

Abstract
Background
Many pathogens secrete toxins that target key host processes resulting in the activation of immune pathways. The secreted Pseudomonas aeruginosa toxin Exotoxin A (ToxA) disrupts intestinal protein synthesis, which triggers the induction of a subset of P. aeruginosa-response genes in the nematode Caenorhabditis elegans.
Results
We show here that one ToxA-induced C. elegans gene, the Tribbles pseudokinase ortholog nipi-3, is essential for host survival following exposure to P. aeruginosa or ToxA. We find that NIPI-3 mediates the post-developmental expression of intestinal immune genes and proteins and primarily functions in parallel to known immune pathways, including p38 MAPK signaling. Through mutagenesis screening, we identify mutants of the bZIP C/EBP transcription factor cebp-1 that suppress the hypersusceptibility defects of nipi-3 mutants.
Conclusions
NIPI-3 is a negative regulator of CEBP-1, which in turn negatively regulates protective immune mechanisms. This pathway represents a previously unknown innate immune signaling pathway in intestinal epithelial cells that is involved in the surveillance of cellular homeostasis. Because NIPI-3 and CEBP-1 are also essential for C. elegans development, NIPI-3 is analogous to other key innate immune signaling molecules such as the Toll receptors in Drosophila that have an independent role during development.
Electronic supplementary material
The online version of this article (doi:10.1186/s12915-016-0334-6) contains supplementary material, which is available to authorized users.
Keywords: Surveillance immunity, Tribbles-like kinase, C/EBP, Pseudomonas aeruginosa, Exotoxin A, Translational inhibition, Caenorhabditis elegans, Innate epithelial immunity, Lifespan machine
Background
A fundamental problem for all multicellular animals is that they must respond to invading pathogens while simultaneously tolerating or facilitating the growth of commensal microbes. Evidence has been mounting that metazoans can recognize pathogens by detecting the activity of so-called pathogen-encoded virulence effectors [1]. Although significant advances have been made in understanding how these immune triggers are sensed, it is poorly understood how they activate the defense responses that lead to the protection of host tissues from pathogen-inflicted damage and ultimately to the resolution of infection.
To investigate these issues, we study the nematode Caenorhabditis elegans which, when exposed to human pathogens, activates multiple discrete immune signaling pathways including an evolutionary conserved p38 MAPK pathway that is also critical for mammalian immunity [2]. For C. elegans infected with the gram-negative nosocomial pathogen Pseudomonas aeruginosa, deployment of these signaling pathways and resulting gene induction is directly correlated with bacterial virulence [3, 4], leading us to hypothesize that P. aeruginosa virulence factors may themselves trigger host immune gene expression. To test this theory, in previously published work, we screened for individual P. aeruginosa effectors that are capable of inducing a host immune response and discovered that exposure to Exotoxin A (ToxA) upregulates C. elegans immune genes [5]. ToxA is an extremely potent toxin of the AB class that inhibits protein translation by catalyzing the ADP-ribosylation of elongation factor 2, the same reaction catalyzed by diphtheria toxin from Corynebacterium diphtheriae and cholix toxin from Vibrio cholerae [6, 7]. The high level of toxicity of these enzymes has enabled their use as immunotoxins to treat a variety of cancers [8].
We determined that C. elegans recognizes ToxA independently of ToxA per se by detecting its enzymatic activity, translational inhibition [5]. Significantly, this immune activation is independent of physical microbial features called microbe- or pathogen-associated molecular patterns (MAMPs/PAMPs) or pattern recognition receptors, which are the traditionally studied mechanisms of pathogen recognition. Dunbar et al. [9] similarly discovered that inhibiting host translation stimulates the MAMP/PAMP-independent upregulation of the C. elegans ZIP-2 transcription factor resulting in zip-2-dependent gene induction. Mammalian cells are also able to sense pathogens by recognizing protein synthesis abnormalities; translational inhibitors secreted by Legionella pneumophila activate NF-κB and MAP kinase signaling and trigger the transcription of their target genes [10, 11], a subset of which are also upregulated at the protein level [12, 13]. Additional cellular processes commonly targeted by bacterial effectors are monitored through similar surveillance mechanisms [1, 14]. While a commonality of all these effector-triggered mechanisms is that they require either injury or modification to the host, the host genetic circuits that respond to these insults and act to protect against subsequent effector-mediated damage are only beginning to be understood. We therefore used the C. elegans/ToxA system to identify and characterize new components of surveillance signaling pathways.
Here, we investigate the genetic pathways that enable nematodes to mitigate the damaging effects of ToxA-mediated translational inhibition. Using an automated C. elegans Lifespan Machine [15], we show that the C. elegans nipi-3 gene is required for animals to survive exposure to ToxA as well as to P. aeruginosa. NIPI-3 is a member of the highly conserved, functionally diverse Tribbles protein family, which, when mutated, has been linked to a variety of disorders related to cell signaling, immunity, metabolism, and cancer [16]. NIPI-3 has been previously shown to function upstream of p38 MAPK in the epidermis during fungal attacks [17] and, more recently, it has been determined to be required in multiple tissues during development (Kim et al., accompanying paper). Through partial loss-of-function and rescue studies, we find here that the C. elegans immune response against ToxA and P. aeruginosa is mediated in adult animals by intestinal NIPI-3 which, in contrast to epidermal NIPI-3, does not directly function in known C. elegans immune pathways but instead represses the activity of the bZIP C/EBP transcription factor CEBP-1.
Results
nipi-3 mutants are hypersusceptible to translational inhibitors and Pseudomonas aeruginosa
We previously found that ToxA does not irreparably damage healthy C. elegans as wild type animals have the same longevity when feeding on an E. coli strain expressing P. aeruginosa PA14 ToxA as on control bacteria [5]. However, nematodes defective in immune signaling pathways, such as the p38 MAPK pathway, die rapidly when fed ToxA E. coli [5], implying that C. elegans normally resist this highly toxic enzyme through an effective host defense. We reasoned that genes upregulated in response to ToxA might be required to protect against or recover from toxin-induced damage and allow wild type worms to survive. Through genome-wide transcriptional profiling using Affymetrix GeneChips©, we previously identified 144 genes that were upregulated in wild type N2 C. elegans fed ToxA [5].
Using RNAi or mutant alleles corresponding to 125 of the most highly upregulated genes (Additional file 1: Table S1), we assayed for premature lethality in worms fed ToxA and found that the nipi-3(fr4) mutant exhibited the most significant reduction in lifespan (data not shown). In our previous microarray analysis, nipi-3 was upregulated 4.3-fold in worms exposed to ToxA for 24 hours and was one of the two most highly induced kinases [5]. Whereas nipi-3 null mutants arrest by larval development stage L3 (Kim et al., accompanying manuscript), nipi-3(fr4) is a fully viable hypomorphic allele, which, as described below, has allowed us to selectively address NIPI-3’s role in pathogen defense. nipi-3(fr4) contains a single mutation (I307N) in a conserved residue in the kinase domain [17].
Using a C. elegans Lifespan Machine, a modified commercial flatbed scanner in conjunction with automated image analysis software [15], we analyzed the survival of the nipi-3(fr4) mutant feeding on ToxA and found that nipi-3(fr4) had a dramatically reduced lifespan on this food compared to wild type animals (Fig. 1a; P < 0.0001). Although nipi-3(fr4) has been reported to have a shortened lifespan [17], its longevity on ToxA was significantly shorter than on a control, non-pathogenic BL21 E. coli strain (Additional file 2: Figure S1a; P < 0.001), whereas the lifespans of wild type N2 worms on control BL21 E. coli or on ToxA were equivalent (Additional file 2: Figure S1a; P > 0.05) [5].
Fig. 1.
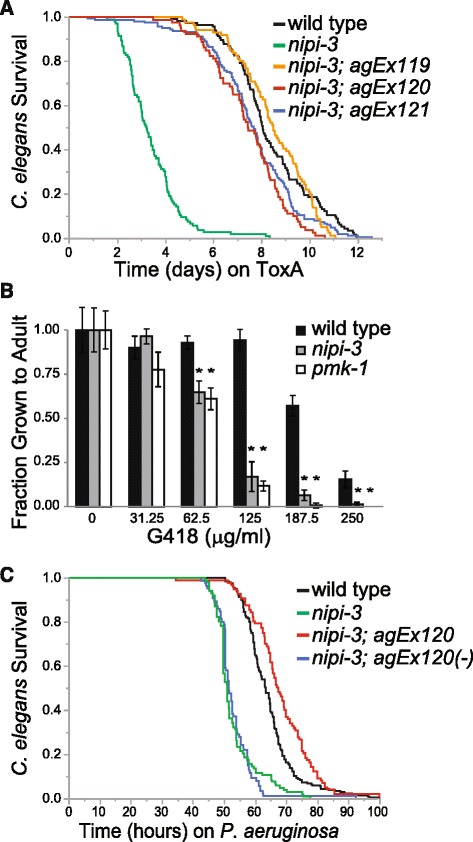
nipi-3(fr4) mutants have reduced resistance to ToxA, G418 and Pseudomonas aeruginosa. a Lifespans of wild type N2, nipi-3(fr4), and three independent lines of nipi-3(fr4) expressing wild type nipi-3p::nipi-3 fed E. coli expressing ToxA starting at the L4 stage. P < 0.0001 comparing nipi-3(fr4) and wild type (log-rank test). b Fraction of synchronized L1 worms that grew to at least young adult stage after 3 days at 20 °C on plates containing the indicated G418 concentration. Results shown are an average of four biological replicates. Error bars represent SD. *P < 0.05 compared to wild type animals at the given concentration (Student’s unpaired t-test). c Lifespans of wild type N2, nipi-3(fr4), and nipi-3(fr4) expressing wild type nipi-3p::nipi-3 fed on P. aeruginosa PA14. nipi-3(fr4);agEx120(-) indicates non-transgenic offspring of nipi-3p::nipi-3 transgenic worms. P < 0.001 comparing nipi-3(fr4) and wild type (log-rank test). Number of animals scored for each condition was ≥ 80 (547 total; a) and > 85 (487 total; c). These are representative experiments of two independent experiments. Primary data for panel b are provided in Additional file 15
We previously showed that C. elegans immune-related signaling pathways, including the PMK-1 p38 MAPK pathway, are activated by ToxA-mediated translational inhibition rather than by ToxA itself. We therefore tested whether nipi-3 mutants are also defective in their response to the protein synthesis inhibitor G418. Similar to pmk-1 mutants, nipi-3(fr4) animals were more sensitive to G418 than wild type animals, indicating that nipi-3 protects the host against translational inhibition and not an unrelated aspect of ToxA intoxication (Fig. 1b). Finally, we reasoned that nipi-3 is likely to be required for C. elegans defense against P. aeruginosa, which produces ToxA. Indeed, nipi-3(fr4) mutants were hypersusceptible to P. aeruginosa in large-lawn automated assays in which C. elegans animals were unable to avoid being in contact with the bacterial lawn (Fig. 1c; P < 0.001). Reintroduction of wild type nipi-3 expressed by its own promoter rescued the hypersusceptibility of nipi-3(fr4) to ToxA and P. aeruginosa (Fig. 1a, c).
To confirm that nipi-3(fr4) is not merely sensitive to any stressor, but is distinctly susceptible to translational inhibitors, we compared the ability of wild type and nipi-3(fr4) worms to recover from prolonged developmental arrest. Animals were arrested at the first larval stage and then starved for up to 14 days (L1 arrest or L1 diapause). Longer starvation reduces the number of animals that can recover upon feeding and grow to adults. Escaping this starvation-induced arrest requires the coordination of multiple pathways, including those required for general lifespan and stress responses [18]. In this assay, nipi-3(fr4) recovery was equivalent to wild type (Additional file 2: Figure S1b), indicating that nipi-3(fr4) is not broadly susceptible to any stressful condition.
Immune genes are misregulated in nipi-3(fr4) mutants
We previously found that ToxA-sensitive mutants, such as pmk-1(km25), misregulate pathogen-responsive genes [5], suggesting that nipi-3(fr4) animals will show similar transcriptional defects. However, unlike for pmk-1, complete loss of nipi-3 function causes animals to arrest at the L2/L3 larval stage (Kim et al., accompanying paper) indicating that nipi-3 is critical to both development and immunity. Thus, to understand the potential relationship between these processes and to dissect apart nipi-3’s apparent roles in both development and immunity, we performed genome-wide transcriptional profiling assays using Affymetrix GeneChips® comparing wild type, nipi-3(fr4), and pmk-1(km25) animals that were fed control OP50 E. coli. Surprisingly, we found that a large number of genes were significantly upregulated in nipi-3(fr4) mutants: 282 genes were upregulated in nipi-3(fr4), whereas, for comparison, only 8 genes were induced in pmk-1(km25) (Additional file 3: Table S2). Gene ontology (GO) term analysis showed that transcripts upregulated in nipi-3 mutants were enriched for processes involved in immunity, similar to the GO terms enriched among the 43 genes downregulated in pmk-1(km25) animals (Additional file 3: Table S2), suggesting nipi-3 may be a negative immune regulator. The only other overrepresented GO terms among the nipi-3(fr4)-upregulated genes involved flavonoid processes (Additional file 3: Table S2) due to the presence of 9 UDP-glucuronosyltransferases which, in addition to modifying flavonoids in plants, metabolize and detoxify xenobiotic compounds in metazoans [19, 20]. There were no enriched GO categories for the 71 genes downregulated in nipi-3(fr4) or the 8 genes upregulated in pmk-1(km25) animals. These gene expression results indicate that, similar to pmk-1(km25), the nipi-3(fr4) allele is specifically defective in immune processes and that we can use nipi-3(fr4) to study the role of nipi-3 in pathogen defense separately from its role in development.
To expand on the microarray results and test the effect of nipi-3(fr4) on immune gene expression under different conditions, we utilized a NanoString© codeset containing 118 C. elegans genes involved in immune- and stress-related responses (Additional file 4: Table S3). NanoString analysis recapitulated the microarray gene expression changes observed between wild type and nipi-3 mutant animals under normal growth conditions. We confirmed that 26 of the 27 codeset genes predicted to be nipi-3-dependent from the microarray data were similarly affected in the NanoString analysis; the remaining gene was upregulated 1.9× (false discovery rate (FDR) < 1 × 10–2), falling just below the two-fold cutoff. An additional 13 NanoString codeset genes that were not identified as being differentially regulated by microarray analysis were also statistically significantly up- or downregulated in nipi-3(fr4), suggesting that the microarray analysis is either less sensitive or more stringent than NanoString.
We next asked whether immune- and stress-related genes are misregulated in nipi-3(fr4) mutants fed P. aeruginosa or ToxA, potentially explaining the hypersusceptibility of nipi-3(fr4) to these conditions. Of the 118 codeset genes, 49 were differentially expressed when comparing nipi-3(fr4) and wild type animals exposed to P. aeruginosa and 27 were affected in nipi-3(fr4) compared to wild type when fed ToxA (Additional file 4: Table S3). In general, genes regulated by nipi-3 were differentially expressed under multiple conditions (Additional files 4, 5: Table S3, Figure S2a); for example, 77% of the genes altered in nipi-3(fr4) on OP50 were also altered in nipi-3(fr4) on P. aeruginosa. We confirmed the microarray and NanoString gene expression changes for a subset of genes using qRT-PCR to analyze wild type and nipi-3(fr4) animals fed P. aeruginosa, ToxA, or their respective control bacteria (Additional file 6: Figure S3a, b).
In support of the hypothesis that nipi-3 is an immune regulator, 44% (156) of the genes up or down regulated in nipi-3(fr4) animals on control OP50 bacteria were also responsive to ToxA or the translational inhibitor Hygromycin B (Additional file 5: Figure S2b; P < 1 × 10–95) as shown by comparing the current microarray dataset with our previous characterization of the wild type ToxA/Hygromycin B response [5]. In addition, genes differentially expressed in nipi-3(fr4) fed P. aeruginosa or ToxA compared to wild type animals on the same food were enriched for pathogen-response genes (Additional files 4, 5, 7: Tables S3, Figure S2c, and Table S4; P < 5 × 10–6). Taken together, these data demonstrate that nipi-3 is necessary for the proper expression of immune genes in both unstressed and infected animals.
ToxA induces defense proteins in wild type but not in nipi-3(fr4) animals
We next sought to determine the role of NIPI-3 on the host’s protein response to translational inhibitors since the extent to which mRNA up or downregulation affects protein expression under these conditions is unknown. We performed a large-scale quantitative proteomic analysis comparing wild type and nipi-3(fr4) animals fed on ToxA or control BL21 E. coli. This analysis detected a total of 7535 expressed proteins that map to 7444 unique C. elegans genes, representing approximately half of the genes expressed in adult animals [21].
We first characterized the protein changes that occur in wild type animals under translational stress. Of the 50 proteins that significantly changed in wild type upon ToxA exposure, 40 were upregulated and related to immunity based on GO term analysis (Additional file 8: Table S5). The induction of immune-related factors by ToxA was consistent with our previously published analysis of ToxA-mediated RNA changes [5]. However, unlike wild type animals, exposing nipi-3(fr4) mutants to ToxA caused more proteins to be downregulated than upregulated, 26 versus 17, respectively (Additional file 8: Table S5). There was almost no overlap between the nipi-3(fr4) and wild type responses to ToxA: only one protein was downregulated in both strains (red arrow, Fig. 2) and there were no common upregulated proteins. There were no enriched GO terms among the proteins up or downregulated by ToxA in nipi-3(fr4) consistent with the nipi-3(fr4) mutant mounting an uncoordinated, and ultimately ineffective, response to the toxin.
Fig. 2.
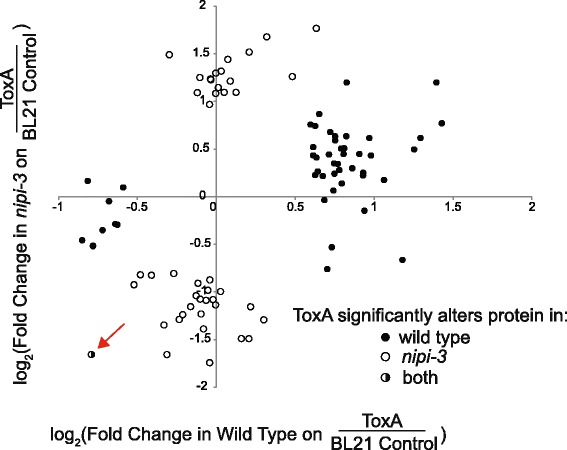
Different proteins are up and downregulated in wild type N2 and nipi-3(fr4) animals following ToxA exposure. Change of protein abundance in wild type N2 and nipi-3(fr4) animals following 24 hours feeding on E. coli expressing ToxA starting at the L4 stage as compared to animals fed control BL21 food. Only proteins significantly up or downregulated in wild type N2 and/or nipi-3(fr4) are included. The red arrow points to the only protein (T15B7.1) significantly altered in both N2 and nipi-3(fr4) animals. Results shown are an average of two biological replicates. Primary data are provided in Additional file 8: Table S5
To directly address the relationship between transcript and protein changes in the presence of ToxA, we compared the proteomic data to the transcriptional profiling analysis that we previously performed using the same ToxA and control strains [5]. Of the 174 transcripts induced or repressed by ToxA in wild type worms, we identified 37 corresponding proteins and, of those, 15 (41%) were significantly affected by ToxA (Additional file 9: Figure S4a). Eight of the 10 RNAs with the highest ToxA induction were also elevated at the protein level. To understand how a protein inhibitor impacts the relationship between RNA and protein expression, we performed the same type of analysis using unstressed conditions lacking translational inhibitors. Specifically, we analyzed transcriptional and proteomic changes in nipi-3(fr4) compared to wild type animals fed on control E. coli (Additional file 10: Table S6) [5]. While 7 of the 10 RNAs with the highest induction in nipi-3(fr4) also had elevated protein levels, overall, there was less correlation between RNA and protein changes as only 12% (23/188) of nipi-3(fr4)-affected RNAs showed significantly altered protein expression. In addition, there were 39 proteins differentially expressed in nipi-3(fr4) that were unaffected at the RNA level (Additional file 9: Figure S4b). One interpretation for the poorer correlation in the nipi-3/wild type control analysis versus the ToxA/BL21 analysis is that RNAs induced during translational disruption have less post-transcriptional regulation than RNAs in unstressed conditions. However, a caveat of the nipi-3 comparison is that different E. coli strains were used as controls in the transcriptomic and proteomic analyses (OP50 vs. BL21, respectively).
Intestinal nipi-3 defends against intestinal pathogens
To confer resistance against the intestinal pathogen P. aeruginosa, nipi-3 must act directly or indirectly in the intestine. NIPI-3 is expressed in multiple tissues in the adult C. elegans and was previously shown to function in the hypodermis but not the intestine to defend against the hypodermal fungal pathogen D. coniospora [17]. To determine whether the ToxA response is mediated by intestinally-expressed NIPI-3 or a systemic signal derived from hypodermally-expressed NIPI-3 and transported to the intestine, we knocked down nipi-3 in either the intestine or hypodermis using tissue-specific RNAi C. elegans strains. We confirmed the tissue-specificity of these strains with bacterial RNAi clones targeting different tissues. We did not observe any silencing phenotypes in the gut-specific RNAi strain for RNAi clones targeting genes in the hypodermis, body wall muscle, or germline, but we did detect low levels of RNAi silencing in non-hypodermal tissues in the hypodermal-specific RNAi strain (Additional file 11: Table S7). Knocking down nipi-3 in a gut-specific RNAi strain made animals hypersusceptible to ToxA and P. aeruginosa (Fig. 3a top; P < 0.001 for each assay), whereas knocking down nipi-3 in the hypodermal RNAi strain did not result in increased ToxA susceptibility compared to control RNAi (Fig. 3a bottom; P > 0.02 for each assay), even though there was a low level of silencing in the intestine hypodermal RNAi strain (Additional file 11: Table S7). To confirm that intestinal nipi-3 is responsible for the gene expression changes observed in Additional files 3, 4, and 6: Tables S2, S3, and Figure S3, we performed qRT-PCR of infection-related genes in either wild type or gut-specific RNAi animals following nipi-3 RNAi. In both strains, five of the transcripts were upregulated and six were downregulated (Fig. 3b). Some transcripts showed a larger difference in wild type animals than in the gut-specific RNAi strain, which was potentially due to a difference in RNAi efficiency or indicates nipi-3 has some activity in a non-gut tissue.
Fig. 3.

Intestinal nipi-3 expression is necessary and sufficient for resistance against ToxA and P. aeruginosa. a Lifespans of gut or hypodermal RNAi strains fed E. coli expressing ToxA (left) or P. aeruginosa PA14 (right) following either nipi-3 or L4440 vector control RNAi. P < 0.001 for gut RNAi assays; P > 0.02 for hyp RNAi assays (log-rank test). b qRT-PCR comparison of L4 animals following nipi-3 RNAi started at the L1 stage. Results shown are an average of four biological replicates, each normalized to the corresponding wild type L4440 control values. Error bars represent SEM. *P < 0.05 compared to corresponding wild type animals (Student’s unpaired t-test). c Lifespans of wild type N2, two independent lines of nipi-3(fr4) expressing intestinal vha-6p::nipi-3, and the non-transgenic offspring of nipi-3(fr4);agEx122 fed E. coli expressing ToxA. Number of animals scored for each condition was > 50 (373 total; a ToxA), ≥ 145 (656 total; a P. aeruginosa), and ≥ 130 (558 total; c). These are representative experiments of four (a ToxA), three (a P. aeruginosa), or two (c) independent experiments. Primary data for panel b are provided in Additional file 15
Finally, we tested whether NIPI-3 is also required in additional tissues by rescuing NIPI-3 only in the intestine of nipi-3(fr4) mutants. Expressing intestinal NIPI-3 was sufficient to rescue the nipi-3(fr4) ToxA defect (Fig. 3c). Taken together, the data in Fig. 3 show that intestinal NIPI-3 is both necessary and sufficient to mediate C. elegans ToxA defense mechanisms. Importantly, in contrast to knocking down nipi-3 systemically, removing nipi-3 only in the intestine did not result in a shortened lifespan on control food, and in fact appeared to enhance the lifespan (Additional file 2: Figure S1c). This latter experiment emphasizes that the role of nipi-3 in pathogen defense can be separated from its lifespan and developmental effects.
Intestinal nipi-3 is not a primary component of known ToxA-response pathways including PMK-1 p38 or KGB-1 JNK-like MAP kinase
We next asked whether intestinal NIPI-3 is a component of known immune signaling pathways. Previous work has shown that, in the hypodermis, NIPI-3 functions upstream of the PMK-1 p38 MAPK pathway and between the protein kinase C TPA-1 and BiP/GRP78 chaperone HSP-3 [22, 23]. However, unlike nipi-3(fr4) animals, tpa-1 and hsp-3 mutants had a normal lifespan on ToxA (Additional file 12: Figure S5; P > 0.4 comparing ToxA and control E. coli for each strain) indicating that NIPI-3 has different functions in the hypodermis and intestine.
In contrast to hsp-3 and tpa-1, pmk-1 mutants die rapidly on ToxA [5] and the mammalian Tribbles homolog Trib2 is required for p38 phosphorylation in mammalian cells [24], suggesting that NIPI-3 and PMK-1 may function together. If NIPI-3 indeed functions solely upstream of or in conjunction with PMK-1, we reasoned that PMK-1 phosphorylation levels should be similar in nipi-3(fr4) animals to the levels observed in sek-1 (MAPKK) mutants [25]. However, whereas PMK-1 phosphorylation is apparently decreased in nipi-3(fr4) compared to wild type worms, it is still present at a significantly higher level than in a sek-1 mutant (Fig. 4a). We also tested whether pmk-1 and nipi-3 regulate common downstream genes as would be predicted if they are components of the same pathway. From our microarray analysis of animals on control OP50 E. coli, there was almost no overlap between nipi-3- and pmk-1-regulated genes (Fig. 4b); only 5 of the 101 genes downregulated in nipi-3(fr4) were repressed in pmk-1(km25) and, of the 282 genes upregulated in nipi-3(fr4), 3 were upregulated and 8 were downregulated in pmk-1(km25) (Additional file 3: Table S2). We additionally analyzed the protein expression of nipi-3(fr4) and pmk-1(km25) mutants exposed to ToxA and detected no proteins in common between the 43 upregulated in nipi-3(fr4) and the 7 induced in pmk-1(km25) when comparing each to wild type worms on ToxA (Additional file 10: Table S6). Somewhat surprisingly, 16 proteins were repressed in both nipi-3(fr4) and pmk-1(km25) from a total of 106 pmk-1(km25)-downregulated and 25 nipi-3(fr4)-downregulated proteins.
Fig. 4.
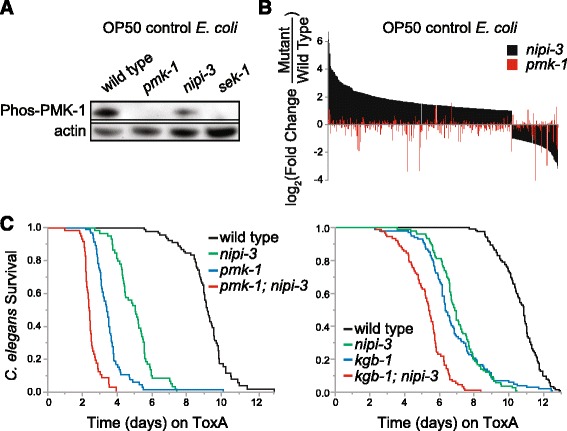
NIPI-3 is not immediately up or downstream of PMK-1 p38 MAPK or KGB-1 JNK-like pathways. a Western blot analysis of phosphorylated PMK-1 in L4 animals of the indicated genotype raised on control OP50 E. coli. b Transcript fold changes determined by microarray analysis of nipi-3(fr4) or pmk-1(km25) animals as compared to wild type N2 raised on OP50 E. coli. Only genes significantly altered in nipi-3(fr4) are shown. c Lifespans of wild type N2 and pmk-1(km25) (left) or kgb-1(km21) (right) fed E. coli expressing ToxA following either nipi-3 or L4440 vector control RNAi. P < 0.001 comparing pmk-1 and pmk-1, nipi-3 and comparing kgb-1 and kgb-1; nipi-3 (log-rank test). Number of animals scored for each condition was ≥ 55 (278 total; c right) and > 90 (424 total; c left). These are representative experiments of two (c left) or three (c right) independent experiments. Primary data for panel b are provided in Additional file 3: Table S2
Finally, consistent with NIPI-3 and PMK-1 mediating separate signaling pathways, we found that animals lacking both nipi-3 and pmk-1 had an increased susceptibility to ToxA as compared to loss of either protein alone (Fig. 4c left; P < 0.001 for pmk-1 and pmk-1;nipi-3). A caveat of this experiment, however, is that the lifespan of the pmk-1;nipi-3 animals on control food was also shortened (Additional file 13: Figure S6a). Taken together, while there may be some cross-talk between the NIPI-3 and PMK-1 pathways, our gene expression and pathogen-susceptibility data are not consistent with a model in which NIPI-3 functions directly up or downstream of PMK-1.
We next focused on the KGB-1 JNK-like MAPK pathway since, like nipi-3, kgb-1 is required for resistance to P. aeruginosa and acts in parallel to pmk-1 (Fig. 6) [25]. In addition, mammalian Trib2 has a role in JNK phosphorylation [24]. While we found that kgb-1 mutants were hypersusceptible to ToxA, loss of both kgb-1 and nipi-3 resulted in an additive ToxA effect compared to loss of either gene alone (Fig. 4c right; P < 0.001 comparing kgb-1;nipi-3 and kgb-1 or nipi-3). However, as with the pmk-1 analysis, the lifespan for kgb-1;nipi-3 on control E. coli was attenuated compared to loss of only kgb-1 or nipi-3 (Additional file 13: Figure S6b). Therefore, we asked whether kgb-1(km21) and nipi-3(fr4) regulate the same set of downstream genes. We determined that none of the eight ToxA-response genes assayed by qRT-PCR were regulated by both kgb-1 and nipi-3 on control E. coli and ToxA (Additional file 6: Figure S3b), consistent with KGB-1 and NIPI-3 acting in parallel.
Fig. 6.
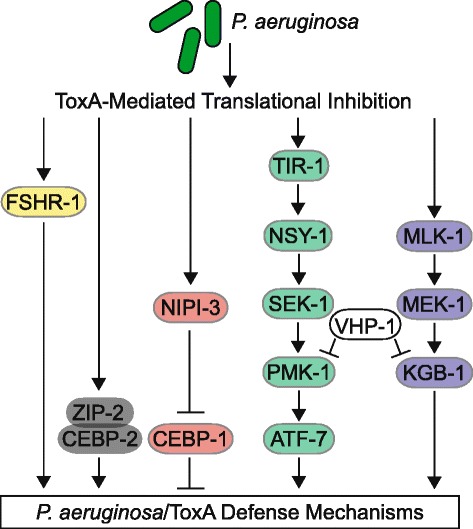
Model of major C. elegans pathways involved in ToxA defenses
Finally, the bzip transcription factor ZIP-2 and G-protein coupled receptor FSHR-1 regulate different subsets of ToxA-response genes and function separately from the PMK-1 and KGB-1 pathways (Fig. 6) [5]. As was the case with pmk-1 and kgb-1, there was no overlap between the transcripts significantly altered in zip-2 or fshr-1 and nipi-3 mutants on control E. coli and P. aeruginosa (Additional file 6: Figure S3a). In addition, zip-2 and fshr-1 mutants were much less sensitive to ToxA than nipi-3(fr4) (Additional file 6: Figure S3c; data not shown for fshr-1), making it unlikely that either ZIP-2 or FSHR-1 are in a linear pathway with NIPI-3.
nipi-3 genetically interacts with the C/EBP bZIP transcription factor cebp-1 to promote ToxA resistance
Based on the epistasis analyses, NIPI-3 represents either the first identified factor of a new signaling pathway acting in parallel to PMK-1 or it functions to modulate multiple pathways and coordinate the downstream effects of different initial inputs. If NIPI-3 represents a separate branch of immune signaling, we reasoned that we should be able to identify distinct factors functioning downstream of nipi-3. To test this, we mutagenized nipi-3(fr4) animals with EMS and identified 22 mutants with increased resistance to ToxA (see Methods for details). We selected two mutants with the strongest phenotypes from two independent pools for further analysis. Through whole genome sequencing, we found that both mutants contained an A246V mutation in the DNA binding domain of the C/EBP bZIP transcription factor CEBP-1. When exposed to ToxA or P. aeruginosa, both the newly-discovered cebp-1(ag33) allele as well as a previously-isolated cebp-1 deletion allele (tm2807), completely suppressed the nipi-3(fr4) defect in both infection conditions as compared to wild type animals (Fig. 5a). However, the single cebp-1 mutants survived longer on ToxA than wild type worms and the double cebp-1, nipi-3 mutants were slightly shorter lived on ToxA than their single cebp-1 mutant counterparts (Fig. 5a left; P < 0.001) suggesting that nipi-3 may have a minor additional function in a non-cebp-1 pathway to protect against ToxA. cebp-1 mutants also suppress the nipi-3(fr4) lifespan defect as the lifespans of the single cebp-1 and double cebp-1, nipi-3 mutants were equivalent on control E. coli (Fig. 5b; P > 0.05).
Fig. 5.
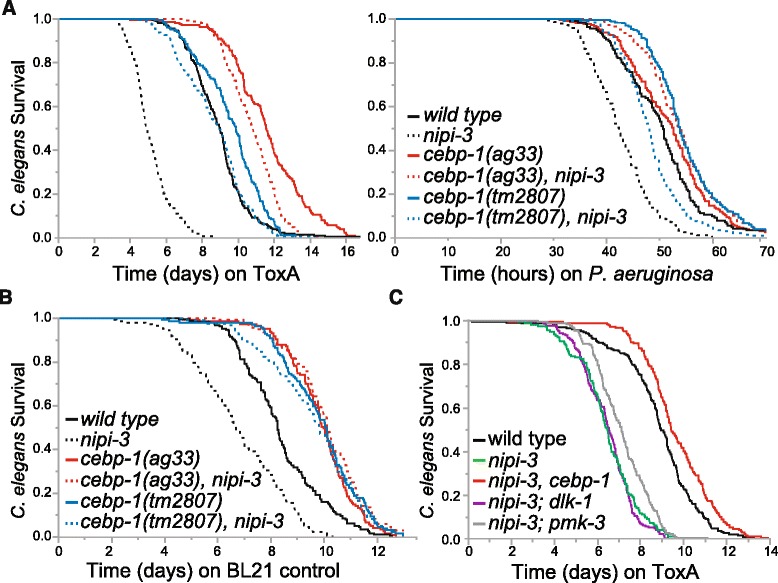
nipi-3(fr4) survival defects are suppressed by loss of cebp-1 but not loss of pmk-3 or dlk-1. Lifespans of the indicated strains on E. coli expressing ToxA (a left, c), P. aeruginosa PA14 (a right), or control BL21 E. coli (b). nipi-3(fr4) was used for all assays. cebp-1, dlk-1, and pmk-3 were inhibited by RNAi in c. Number of animals scored for each condition was > 160 (1178 total; a right), > 140 (1239 total; a left), > 120 (875 total; b), and > 160 (937 total; c). These are representative experiments of two independent experiments
CEBP-1 is expressed in pharyngeal, neuronal and intestinal cells [26], and is known to function in neurons during axon regeneration [27]. Therefore, we asked whether cebp-1 acts in the intestine to mediate ToxA defenses. To test this, we used the gut-specific RNAi strain described above and compared its susceptibility to ToxA following knockdown of either nipi-3 or cebp-1 or of both genes simultaneously. While loss of intestinal nipi-3 resulted in ToxA hypersusceptibility, animals with co-knockdown of intestinal nipi-3 and cebp-1 had an equivalent lifespan on ToxA as those lacking only intestinal cebp-1 (Additional file 14: Figure S7a; for nipi-3, cebp-1 vs. cebp-1 RNAi, P = 0.95 by log-rank test and 0.4 by Wilcoxon test; for nipi-3 vs. cebp-1 RNAi, P = 0.089 by long-rank test and 0.0004 by Wilcoxon test), indicating that CEBP-1 mediates the ToxA defense by functioning in the intestine.
In neurons, CEBP-1 acts downstream of the DLK-1/PMK-3 p38 MAPK pathway [27] and so we determined whether dlk-1 and pmk-3 are also involved in the NIPI-3 intestinal immune pathway. We found that the PMK-3 pathway is not necessary for the ToxA defense since, unlike RNAi-mediated knockdown of cebp-1, RNAi of dlk-1 or pmk-3 did not suppress the ToxA sensitivity of nipi-3(fr4) (Fig. 5c).
Finally, since loss of nipi-3 results in immune gene misexpression (Additional files 3, 4, 6: Tables S2, S3 and Figures S3), we tested whether cebp-1 rescues nipi-3’s pathogen survival phenotype by restoring wild type gene expression levels during ToxA intoxication. Using qRT-PCR, we compared wild type animals with single nipi-3 and cebp-1 mutants as well as double nipi-3, cebp-1 mutants. For some genes, such as T24B8.5, the nipi-3(fr4) defect was partially rescued in the double nipi-3, cebp-1 mutants but, for others, such as F11D11.3, the double mutants showed stronger misregulation than nipi-3(fr4) alone (Additional file 14: Figure S7b). A major limitation of this experiment, however, is that it is unknown which nipi-3-dependent genes are essential for the ToxA response. We attempted to identify critical effector proteins acting downstream of NIPI-3/CEBP-1 by asking whether any of the individual proteins differentially regulated in nipi-3(fr4) could account for the pathogen sensitivity of these mutants. To mimic the defects in nipi-3(fr4), for proteins downregulated in nipi-3(fr4), we knocked down the corresponding genes by RNAi in wild type worms and assayed for worms that phenocopied the nipi-3(fr4) ToxA susceptibility. We similarly inhibited the proteins that were upregulated in nipi-3(fr4) using RNAi in nipi-3(fr4) mutants and examined whether these animals survived longer on ToxA. However, we found no major changes in ToxA sensitivity following these knockdowns (data not shown).
Discussion
In this study, we find that NIPI-3 is an essential component of the C. elegans defense against the translational inhibitor ToxA as well as P. aeruginosa, the bacterial pathogen that secretes ToxA. Unlike other previously described immune signaling genes, including pmk-1, nipi-3 is upregulated by ToxA. Perhaps counterintuitively, loss of nipi-3 results in the upregulation of many ToxA-inducible genes in the absence of translational inhibition. This does not occur for the ToxA-sensitive pmk-1 mutant in which immune and stress genes are overall downregulated compared to wild type animals and suggests that NIPI-3 may have a specialized inhibitory role in responding to translational inhibitors.
NIPI-3 plays critical roles in both development and immunity, analogous to other key innate immune signaling molecules such as the Toll receptors in Drosophila [28] and bar-1 β-catenin in C. elegans [29]. While null mutations in nipi-3 cause animals to arrest by the L3 stage (Kim et al., accompanying paper), the use of the fully viable nipi-3(fr4) hypomorphic allele has allowed us to specifically investigate NIPI-3’s role in pathogen defense. Through tissue-specific RNAi and rescue experiments, we determined that NIPI-3-mediated ToxA and P. aeruginosa defenses are controlled in adult animals by intestinally expressed nipi-3, consistent with P. aeruginosa being a C. elegans intestinal pathogen. Previous reports show that hypodermal but not intestinal expression of nipi-3 is important for survival against the hypodermal pathogen D. coniospora [17]. A simple explanation of these data is that, in late larval stage and adult animals, following the requirement for NIPI-3 in development, NIPI-3 is only necessary in the tissue that is under pathogenic attack, which implies that it functions autonomously to mediate local immune responses. Alternatively, it is also possible that intestinally- and hypodermally-expressed nipi-3 regulate different immune mechanisms, with the intestinal responses effective against ToxA-like molecules and the hypodermal responses effective against distinct pathogenic mechanisms of D. coniospora. Supporting the hypothesis that nipi-3 may have unique functions in different tissues, we show here that the immune signaling molecules hsp-3 and tpa-1, which function in the hypodermal NIPI-3 pathway [22, 23], are dispensable for mounting ToxA resistance in the intestine, even though, as with nipi-3, they are expressed in the intestine [30, 31].
In contrast to hsp-3 and tpa-1, pmk-1 mutants are hypersensitive to ToxA and the translational inhibitor G418 but nipi-3 does not appear to function strictly upstream or downstream of p38 MAPK in response to ToxA. Microarray analysis of pmk-1 and nipi-3(fr4) L4 animals, the stage exposed to ToxA in our standard assay, revealed that the gene expression signature in these two mutants is remarkably different on control OP50 bacteria. However, there may be some interplay between nipi-3 and pmk-1 because the overall level of activated PMK-1 was reduced in L4 nipi-3(fr4) animals. Upon exposure to ToxA, most of the proteins that changed in the young adult nipi-3(fr4) mutant animals exhibited an increase over wild type, whereas the opposite was true for the pmk-1 mutant animals, where many more proteins were downregulated. While the majority of overall proteins differentially expressed in nipi-3(fr4) or pmk-1(km25) on ToxA did not overlap, a subset of proteins downregulated in nipi-3(fr4) mutant animals were also reduced in the pmk-1 mutant, suggesting that either these are a signature of worms dying from ToxA exposure or there is a commonality in the nipi-3 and pmk-1 responses to ToxA. However, we argue that nipi-3 and pmk-1 primarily function in parallel for their response to the toxin because reduction of nipi-3 activity by RNAi in the pmk-1 null mutant resulted in increased hypersusceptibility to ToxA. Interestingly, and in contrast to our P. aeruginosa data, nipi-3 acts upstream of the p38 MAPK signaling cassette (tir-1/nsy-1/sek-1/pmk-1) in the hypodermis to regulate the induction of anti-microbial peptides in response to D. coniospora [17, 22, 32]. In addition, mutations in the p38 PMK-1 MAPK pathway suppress nipi-3-dependent developmental arrest and nipi-3 null mutant animals hyperactive PMK-1 at early larval stages (Kim et al., accompanying paper). Combining these studies, it is clear that there are multiple regulatory interactions between nipi-3 and pmk-1 across different developmental stages and tissues and these likely contribute to the varied functions of NIPI-3.
For adult nipi-3(fr4) animals exposed to P. aeruginosa or ToxA, their shortened lifespan can be completely suppressed by loss of the C/EBP bZIP transcription factor cebp-1. Removal of cebp-1 does not cause C. elegans to be hypersusceptible to ToxA, suggesting it normally functions as a negative regulator. Therefore, the explanation for nipi-3(fr4) lethality on ToxA is that CEBP-1 function is now increased, making NIPI-3 a negative regulator of CEBP-1 (Fig. 6). In support of this model, Kim et al. (accompanying paper) found that cebp-1 transcription is increased in the nipi-3 mutant and there is a remarkable overlap in the gene classes enriched in cebp-1 targets and those repressed by nipi-3, specifically in the “stress cluster” (Kim et al., accompanying paper). Finally, other Tribbles proteins are also known to promote degradation of target proteins, including MAPKs and C/EBP proteins [16]. It should be noted that cebp-1 mRNA and protein were upregulated in nipi-3(fr4) animals, although these inductions were not statistically significant.
While the cebp-1 ag33 allele results in a relatively minor change in CEBP-1, this mutation has been isolated independently at least three times: twice in this study and once by Bounoutas et al. [33] in a neuronal mutagenesis screen that identified cebp-1(u819) as a suppressor of the microtubule disrupter colchicine, which normally causes a general decrease in protein expression in C. elegans touch receptor neurons. Unlike in our study, mutations in the PMK-3 p38 MAPK phenocopy the loss of neuronal cebp-1. For both our study and that of Bournoutas et al. [33], regardless of the upstream pathway, CEBP-1 is detrimental to mitigating the effects of translational inhibitors, either ToxA or colchicine. Interestingly, another C/EBP transcription factor, cebp-2, has also recently been shown to play an important role in C. elegans translational defense. However, unlike CEBP-1, CEBP-2 promotes ToxA and P. aeruginosa protective mechanisms by acting as a heterodimeric transcription factor with ZIP-2 [34]. The ability of CEBP proteins to both promote and repress protective host defenses against the same pathogen demonstrates the range of CEBP immune functions and raises the question of whether, for example, CEBP-1 could be a positive effector under a different condition.
The mechanism by which translational inhibitors activate the NIPI-3/CEBP-1 pathway remains an open question. Dunbar et al. [9] discovered that ZIP-2 accumulation during a P. aeruginosa infection is mediated by open reading frames in the zip-2 5’UTR (uORFs). This pathogen-mediated increase in translation is potentially similar to the response of yeast GCN4 and mammalian ATF4 to nutritional stresses and translational inhibition [35]. However, we did not observe uORFs in the nipi-3 5’UTR. Moreover, proteins induced by ToxA in wild type worms were not enriched for uORFs, implying that additional activation mechanisms must be present. One such strategy may be linked to transcript abundance; Ivanov and Roy [13] found that the most abundant transcripts could bypass the general protein synthesis block caused by L. pneumophila translational inhibitors. However, a similar study by Asrat et al. [12] found that translation of the IL-1β cytokine was independent of its transcript stability, suggesting that increasing a transcript’s concentration is not sufficient to ensure its protein expression. Another mechanism of post-transcriptional regulation could be mediated by a second protein such as a labile repressor that is not synthesized when translation is disrupted. This is observed at the transcriptional level in mammalian macrophages when L. pneumophila translational inhibitors block the production of new NFκB repressor IκB, resulting in increased gene expression [10]. It is possible that, if it exists, such a suppressor could be identified in further C. elegans screening.
Future studies are also likely to identify additional mechanisms for activating surveillance immune pathways and may identify strategies employed by the pathogens to suppress them. A striking finding from this study is the approximately 40% correlation between transcript and protein changes in the worms following ToxA exposure, which is comparable to other published studies analyzing a variety of conditions [36] and suggests that, after a 24 hour exposure, ToxA does not globally affect post-transcriptional processing. However, ToxA expressed by the P. aeruginosa strain PA01 is able to broadly reduce C. elegans protein synthesis [37], which may indicate that P. aeruginosa expresses effectors to inhibit the nematode’s ToxA defense.
Conclusions
We have shown here that nipi-3 enables hosts to survive pathogen-mediated damage to protein translation. NIPI-3 mediates post-developmental immunity in the intestine by acting as a negative regulator of the C/EBP transcription factor CEBP-1, which in turn negatively regulates protective immune processes. This pathway represents a new branch of intestinal innate immune signaling.
Methods
Strains
C. elegans were maintained using standard methods. C. elegans strains used in this study were: N2 (wild type), IG544 nipi-3(fr4), KU25 pmk-1(km25), MJ563 tpa-1(k530), RB1104 hsp-3(ok1083), AU0067 kgb-1(km21), RB911 fshr-1(ok778), ERT61 zip-2(tm4248), KU4 sek-1(km4), MGH167 sid-1(qt9);alxIs6[vha-6p::SID-1::SL2::GFP], JM43 rde-1(ne219);Is[wrt-2p::RDE-1], CZ8920 cebp-1(tm2807), KU12 dlk-1(km12), BS33830 pmk-3(ok169), and CZ8920 cebp-1(tm2807). The C. elegans strains created for this study were: AU0329 nipi-3(fr4); agEx119(myo-2p::MCherry, nipi-3p::NIPI-3), AU0330 nipi-3(fr4);agEx120(myo-2p::MCherry, nipi-3p::NIPI-3), AU0331 nipi-3(fr4);agEx121(myo-2p::MCherry, nipi-3p::NIPI-3), AU0332 nipi-3(fr4);agEx122(myo-2p::MCherry, vha-6p::NIPI-3), AU0333 nipi-3(fr4);agEx123(myo-2p::MCherry, vha-6p::NIPI-3), AU0350 nipi-3(fr4) cebp-1(ag33), AU0352 cebp-1(ag33), and AU0351 nipi-3(fr4) cebp-1(tm2807).
nipi-3p::NIPI-3 was amplified from the fosmid UBC_f80B0723Q (source bioscience UK limited) with the primers 5’-TGTTACCTGAAAGTTGCGGA and 5’-CCCGATTCAACTGTTTCAGG. For vha-6p::NIPI-3, NIPI-3 was amplified from the nipi-3p::NIPI-3 fusion using the primers 5’- CTAAACTAGTGGGTATGGCTCGTACAAAATGC and 5’-CCCGATTCAACTGTTTCAGG and vha-6p was amplified from N2 genomic DNA with the primers 5’-GATATTGCCAGCATGCTCAACG and 5’-TCTAGATATGGGTTTTGGTAGGTTTTAGTCG. The NIPI-3 and vha-6p fragments were ligated together in the presence of SpeI, XbaI, and T4 DNA ligase (NEB) and the expected sized fragment was gel purified with the QIAquick Gel Extraction kit (Qiagen). Constructs were injected into worms with the co-injection marker myo-2p::MCherry, and 1 KB ladder (NEB).
ToxA and BL21 E. coli control (an empty pet100 vector control) have been previously described [5]. All P. aeruginosa assays used the clinical isolate PA14 [38]. RNAi clones were obtained from the Ahringer RNAi library [39] and confirmed by sequencing.
C. elegans lifespan/killing assays
nipi-3 RNAi experiments were started from the L1 stage and mimicked the survival phenotype of nipi-3(fr4) mutants. All lifespan and killing assays were performed using the C. elegans Lifespan Machine [15] using worms and bacteria prepared as described [5], with the modification that E. coli plates were incubated for 24 hours at room temperature before use. All P. aeruginosa experiments utilized full lawn assays. The automated, high-resolution P. aeruginosa strain PA14 killing assays required significant modification of the image analysis software described in [15] to account for the disappearance, or ghosting, of dead animals, the relative opaqueness of the PA14 lawn, and a different plate type.
Multiplexed quantitative proteomics
Approximately 10,000 L4 worms were plated per condition and collected after 24 hours at 25 °C. Worms were washed with M9, incubated for 15 minutes at room temperature to remove intestinal bacteria, washed again and flash frozen in liquid nitrogen. Worms were ground using a mortar and pestle on dry ice and resuspended in 50 mM HEPES pH 8.5, 3% SDS, and EDTA-free complete protease-inhibitor (Roche). Samples were spun down to collect the supernatant. Multiplexed quantitative proteomics was performed using TMT reagents and an Orbitrap Fusion mass spectrometer applying Simultaneous Precursor Selection-MS3 supported quantification [40, 41], see below. Differentially regulated proteins were determined by QSPEC (http://www.nesvilab.org/qspec.php/; version 1.2.2) [42] using a FDR < 0.05 and log2(Fold Change) > 0.585 or < –0.585.
For the mass spectrometer analysis, cells were lysed in a buffer containing 75 mM NaCl, 50 mM HEPES (pH 8.5), 10 mM sodium pyrophosphate, 10 mM NaF, 10 mM β-glycerophosphate, 10 mM sodium orthovanadate, 10 mM phenylmethanesulfonylfluoride, Roche Complete Protease Inhibitor EDTA-free tablets, and 3% sodium dodecyl sulfate. Lysis was achieved by passing cells 10 times through a 21-gauge needle. Lysates were further processed through reduction and thiol alkylation was followed by purifying the proteins using MeOH/CHCl3 precipitation. Protein digestion was performed with Lys-C and trypsin. Peptides were labeled with TMT-10plex reagents (Thermo Scientific) [43] and fractionated by basic pH reversed phase chromatography as described elsewhere [44]. Multiplexed quantitative proteomics was performed on an Orbitrap Fusion mass spectrometer (Thermo Scientifc) using a Simultaneous Precursor Selection-based MS3 method [41]. MS2 spectra were assigned using a SEQUEST-based [45] proteomics analysis platform [46]. Based on the target-decoy database search strategy [47] and employing linear discriminant analysis and posterior error histogram sorting, peptide and protein assignments were filtered to FDR of < 1% [46]. Peptides with sequences that were contained in more than one protein sequence from the UniProt database were assigned to the protein with most matching peptides [46]. TMT reporter ion intensities were extracted as that of the most intense ion within a 0.03 Th window around the predicted reporter ion intensities in the collected MS3 spectra. Only MS3 with an average signal-to-noise value of larger than 40 per reporter ion as well as with an isolation specificity [40] of larger than 0.75 were considered for quantification. A two-step normalization of the protein TMT-intensities was performed by first normalizing the protein intensities over all acquired TMT channels for each protein based to the median average protein intensity calculated for all proteins. To correct for slight mixing errors of the peptide mixture from each sample, a median of the normalized intensities was calculated from all protein intensities in each TMT channel and the protein intensities were normalized to the median value of these median intensities.
Gene expression analyses
Microarray samples were prepared as previously described [5]. RMA normalized gene expression values were calculated using the R package ‘affy’ (Release 3.3) [48], while differential expression analysis was performed with the package ‘limma’ [49]. NanoString probe hybridization and data acquisition were performed according to manufacturer’s protocols and differentially regulated transcripts were determined by edgeR [50]. For the microarray and NanoString analyses, genes were considered differentially regulated at FDR < 0.05 and log2(Fold Change) > 1 or < –1. Quantitative RT-PCR was performed and analyzed as described [5] and P values were determined with an unpaired, two-tailed Student t-test.
GO term analysis
Data was analyzed through the Gene Ontology Consortium (www.geneontology.org) [51] [52]. Terms were considered enriched if P < 1 × 10–3 and Fold Enrichment > 5.
Immunoblot analysis
L4 worms raised on OP50 were washed with M9 to remove bacteria and resuspended in Laemmli sample buffer (BioRad). Samples were boiled, spun down, and supernatants were flash frozen in liquid nitrogen. Proteins were resolved on a 4–12% Bis-Tris SDS gel, transferred to a nitrocellulose membrane, and probed with anti-phospho-PMK-1 (Promega) or anti-actin (Abcam) antibodies.
Larval starvation assay
L1 stage animals were synchronized by hypochlorite treatment and overnight hatching in M9 at room temperature. At the indicated day, approximately 100 animals were transferred to NGM plates seeded with OP50 bacteria and counted. Plates were incubated at room temperature for 2 or 3 days and scored for growth.
Mutagenesis
Ethyl methane sulfonate mutagenesis of nipi-3(fr4) animals was carried out using standard methodology [53]. Fortuitously, we found that feeding nipi-3(fr4) on HT115 E. coli expressing vhp-1 RNAi led to early stage larval arrest, animals were of undetermined developmental age but not larger than wild type L2 animals, and the arrested animals subsequently died within several days. This is in contrast to N2 wild type animals fed the same vhp-1 RNAi expressing strain that showed delayed development compared to the control HT115 L4440 strain but matured to gravid adults within 2–3 days at 25 °C. F2 generation eggs from approximately 25,000 mutagenized nipi-3(fr4) haploid genomes were isolated by hypochlorite treatment and dropped directly onto vhp-1 RNAi food. Animals were incubated at 25 °C for 2 days and L4, young adult or gravid adult animals were picked from the vhp-1 RNAi to standard OP50 E. coli food. The recovered putative nipi-3 suppressors were re-screened for sensitivity to ToxA in order to eliminate (1) RNAi defective mutants and (2) known suppressors of vhp-1, such as mlk-1, mek-1, kgb-1 [54], and pmk-3 [55], which have been previously shown to suppress the larval arrest of vhp-1 mutants but do not suppress the ToxA sensitivity of nipi-3. For this ToxA secondary test, between 20 and 45 L4 progeny from each putative nipi-3 suppressor were picked to ToxA and their survival at 25 °C was followed until all the un-mutagenized nipi-3(fr4) animals were dead, at which point 100% of N2 wild type animals were still alive. Two suppressors from independent pools of mutagenized animals were identified that restored nipi-3(fr4) mutant animals to wild type levels of ToxA resistance. Each of the two suppressors was back crossed to nipi-3(fr4) and at least 50 F2 recombinants which grew to maturity on vhp-1 RNAi and exhibited wild type levels of ToxA resistance were pooled for DNA isolation. The corresponding genetic lesions were identified by next generation sequencing technology as previously described [56]. Methods were modified for sequencing on Illumina MiSeq according to the manufacturer’s instructions. The nipi-3(fr4) strain was also sequenced and used as the reference strain for identification of homozygous variants. During the process of backcrossing, the nipi-3(fr4) suppressors were both shown to be X-linked, which aided in the identification of relevant sequence variants.
Acknowledgments
We are grateful to the Caenorhabditis Genetics Center (CGC), Alex Soukas, Jonathan Ewbank, and Martin Chalfie for C. elegans strains, Nicolas Lehrbach and Robert Dowen for EMS/NGS advice, and Blake Parsons and Meera Ramakrishnan for their help with the cebp-1 screen. We also thank Nathalie Pujol for helpful discussions and Kyung Won Kim, and Yishi Jin for sharing unpublished data.
Funding
This work was supported by National Institutes of Health grants F32 AI098307 awarded to DM, R01 AI085581 and P30 DK040561 awarded to FMA, and Glenn Award from the Glenn Foundation for Medical Research to NS.
Availability of data and materials
The microarray dataset supporting the conclusions of this article is available in the National Center for Biotechnology Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo). The accession number for these data is GSE87052. The other datasets supporting the conclusions of this article are included within the article and its additional files.
Authors’ contributions
Conceived and designed the experiments: DLM, RLF, FMA. Performed the experiments: DLM, RLF. Developed the automated P. aeruginosa assays: NS, DM, ALC. Performed the multiplexed quantitative proteomics: WH. Analyzed proteomic and transcriptomic datasets: DLM, AA, RS. Wrote the manuscript: DLM, RLF, FMA. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Abbreviations
- FDR
False Discovery Rate
- GO
Gene Ontology
- ToxA
Exotoxin A
Additional files
Testing whether ToxA-responsive genes defend against ToxA. This table lists the genes that were previously found to be significantly induced by E. coli expressing ToxA [5] and were inhibited either by RNAi or with mutant alleles to determine whether they are necessary for ToxA resistance. (XLSX 15 kb)
The ToxA defect of nipi-3(fr4) is not a result of general strain health. a. Lifespans of nipi-3(fr4) and wild type N2 fed E. coli expressing ToxA or the BL21 control. P < 0.001 comparing nipi-3(fr4) ToxA and BL21; P > 0.05 comparing N2 ToxA and BL21 (log-rank test). b. Fraction of nipi-3(fr4) or wild type N2 animals that grow within 2 or 3 days of being fed OP50 following starvation at room temperature for the indicated time. Results shown are an average of six biological replicates. Error bars represent SD. c. Lifespans of wild type N2 and MGH167 (gut RNAi) animals fed on BL21 control E. coli following either nipi-3 or L4440 vector control RNAi. Number of animals scored for each condition was > 65 (286 total; a) and > 65 (332 total; c). These are representative experiments of four (a), three (c), or two (b) independent experiments. (PDF 435 kb)
Affymetrix microarray analysis of wild type N2, nipi-3(fr4), and pmk-1(km25) gene expression. This table lists the genes differentially expressed in wild type N2 and nipi-3(fr4) (first tab) or pmk-1(km25) (second tab) animals fed control OP50 E. coli at 20 °C until the L4 stage. Expression values are the average of three independent replicates. Gene ontology terms enriched in each category are listed in the third tab. (XLSX 59 kb)
NanoString analysis of wild type N2 and nipi-3(fr4) gene expression. This table lists the gene expression fold changes between wild type N2 and nipi-3(fr4) animals fed P. aeruginosa PA14 or control OP50 E. coli for 6 hours (first tab) or fed E. coli expressing ToxA or BL21 control for 24 hours (second tab). Expression values are the average of three (PA14 and OP50) or two (ToxA and BL21 control) independent replicates and normalized to three control genes. Primary data are provided in Additional file 15. (XLSX 33 kb)
Genes differentially expressed in nipi-3(fr4) are enriched for translational inhibitor- and pathogen-response genes. a. Overlaps between genes differentially expressed in nipi-3(fr4) versus wild type animals fed the indicated food. Numbers provided for major overlap classes. b. Overlap between genes differentially expressed in nipi-3(fr4) versus wild type animals fed control OP50 E. coli and genes induced/repressed by ToxA or hygromycin versus control bacteria in wild type animals [5]. P < 1 × 10–95 (hypergeometric test). c. Overlap between genes differentially expressed in nipi-3(fr4) versus wild type animals fed P. aeruginosa or ToxA and genes induced/repressed by P. aeruginosa or ToxA versus control bacteria in wild type animals P < 5 × 10–6 (hypergeometric test). Data collected from microarray (b) or NanoString (a, c) analyses. Primary data for panels a and c are provided in Additional file 15. (PDF 403 kb)
Lifespan and gene expression analysis of nipi-3(fr4) and mutants of other immune pathways required for the ToxA response. a. qRT-PCR comparison of wild type N2, nipi-3(fr4), pmk-1(km25), zip-2(tm4248), and fshr-1(ok778) animals following exposure to P. aeruginosa PA14 or OP50 E. coli for 6 hours. Results shown are an average of two (fshr-1) or three (remaining samples) biological replicates. b. qRT-PCR comparison of wild type N2, nipi-3(fr4), and kgb-1(km21) animals following exposure to E. coli expressing ToxA or the BL21 control for 24 hours. Results shown are an average of two biological replicates. For a and b, results are normalized to the value of wild type worms on control E. coli for the given gene. Error bars represent SEM. *P < 0.05 (Student’s t-test) when compared to the corresponding wild type animals. c. Lifespans of wild type N2, pmk-1(km25), zip-2(tm4248), and nipi-3(RNAi) fed E. coli expressing ToxA. Number of animals scored for each condition was ≥ 55 (257 total). This is a representative experiment of three independent experiments. Primary data for panels a and b are provided in Additional file 15. (PDF 463 kb)
NanoString analysis of wild type N2 gene expression on P. aeruginosa or ToxA. This table lists the gene expression fold changes between wild type N2 animals fed P. aeruginosa PA14 and animals fed control OP50 E. coli for 6 hours or between N2 animals fed E. coli expressing ToxA and animals fed the BL21 control for 24 hours. Expression values are the average of three (PA14 and OP50) or two (ToxA and BL21 control) independent replicates and normalized to three control genes. Primary data are provided in Additional file 15. (XLSX 22 kb)
ToxA-induced protein changes. This table lists the proteins differentially expressed in wild type N2 (first tab) or nipi-3(fr4) (second tab) animals fed E. coli expressing ToxA or the BL21 control for 24 hours. Relative expression levels are an average of two independent replicates. Primary data are provided (third tab). (XLSX 1584 kb)
Comparing RNA and protein changes in animals exposed to ToxA or control bacteria. Changes of protein and RNA abundances in wild type N2 animals following a 24 hour exposure to E. coli expressing ToxA as compared to animals fed a control BL21 E. coli (top) or in nipi-3(fr4) animals fed control bacteria as compared to similarly treated wild type animals (bottom). Only values with significant protein and/or RNA changes are included. Results shown are an average of two (protein) or three (RNA) biological replicates. Primary data are provided in Additional file 8: Table S5. (PDF 423 kb)
Proteins misregulated in nipi-3(fr4) or pmk-1(km25) as compared to wild type worms. This table lists the proteins differentially expressed in wild type N2 and nipi-3(fr4) animals fed BL21 control bacteria (first tab) or E. coli expressing ToxA (second tab) for 24 hours. This table also lists the proteins differentially expressed in wild type N2 and pmk-1(km25) animals fed E. coli expressing ToxA for 24 hours (third tab). Relative expression levels are an average of two independent replicates. Primary data are provided in Additional file 8. (XLSX 29 kb)
Specificity of tissue-restricted RNAi strains. This table lists the results of RNAi feeding experiments using double stranded RNAs targeting genes expressed in the indicated tissue. Results presented as percentage of affected animals and the total number scored. Synchronized L1 worms were dropped onto the RNAi food and, after 3 days, worms were scored as affected if they developmentally arrested (act-5), twitched in 10 mM levamisole (unc-22), had a detached cuticle (bli-1), low mobility (unc-45), sterility (pos-1) or any of the previous phenotypes (L4440 control). (XLSX 10 kb)
Hypodermal immune genes tpa-1 and hsp-3 are not hypersusceptible to ToxA. Lifespans of tpa-1(k530) and hsp-3(ok1083) fed on E. coli expressing ToxA or the BL21 control. Number of animals scored for each condition was > 45 (280 total). This is a representative experiment of four independent experiments. (PDF 360 kb)
Loss of nipi-3 and pmk-1 or kgb-1 results in a shortened lifespan on control food. Lifespans of pmk-1(km25), pmk-1(km25);nipi-3(RNAi) (a) or kgb-1(km21), kgb-1(km21);nipi-3(RNAi) (b) and nipi-3(RNAi) fed on BL21 control E. coli. Number of animals scored for each condition was ≥ 160 (482 total; a) and ≥ 140 (381 total; b). These are representative experiments of two independent experiments. (PDF 388 kb)
cebp-1 acts in the intestine and affects immune gene expression. a. Lifespans of MGH167 (gut RNAi) animals grown on equal mixtures of cebp-1 RNAi and L4440 vector control; nipi-3 RNAi and L4440 vector control; cebp-1 and nipi-3 RNAi; or L4440 vector control alone to the L4 stage. Animals were then transferred to E. coli expressing ToxA. Note that the mixed nipi-3 RNAi showed less ToxA susceptibility than undiluted nipi-3 RNAi (Fig. S1b). P = 0.95 (log-rank test) and 0.4 (Wilcoxon test) for nipi-3, cebp-1 versus cebp-1 RNAi; P = 0.089 (log-rank test) and 0.0004 (Wilcoxon test) for nipi-3 versus cebp-1 RNA. Number of animals scored for each condition was > 65 (426 total). This is a representative experiment of two independent experiments. b. qRT-PCR comparison of the indicated strains exposed to E. coli expressing ToxA for 24 hours. Results shown are an average of two biological replicates and are normalized to the corresponding wild type ToxA value. Error bars represent SEM. nipi-3 refers to nipi-3(fr4). Primary data for panel b are provided in Additional file 15. (PDF 206 kb)
Primary data for Figs. 1b and 3b, Figures S3a, b, and S7b, and Tables S3 and S4. (XLSX 35 kb)
Contributor Information
Deborah L. McEwan, Email: mcewan@molbio.mgh.harvard.edu
Rhonda L. Feinbaum, Email: rfeinbaum@brandeis.edu
Nicholas Stroustrup, Email: nicholas_stroustrup@hms.harvard.edu.
Wilhelm Haas, Email: whaas@mgh.harvard.edu.
Annie L. Conery, Email: aconery@molbio.mgh.harvard.edu
Anthony Anselmo, Email: anselmo@molbio.mgh.harvard.edu.
Ruslan Sadreyev, Email: sadreyev@molbio.mgh.harvard.edu.
Frederick M. Ausubel, Email: ausubel@molbio.mgh.harvard.edu
References
- 1.Colaço HG, Moita LF. Initiation of innate immune responses by surveillance of homeostasis perturbations. FEBS J. 2016;283(13):2448–57. doi: 10.1111/febs.13730. [DOI] [PubMed] [Google Scholar]
- 2.Pukkila-Worley R, Ausubel FM. Immune defense mechanisms in the Caenorhabditis elegans intestinal epithelium. Curr Opin Immunol. 2012;24(1):3–9. doi: 10.1016/j.coi.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Estes KA, Dunbar TL, Powell JR, Ausubel FM, Troemel ER. bZIP transcription factor zip-2 mediates an early response to Pseudomonas aeruginosa infection in Caenorhabditis elegans. Proc Natl Acad Sci. 2010;107(5):2153–8. doi: 10.1073/pnas.0914643107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Irazoqui JE, Troemel ER, Feinbaum RL, Luhachack LG, Cezairliyan BO, Ausubel FM. Distinct pathogenesis and host responses during infection of C. elegans by P. aeruginosa and S. aureus. PLoS Pathog. 2010;6:e1000982. doi: 10.1371/journal.ppat.1000982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McEwan Deborah L, Kirienko Natalia V, Ausubel FM. Host translational inhibition by Pseudomonas aeruginosa Exotoxin A triggers an immune response in Caenorhabditis elegans. Cell Host Microbe. 2012;11(4):364–74. doi: 10.1016/j.chom.2012.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yates SP, Jørgensen R, Andersen GR, Merrill AR. Stealth and mimicry by deadly bacterial toxins. Trends Biochem Sci. 2006;31(2):123. doi: 10.1016/j.tibs.2005.12.007. [DOI] [PubMed] [Google Scholar]
- 7.Jorgensen R, Purdy AE, Fieldhouse RJ, Kimber MS, Bartlett DH, Merrill AR. Cholix toxin, a novel ADP-ribosylating factor from Vibrio cholerae. J Biol Chem. 2008;283(16):10671–8. doi: 10.1074/jbc.M710008200. [DOI] [PubMed] [Google Scholar]
- 8.Weidle UH, Tiefenthaler G, Schiller C, Weiss EH, Georges GUY, Brinkmann U. Prospects of bacterial and plant protein-based immunotoxins for treatment of cancer. Cancer Genomics Proteomics. 2014;11(1):25–38. [PubMed] [Google Scholar]
- 9.Dunbar Tiffany L, Yan Z, Balla Keir M, Smelkinson Margery G, Troemel ER. C. elegans detects pathogen-induced translational inhibition to activate immune signaling. Cell Host Microbe. 2012;11(4):375–86. doi: 10.1016/j.chom.2012.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fontana MF, Banga S, Barry KC, Shen X, Tan Y, Luo Z-Q, Vance RE. Secreted bacterial effectors that inhibit host protein synthesis are critical for induction of the innate immune response to virulent Legionella pneumophila. PLoS Pathog. 2011;7(2) doi: 10.1371/journal.ppat.1001289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fontana MF, Shin S, Vance RE. Activation of host mitogen-activated protein kinases by secreted Legionella pneumophila effectors that inhibit host protein translation. Infect Immun. 2012;80(10):3570–5. doi: 10.1128/IAI.00557-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Asrat S, Dugan AS, Isberg RR. The frustrated host response to Legionella pneumophila is bypassed by MyD88-dependent translation of pro-inflammatory cytokines. PLoS Pathogens. 2014;10(7) doi: 10.1371/journal.ppat.1004229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ivanov SS, Roy CR. Pathogen signatures activate a ubiquitination pathway that modulates the function of the metabolic checkpoint kinase mTOR. Nat Immunol. 2013;14(12):1219–28. doi: 10.1038/ni.2740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Melo Justine A, Ruvkun G. Inactivation of conserved C. elegans genes engages pathogen- and xenobiotic-associated defenses. Cell. 2012;149(2):452–66. doi: 10.1016/j.cell.2012.02.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stroustrup N, Ulmschneider BE, Nash ZM, Lopez-Moyado IF, Apfeld J, Fontana W. The Caenorhabditis elegans lifespan machine. Nat Methods. 2013;10(7):665–70. doi: 10.1038/nmeth.2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lohan R, Keeshan K. The functionally diverse role of tribbles. Biochem Soc Trans. 2013;41:1096–100. doi: 10.1042/BST20130105. [DOI] [PubMed] [Google Scholar]
- 17.Pujol N, Cypowyj S, Ziegler K, Millet A, Astrain A, Goncharov A, Jin Y, Chisholm AD, Ewbank JJ. Distinct innate immune responses to infection and wounding in the C. elegans epidermis. Curr Biol. 2008;18(7):481–9. doi: 10.1016/j.cub.2008.02.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baugh LR. To grow or not to grow: nutritional control of development during Caenorhabditis elegans L1 arrest. Genetics. 2013;194(3):539–55. doi: 10.1534/genetics.113.150847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lindblom TH, Dodd AK. Xenobiotic detoxification in the nematode Caenorhabditis elegans. J Exp Zool A Comp Exp Biol. 2006;305(9):720–30. doi: 10.1002/jez.a.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shao H, He X, Achnine L, Blount JW, Dixon RA, Wang X. Crystal structures of a multifunctional triterpene/flavonoid glycosyltransferase from Medicago truncatula. Plant Cell. 2005;17(11):3141–54. doi: 10.1105/tpc.105.035055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Spencer WC, Zeller G, Watson JD, Henz SR, Watkins KL, McWhirter RD, Petersen S, Sreedharan VT, Widmer C, Jo J, et al. A spatial and temporal map of C. elegans gene expression. Genome Res. 2011;21(2):325–41. doi: 10.1101/gr.114595.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ziegler K, Kurz CL, Cypowyj S, Couillault C, Pophillat M, Pujol N, Ewbank JJ. Antifungal innate immunity in C. elegans: PKC[delta] links G protein signaling and a conserved p38 MAPK cascade. Cell Host Microbe. 2009;5(4):341. doi: 10.1016/j.chom.2009.03.006. [DOI] [PubMed] [Google Scholar]
- 23.Couillault C, Fourquet P, Pophillat M, Ewbank JJ. A UPR-independent infection-specific role for a BiP/GRP78 protein in the control of antimicrobial peptide expression in C. elegans epidermis. Virulence. 2012;3(3):299–308. doi: 10.4161/viru.20384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wei S-C, Rosenberg IM, Cao Z, Huett AS, Xavier RJ, Podolsky DK. Tribbles 2 (Trib2) is a novel regulator of toll-like receptor 5 signaling. Inflamm Bowel Dis. 2012;18(5):877–88. doi: 10.1002/ibd.22883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim DH, Liberati NT, Mizuno T, Inoue H, Hisamoto N, Matsumoto K, Ausubel FM. Integration of Caenorhabditis elegans MAPK pathways mediating immunity and stress resistance by MEK-1 MAPK kinase and VHP-1 MAPK phosphatase. Proc Natl Acad Sci U S A. 2004;101(30):10990–4. doi: 10.1073/pnas.0403546101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reece-Hoyes JS, Shingles J, Dupuy D, Grove CA, Walhout AJ, Vidal M, Hope IA. Insight into transcription factor gene duplication from Caenorhabditis elegans Promoterome-driven expression patterns. BMC Genomics. 2007;8:27. doi: 10.1186/1471-2164-8-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yan D, Wu Z, Chisholm AD, Jin Y. The DLK-1 Kinase promotes mRNA stability and local translation in C. elegans synapses and axon regeneration. Cell. 2009;138(5):1005–18. doi: 10.1016/j.cell.2009.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Valanne S, Wang J-H, Rämet M. The Drosophila Toll signaling pathway. J Immunol. 2011;186(2):649–56. doi: 10.4049/jimmunol.1002302. [DOI] [PubMed] [Google Scholar]
- 29.Irazoqui JE, Ng A, Xavier RJ, Ausubel FM. Role for β-catenin and HOX transcription factors in Caenorhabditis elegans and mammalian host epithelial-pathogen interactions. Proc Natl Acad Sci U S A. 2008;105(45):17469–74. doi: 10.1073/pnas.0809527105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Caruso M-E, Jenna S, Bouchecareilh M, Baillie DL, Boismenu D, Halawani D, Latterich M, Chevet E. GTPase-mediated regulation of the unfolded protein response in Caenorhabditis elegans is dependent on the AAA+ ATPase CDC-48. Mol Cell Biol. 2008;28(13):4261–74. doi: 10.1128/MCB.02252-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dupuy D, Bertin N, Hidalgo CA, Venkatesan K, Tu D, Lee D, Rosenberg J, Svrzikapa N, Blanc A, Carnec A, et al. Genome-scale analysis of in vivo spatiotemporal promoter activity in Caenorhabditis elegans. Nat Biotech. 2007;25(6):663–8. doi: 10.1038/nbt1305. [DOI] [PubMed] [Google Scholar]
- 32.Dierking K, Polanowska J, Omi S, Engelmann I, Gut M, Lembo F, Ewbank Jonathan J, Pujol N. Unusual regulation of a STAT protein by an SLC6 family transporter in C. elegans epidermal innate immunity. Cell Host Microbe. 2011;9(5):425–35. doi: 10.1016/j.chom.2011.04.011. [DOI] [PubMed] [Google Scholar]
- 33.Bounoutas A, Kratz J, Emtage L, Ma C, Nguyen KC, Chalfie M. Microtubule depolymerization in Caenorhabditis elegans touch receptor neurons reduces gene expression through a p38 MAPK pathway. Proc Natl Acad Sci U S A. 2011;108(10):3982–7. doi: 10.1073/pnas.1101360108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reddy KC, Dunbar TL, Nargund AM, Haynes CM, Troemel ER. The C. elegans CCAAT-enhancer-binding protein gamma is required for surveillance immunity. Cell Reports. 2016;14(7):1581–9. doi: 10.1016/j.celrep.2016.01.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sonenberg N, Hinnebusch AG. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell. 2009;136(4):731–45. doi: 10.1016/j.cell.2009.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vogel C, Marcotte EM. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat Rev Genet. 2012;13(4):227–32. doi: 10.1038/nrg3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Balasubramanian V, Sellegounder D, Suman K, Krishnaswamy B. Proteome analysis reveals translational inhibition of Caenorhabditis elegans enhances susceptibility to Pseudomonas aeruginosa PAO1 pathogenesis. J Proteomics. 2016;145:141–52. doi: 10.1016/j.jprot.2016.04.025. [DOI] [PubMed] [Google Scholar]
- 38.Rahme LG, Stevens EJ, Wolfort SF, Jing S, Tompkins RG, Ausubel FM. Common virulence factors for bacterial pathogenicity in plants and animals. Science. 1995;268(5219):1899–902. doi: 10.1126/science.7604262. [DOI] [PubMed] [Google Scholar]
- 39.Kamath RS, Ahringer J. Genome-wide RNAi screening in Caenorhabditis elegans. Methods. 2003;30(4):313–21. doi: 10.1016/S1046-2023(03)00050-1. [DOI] [PubMed] [Google Scholar]
- 40.Ting L, Rad R, Gygi SP, Haas W. MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics. Nat Meth. 2011;8(11):937–40. doi: 10.1038/nmeth.1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McAlister GC, Nusinow DP, Jedrychowski MP, Wühr M, Huttlin EL, Erickson BK, Rad R, Haas W, Gygi SP. MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal Chem. 2014;86(14):7150–8. doi: 10.1021/ac502040v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Choi H, Fermin D, Nesvizhskii AI. Significance analysis of spectral count data in label-free shotgun proteomics. Mol Cell Proteomics. 2008;7(12):2373–85. doi: 10.1074/mcp.M800203-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McAlister GC, Huttlin EL, Haas W, Ting L, Jedrychowski MP, Rogers JC, Kuhn K, Pike I, Grothe RA, Blethrow JD, et al. Increasing the multiplexing capacity of TMTs using reporter ion isotopologues with isobaric masses. Anal Chem. 2012;84(17):7469–78. doi: 10.1021/ac301572t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tolonen AC, Haas W. Quantitative proteomics using reductive dimethylation for stable isotope labeling. J Vis Exp. 2014;89:51416. doi: 10.3791/51416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Eng JK, McCormack AL, Yates JR. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom. 1994;5(11):976–89. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 46.Huttlin EL, Jedrychowski MP, Elias JE, Goswami T, Rad R, Beausoleil SA, Villén J, Haas W, Sowa ME, Gygi SP. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell. 2010;143(7):1174–89. doi: 10.1016/j.cell.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Elias JE, Gygi SP. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Meth. 2007;4(3):207–14. doi: 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- 48.Gautier L, Cope L, Bolstad BM, Irizarry RA. affy – analysis of Affymetrix GeneChip data at the probe level. Bioinformatics. 2004;20(3):307–15. doi: 10.1093/bioinformatics/btg405. [DOI] [PubMed] [Google Scholar]
- 49.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–40. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Consortium TGO. Gene Ontology Consortium: going forward. Nucleic Acids Res. 2015;43(D1):D1049–56. doi: 10.1093/nar/gku1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene Ontology: tool for the unification of biology. Nat Genet. 2000;25(1):25–9. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jorgensen EM, Mango SE. The art and design of genetic screens: Caenorhabditis elegans. Nat Rev Genet. 2002;3(5):356–69. doi: 10.1038/nrg794. [DOI] [PubMed] [Google Scholar]
- 54.Mizuno T, Hisamoto N, Terada T, Kondo T, Adachi M, Nishida E, Kim DH, Ausubel FM, Matsumoto K. The Caenorhabditis elegans MAPK phosphatase VHP‐1 mediates a novel JNK‐like signaling pathway in stress response. EMBO J. 2004;23(11):2226–34. doi: 10.1038/sj.emboj.7600226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nix P, Hisamoto N, Matsumoto K, Bastiani M. Axon regeneration requires coordinate activation of p38 and JNK MAPK pathways. Proc Natl Acad Sci. 2011;108(26):10738–43. doi: 10.1073/pnas.1104830108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lehrbach NJ, Ruvkun G. Proteasome dysfunction triggers activation of SKN-1A/Nrf1 by the aspartic protease DDI-1. eLife. 2016;5 doi: 10.7554/eLife.17721. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The microarray dataset supporting the conclusions of this article is available in the National Center for Biotechnology Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo). The accession number for these data is GSE87052. The other datasets supporting the conclusions of this article are included within the article and its additional files.