI118927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Giraud S, Bessette B, Boda C, et al. In vitro apoptotic induction of human glioblastoma cells by Fas ligand plus etoposide and in vivo antitumour activity of combined drugs in xenografted nude rats. Int J Oncol. 2007;30:273–281. [PubMed] [Google Scholar]
- 11.Lin YM, Jan HJ, Lee CC, et al. Dexamethasone reduced invasiveness of human malignant glioblastoma cells through a MAPK phosphatase-1 (MKP-1) dependent mechanism. Eur J Pharmacol. 2008;593:1–9. doi: 10.1016/j.ejphar.2008.06.111. [DOI] [PubMed] [Google Scholar]
- 12.Villeneuve J, Galarneau H, Beaudet MJ, et al. Reduced glioma growth following dexamethasone or anti-angiopoietin 2 treatment. Brain Pathol. 2008;18:401–414. doi: 10.1111/j.1750-3639.2008.00139.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wong ET, Lok E, Gautam S, et al. Dexamethasone exerts profound immunologic interference on treatment efficacy for recurrent glioblastoma. British journal of cancer. 2015;113:232–241. doi: 10.1038/bjc.2015.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reya T, Morrison SJ, Clarke MF, et al. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–111. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 15.Singh SK, Hawkins C, Clarke ID, et al. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 16.Hossain A, Gumin J, Gao F, et al. Mesenchymal Stem Cells Isolated From Human Gliomas Increase Proliferation and Maintain Stemness of Glioma Stem Cells Through the IL-6/gp130/STAT3 Pathway. Stem Cells. 2015;33:2400–2415. doi: 10.1002/stem.2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shannon S, Vaca C, Jia D, et al. Dexamethasone-Mediated Activation of Fibronectin Matrix Assembly Reduces Dispersal of Primary Human Glioblastoma Cells. PLoS One. 2015;10:e0135951. doi: 10.1371/journal.pone.0135951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Campana D, Manabe A, Evans WE. Stroma-supported immunocytometric assay (SIA): a novel method for testing the sensitivity of acute lymphoblastic leukemia cells to cytotoxic drugs. Leukemia. 1993;7:482–488. [PubMed] [Google Scholar]
- 19.Cheema TA, Kanai R, Kim GW, et al. Enhanced antitumor efficacy of low-dose Etoposide with oncolytic herpes simplex virus in human glioblastoma stem cell xenografts. Clin Cancer Res. 2011;17:7383–7393. doi: 10.1158/1078-0432.CCR-11-1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheadle C, Cho-Chung YS, Becker KG, et al. Application of z-score transformation to Affymetrix data. Appl Bioinformatics. 2003;2:209–217. [PubMed] [Google Scholar]
- 21.Fu J, Khaybullin R, Liang X, et al. Discovery of Gene Regulation Pattern in Lung Cancer by Gene Expression Profiling Using Human Tissues. Genom Data. 2015;3:112–115. doi: 10.1016/j.gdata.2014.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cheadle C, Vawter MP, Freed WJ, et al. Analysis of microarray data using Z score transformation. J Mol Diagn. 2003;5:73–81. doi: 10.1016/S1525-1578(10)60455-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moroz MA, Huang R, Kochetkov T, et al. Comparison of corticotropin-releasing factor, dexamethasone, and temozolomide: treatment efficacy and toxicity in U87 and C6 intracranial gliomas. Clin Cancer Res. 2011;17:3282–3292. doi: 10.1158/1078-0432.CCR-10-3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Srinivasan S, Patric IR, Somasundaram K. A ten-microRNA expression signature predicts survival in glioblastoma. PLoS One. 2011;6:e17438. doi: 10.1371/journal.pone.0017438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Celiku O, Johnson S, Zhao S, et al. Visualizing molecular profiles of glioblastoma with GBM-BioDP. PLoS One. 2014;9:e101239. doi: 10.1371/journal.pone.0101239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lamb J, Crawford ED, Peck D, et al. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006;313:1929–1935. doi: 10.1126/science.1132939. [DOI] [PubMed] [Google Scholar]
- 27.Wei G, Twomey D, Lamb J, et al. Gene expression-based chemical genomics identifies rapamycin as a modulator of MCL1 and glucocorticoid resistance. Cancer Cell. 2006;10:331–342. doi: 10.1016/j.ccr.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 28.Hieronymus H, Lamb J, Ross KN, et al. Gene expression signature-based chemical genomic prediction identifies a novel class of HSP90 pathway modulators. Cancer Cell. 2006;10:321–330. doi: 10.1016/j.ccr.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 29.Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee JK, Joo KM, Lee J, et al. Targeting the epithelial to mesenchymal transition in glioblastoma: the emerging role of MET signaling. Onco Targets Ther. 2014;7:1933–1944. doi: 10.2147/OTT.S36582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wen PY, Schiff D, Cloughesy TF, et al. A phase II study evaluating the efficacy and safety of AMG 102 (rilotumumab) in patients with recurrent glioblastoma. Neuro Oncol. 2011;13:437–446. doi: 10.1093/neuonc/noq198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bunn HF, Poyton RO. Oxygen sensing and molecular adaptation to hypoxia. Physiol Rev. 1996;76:839–885. doi: 10.1152/physrev.1996.76.3.839. [DOI] [PubMed] [Google Scholar]
- 33.Maxwell PH, Wiesener MS, Chang GW, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271–275. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- 34.Dreyfuss JM, Johnson MD, Park PJ. Meta-analysis of glioblastoma multiforme versus anaplastic astrocytoma identifies robust gene markers. Mol Cancer. 2009;8:71. doi: 10.1186/1476-4598-8-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang SD, Leung KL, McCrudden CM, et al. The Prognostic Significance of Combining VEGFA, FLT1 and KDR mRNA Expressions in Brain Tumors. J Cancer. 2015;6:812–818. doi: 10.7150/jca.11975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kreisl TN, Kim L, Moore K, et al. Phase II trial of single-agent bevacizumab followed by bevacizumab plus irinotecan at tumor progression in recurrent glioblastoma. J Clin Oncol. 2009;27:740–745. doi: 10.1200/JCO.2008.16.3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Friedman HS, Prados MD, Wen PY, et al. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol. 2009;27:4733–4740. doi: 10.1200/JCO.2008.19.8721. [DOI] [PubMed] [Google Scholar]
- 38.Liu C, Zhang Y, Zhang K, et al. Expression of estrogen receptors, androgen receptor and steroid receptor coactivator-3 is negatively correlated to the differentiation of astrocytic tumors. Cancer Epidemiol. 2014;38:291–297. doi: 10.1016/j.canep.2014.03.001. [DOI] [PubMed] [Google Scholar]
- 39.Harmalkar M, Upraity S, Kazi S, et al. Tamoxifen-Induced Cell Death of Malignant Glioma Cells Is Brought About by Oxidative-Stress-Mediated Alterations in the Expression of BCL2 Family Members and Is Enhanced on miR-21 Inhibition. J Mol Neurosci. 2015 doi: 10.1007/s12031-015-0602-x. [DOI] [PubMed] [Google Scholar]
- 40.He W, Liu R, Yang SH, et al. Chemotherapeutic effect of tamoxifen on temozolomide-resistant gliomas. Anticancer Drugs. 2015;26:293–300. doi: 10.1097/CAD.0000000000000197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chowdhury UR, Samant RS, Fodstad O, et al. Emerging role of nuclear protein 1 (NUPR1) in cancer biology. Cancer Metastasis Rev. 2009;28:225–232. doi: 10.1007/s10555-009-9183-x. [DOI] [PubMed] [Google Scholar]
- 42.Jiang WG, Davies G, Martin TA, et al. Com-1/p8 acts as a putative tumour suppressor in prostate cancer. Int J Mol Med. 2006;18:981–986. [PubMed] [Google Scholar]
- 43.Bastien JI, McNeill KA, Fine HA. Molecular characterizations of glioblastoma, targeted therapy, and clinical results to date. Cancer. 2015;121:502–516. doi: 10.1002/cncr.28968. [DOI] [PubMed] [Google Scholar]
- 44.Furnari FB, Fenton T, Bachoo RM, et al. Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev. 2007;21:2683–2710. doi: 10.1101/gad.1596707. [DOI] [PubMed] [Google Scholar]
- 45.Chai KM, Wang CY, Liaw HJ, et al. Downregulation of BRCA1-BRCA2-containing complex subunit 3 sensitizes glioma cells to temozolomide. Oncotarget. 2014;5:10901–10915. doi: 10.18632/oncotarget.2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Roth P, Wick W, Weller M. Steroids in neurooncology: actions, indications, side-effects. Curr Opin Neurol. 2010;23:597–602. doi: 10.1097/WCO.0b013e32833e5a5d. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Glioblastoma stem cells harbor characteristics of cancer stem cells. (A) Histograms show FACS data for GSC1 (i), GSC3 (ii), and GSC6 (iii) labeled with antibodies against the stemness markers SOX2 and NES and the proliferation marker KI67. Percentages of positive cells for specific markers are shown under the plots in brackets. Isotype control peaks are shown as secondary antibody control. (B) Scans of Western blots of whole-cell extracts of GSC1, GSC3, and GSC6 that were cultured under proliferative or differentiation conditions and probed with antibodies against SOX2, NES, and KI67. GAPDH is loading control. (C) Micrographs of immunofluorescence staining for differentiation marker GFAP (red) in GSC1 (i), GSC3 (ii), and GSC6 (iii) after differentiation. Nuclei are shown in blue. Scale bar = 250 µm. (D) Micrographs of H & E–stained mouse brains bearing orthotopic xenograft tumors arising from implantation of GSC1 (i), GSC3 (ii), or GSC6 (iii). Histologic examination of the tumors revealed high cellularity (purple), hypervascularity (endothelial proliferation: dark red), and necrosis (N). Scale bar = 200 µm.
The transcription factor C/EBPbeta (CEBPB), a master regulator of mesenchymal transformation in glioblastoma is activated upon dexamethasone exposure (activation z-score 1.69, P = 7.33E-043). The schematic representation shows a network analysis for CEBPB as upstream regulator affected by dexamethasone upon in vitro exposure of 3 GSC lines with 50 µM dexamethasone for 6 days.
Viability percentages of camptothecin exposed GSCs for 24 and 48 hours. At 24-hour time point the difference in viability of GSC1 and GSC3 were insignificant (p=0.058), whereas that at 48 hour time point was significantly different (p=1.0314e-006).