


Abstract
The Schizosaccharomyces pombe pfh1+ gene (PIF1 homolog) encodes an essential enzyme that has both DNA helicase and ATPase activities and is implicated in lagging strand DNA processing. Mutations in the pfh1+ gene suppress a temperature-sensitive allele of cdc24+, which encodes a protein that functions with Schizosaccharomyces pombe Dna2 in Okazaki fragment processing. In this study, we describe the enzymatic properties of the Pfh1 helicase and the genetic interactions between pfh1 and cdc24, dna2, cdc27 or pol 3, all of which are involved in the Okazaki fragment metabolism. We show that a full-length Pfh1 fusion protein is active as a monomer. The helicase activity of Pfh1 displaced only short (<30 bp) duplex DNA regions efficiently in a highly distributive manner and was markedly stimulated by the presence of a replication-fork-like structure in the substrate. The temperature-sensitive phenotype of a dna2-C2 or a cdc24-M38 mutant was suppressed by pfh1-R20 (a cold-sensitive mutant allele of pfh1) and overexpression of wild-type pfh1+ abolished the ability of the pfh1 mutant alleles to suppress dna2-C2 and cdc24-M38. Purified Pfh1-R20 mutant protein displayed significantly reduced ATPase and helicase activities. These results indicate that the simultaneous loss-of-function mutations of pfh1+ and dna2+ (or cdc24+) are essential to restore the growth defect. Our genetic data indicate that the Pfh1 DNA helicase acts in concert with Cdc24 and Dna2 to process single-stranded DNA flaps generated in vivo by pol δ-mediated lagging strand displacement DNA synthesis.
INTRODUCTION
The synthesis and subsequent maturation of Okazaki fragments on the lagging strand template during eukaryotic DNA replication requires the coordinated action of many replication proteins (1–7) and proceeds in the following manner. First, 10–15 nt RNA primers are synthesized, followed by coupled short (20–30 nt) DNA synthesis by the polymerase (pol) α-primase complex (8,9). The RNA–DNA primer is then extended by DNA pol δ to full-length Okazaki fragments (6). The primer RNA can be completely removed by the combined action of RNase HI and Fen1 while the Okazaki fragments remain hybridized to the lagging strand template (5). Recently, it was shown that pol δ displaces the 5′ end region of downstream Okazaki fragments during extension of the upstream Okazaki fragments (10,11). This results in the generation of single-stranded DNA (ssDNA) flaps that can be efficiently processed by the combined action of Dna2 and Fen1 (11). During this processing step, replication protein A (RPA) plays a critical role in regulating the sequential action of Dna2 and Fen1. Dna2 is responsible for the removal of most of the flap, leaving a short (6 nt) flap region that can be removed by Fen1. Fen1 and other nucleases ultimately generate nicks that are sealed by DNA ligase I. Although it is well accepted that Dna2 is an essential nuclease involved in Okazaki fragment processing, it is not clear what fraction of Okazaki fragments is processed in a Dna2-dependent manner. The frequency of the involvement of Dna2 is probably directly related to the size distribution of the flaps generated in vivo by pol δ-catalyzed displacement DNA synthesis.
In addition, other factors (and their homologs in other organisms) such as Werner helicase and Cdc24 from mouse and S.pombe, respectively, are likely to be involved in Okazaki fragment processing by virtue of their functional interactions with Fen1 and Dna2. The roles of these proteins, however, are not clear in Okazaki fragment processing. The Werner helicase, which possesses both 3′ to 5′ helicase and 3′ to 5′ exonuclease activities, greatly stimulates Fen1-mediated cleavage of flap or nicked duplex (12,13). However, neither the helicase nor the exonuclease of the Werner protein is required for this stimulation, which depends on direct protein–protein interactions (13).
In fission yeast, cdc24+ has been implicated in Okazaki fragment processing because of its genetic and physical interactions with dna2+ (14,15). The cdc24 mutation resulted in a similar phenotype to that of a dna2 mutant (14). Overexpression of Dna2 rescued the temperature-sensitive phenotype of the cdc24-G1 mutation (14). Moreover, pfh1 mutations suppressed the temperature sensitivity of the cdc24-M38 mutation and conferred cold sensitivity (17). The pfh1+ gene of S.pombe is homologous to Saccharomyces cerevisiae PIF1 that has dual roles in vivo; it is required for the stable maintenance of mitochondrial DNA (18) and telomere integrity (19,20). In addition, the temperature-dependent lethality of dna2-C2 mutants was suppressed by overexpression of cdc1+ or cdc27+, both of which encode subunits of pol δ (16).
Owing to their complex genetic interactions and lack of biochemical data, it is unclear how the cdc24+, pfh1+, dna2+ and pol δ proteins act in concert during Okazaki fragment processing. Nevertheless, the genetic data suggest a close functional relationship among cdc24+, pfh1+, dna2+ and pol δ. Although Okazaki fragment processing in eukaryotes is highly conserved, the specific protein factors required for processing varies from organism to organism. For example, S.pombe Dna2 displays no detectable DNA helicase activity (unpublished data). Similarly, Xenopus laevis Dna2 has negligible ATPase activity (21). This is in contrast to the observation that the DNA unwinding activity of Dna2 aids its endonuclease activity by resolving any potential secondary structure present in the flap (22). Under these circumstances, it is conceivable that DNA helicase activity is provided by another cellular DNA helicase that interacts with the Dna2 protein. One such candidate is Pfh1. One prediction is that a Dna2-associated helicase, if it exists, may possess biochemical properties similar to those of the S.cerevisiae Dna2 helicase. Alternatively, in vivo displacement of DNA synthesis by S.pombe and X.laevis pol δ may not produce long flaps from the 5′ end regions of Okazaki fragments. If this were the case, these organisms would be less dependent on or entirely independent of a requirement for the DNA helicase activity of Dna2.
Like yeast Dna2, Pfh1 unwinds duplex DNA in the 5′ to 3′ direction in an ATP-dependent fashion (17,20). However, other properties of Pfh1 required to understand its precise function remain unclear. Expression of the full-length Pfh1 protein in Escherichia coli and other systems, including yeasts and insect cells, resulted in insoluble aggregates and extensively degraded protein preparations, which has made it difficult to examine its biochemical properties (16,22) (see also Results). As a part of our ongoing study of Okazaki fragment processing, we have isolated the full-length S.pombe Pfh1 enzyme. We have also analyzed the genetic interactions of pfh1 with the cdc24 and the dna2 genes in order to gain more insight into their function in vivo. In this study, we describe the biochemical properties of Pfh1 and show that it interacts with dna2+ and cdc24+. Loss-of-function mutation of pfh1+ abrogated the requirements for a fully functional dna2+ and cdc24+, indicating that dna2+ (or cdc24+) should remain active as long as pfh1+ is functional. Based on its biochemical properties and genetic interactions determined by this study, the role of pfh1+ in Okazaki fragment processing is discussed.
MATERIALS AND METHODS
Fission yeast strains, media and vectors
The strains of S.pombe used in this study are as follows: the two pfh1 mutant strains 24R20 (h− cdc24-M38 pfh1-R20 leu1-32) and 24R23 (h− cdc24-M38 pfh1-R23 leu1-32) were described previously (17). GH101-2 (h+ pfh1-R20 leu1-32) was isolated from spores obtained from crosses of sp34 (h+ leu1-32 ade6-M216) (16) and H303-12 (h− pfh1-R20 leu1-32) (17). GH102-6 (h− pfh1-R20 dna2-C2 leu1-32) was isolated from spores obtained from crosses of HK10 (h− dna2-C2 leu1-32 ura4-D18) (16) and GH101-2 above. The two cold-sensitive pol δ mutant strains H231-14 (h− pol3-R18 leu1-32) and H369-5 (h− cdc27-R22 leu1-32) were isolated during this study.
Culture media were prepared as described previously (23,24). The pREP vectors used were described previously (25,26). The ΦX174 and M13 ss circular DNAs (sscDNAs) were purchased from New England Biolabs (MA, USA).
Plasmids for overexpression of wild-type and mutant Pfh1 proteins
To insert a FLAG epitope tag at the C-terminus of NusA-Pfh1, PCR was performed using the two oligonucleotides (5′-CTG CAG TCA CTT GTC ATC GTC GTC CTT GTA GTC TGA ACT TCC CTT AAT TTT GAC TCC CCT CAT-3′ and 5′-TCT GTT AAT GGT TTG CCG-3′) and pET43-Pfh1 (17) as a template DNA. The underlined sequence of the longer oligonucleotide encodes the FLAG epitope. The PstI-XbaI fragment of the resulting PCR product was cloned into the pET43-Pfh1 (17) digested with PstI and XbaI to obtain pET43-Pfh1F that expresses NusA-Pfh1 with a FLAG epitope at its C-terminus (NusA-Pfh1-F). To construct an expression vector (pET43-Pfh1F-R20) that produces a Pfh1-R20 mutant protein, a single amino acid change (leucine into serine at amino acid position 458) was introduced into the Pfh1 open reading frame (ORF) in pET43-Pfh1F using the Quick-Change™ Site Directed Mutagenesis Kit (Stratagene).
Purification of the recombinant Pfh1 proteins
Purification of the recombinant Pfh1 proteins was carried out using the same procedures as described previously (17) except that we introduced FLAG-affinity column chromatography and a second round of a Superdex 200 HR 10/30 column chromatography. Briefly, the crude extracts (4.2 mg/ml, 30 ml) were prepared from E.coli BL21 harboring either pET43-Pfh1F or pET43-Pfh1F-R20 as described in (17) and loaded onto a Ni2+-charged HiTrap-chelating column (1 ml; Pharmacia Biotech). The eluted fractions from this column were pooled (1.4 mg/ml, 10 ml) and mixed with 400 μl of anti-FLAG M2 Ab-agarose beads (Sigma) equilibrated with buffer T (25 mM Tris–HCl, pH 7.5, 10% glycerol, 1 mM phenylmethylsulfonyl fluoride, 1 mM benzamidine, 1.25 μg/ml leupeptin, 0.625 μg/ml pepstatin A, 0.02% NP-40 and 500 mM NaCl) and incubated at 4°C for 4 h on a rocking platform. After washing four times with 10-bead volumes of buffer T500, bound proteins were eluted three times by incubation at 4°C for 30 min with 400 μl of buffer T500 containing 0.2 mg/ml of the FLAG peptide. The eluates were pooled and concentrated. Aliquots (0.4 ml) were loaded onto a Superdex 200 HR 10/30 column (24 ml; Pharmacia Biotech). The column was eluted with buffer T500, and the active fractions were loaded again onto a Superdex 200 HR 10/30 column. The eluted fractions were assayed for DNA helicase and ssDNA-dependent ATPase activities (see below). The active fractions (>98% in purity) were stored at −80°C. Protein concentrations were determined by the use of Bradford solution (Bio-Rad, USA) as recommended by the supplier.
Preparation of DNA substrates for helicase and gel mobility shift assays
The oligonucleotides used for the preparation of DNA substrates to examine the DNA unwinding or binding activities of Pfh1 are listed in Table 1 and the substrates were prepared as described previously (11,27). The schematic structures of substrates are shown in each figure, and the oligonucleotides used in each experiment are numbered in a circle.
Table 1. Oligonucleotides used in this study.
| Oligo | Sequence |
|---|---|
| 1 | 5′-CGA ACA ATT CAG CGG CTT TAA CCG GAC GCT CGA CGC CAT TAA TAA TGT TTT C-3′ (52) |
| 2 | 5′-GAA AAC ATT ATT AAT GGC GTC GAG CTA GGC ACA AGG CGA ACT GCT AAC GG-3′ (50) |
| 3 | 5′-TGG GCT CAC GTG GTC GAC GCT GGA GGT GAT CAC CAG ATG ATT GCT AGG CAT GCT TTC CGC AAG AGA ACG GGC GTC TGC GTA CCC GTG CAG-3′ (90) |
| 4 | 5′-CTG CAC GGG TAC GCA GAC GCC-3′ (21) |
| 5 | 5′-TTT TTT TTT TTT TTT TTT TTT TTT TCG GAC GCT CGA CGC CAT TAA TAA TGT TTT C-3′ (55) |
| 6 | 5′-TGA AAA CAT TAT TAA TGG CGT CGA GCG TCC G-3′ (31) |
| 7 | 5′-TGA AAA CAT TAT TAA TGG CGT CGA GCG TCC GTT TTT TTT TTT TTT TTT TTT TTT TT-3′ (56) |
| 8 | 5′-CTG CAC GGG TAC GCA GAC GCC CGT TCT CTT-3′ (30) |
| 9 | 5′-GAT CAC CTC CAG CGT CGA CCA CGT GAG CCC-3′ (30) |
| 10 | 5′-CCG TTA GCA GTT CGC CTT GTG CCT A-3′ (25) |
| 11 | 5′-CGA ACA ATT CAG CGG CTT TAA CCG GAC GCT CGA CGC CAT TAA TAA TGT TTT C-3′ (52) |
| 12 | 5′-TTT TTT TTT TTT TTT TTT TTT CGG ACG CTC GAC GCC ATT AAT AAT GTT TTC-3′ (51) |
| 13 | 5′-TTT TTT TTT TTT TTT TTT TTT CGG CTT TAA CCG GAC GCT CGA CGC CAT TAA TAA TGT TTT C-3′ (61) |
| 14 | 5′-TTT TTT TTT TTT TTT TTT TTT CGA ACA ATT CAG CGG CTT TAA CCG GAC GCT CGA CGC CAT TAA TAA TGT TTT C-3′ (73) |
| 15 | 5′-GCG CAT GTG CGT TCC ATT TGC CGA ACA ATT CAG CGG CTT TAA CCG GAC GCT CGA CGC CAT TAA TAA TGT TTT C-3′ (73) |
| 16 | 5′-CTG CTG CTG CTG CTG CTG CTG CTG CTG CTG CTG CTG CTG CTG CTG CTG GCT CGA CGC CAT TAA TAA TGT TTT C-3′ (73) |
| 17 | 5′-CTC CTC CTC CTC CTC CTC CTC CTC CTC CTC CTC CTC CTC CTC CTC CTC GCT CGA CGC CAT TAA TAA TGT TTT C-3′ (73) |
| 18 | 5′-GCC TGC ACG TGG CGA TCG TT CGA TCG CCA CGT GCA GGC G CTG CTG CTG GCT CGA CGC CAT TAA TAA TGT TTT C-3′ (73) |
| 19 | 5′-GCG GCC TGG CTG CTG CTG CTG CTG CTG CTG CTG CTG CTG CCA GGC CGC GCT CGA CGC CAT TAA TAA TGT TTT C-3′ (73) |
| 20 | 5′-CAG CGT CGA CCA CGT GAG CCC-3′ (21) |
The numbers in parentheses indicate the length of each oligonucleotide. Boldface indicates ribonucleotides.
ATPase and helicase assays
DNA-dependent ATPase activity of Pfh1 was measured in standard reaction mixtures (20 μl) containing 50 mM Tris–HCl (pH 8.5), 50 mM NaCl, 2 mM DTT, 2 mM MgCl2, 0.25 mg/ml BSA, 200 μM cold ATP, 8.25 nM [γ-32P]ATP (>3000 Ci/mmol) and 50 ng of M13 sscDNA when necessary. After incubation at 30°C for 10 min the reaction was stopped with 4 μl of 20 mM EDTA (pH 8.0). An aliquot (2 μl) was spotted onto a polyethyleneimine-cellulose plate (J. T. Baker, USA) and developed in 0.5 M LiCl/1.0 M formic acid. The products were analyzed using a PhosphorImager (Bio-Rad). Helicase activity of Pfh1 was measured in standard reaction mixtures (20 μl) containing 50 mM Tris–HCl (pH 8.5), 100 mM NaCl, 2 mM DTT, 2 mM MgCl2, 2 mM ATP, 0.25 mg/ml BSA and the 5′-32P-labeled partial duplex DNA substrate indicated (15 fmol). Reactions were incubated at 30°C for 10 min and then stopped with 4 μl of 6× stop solution [60 mM EDTA, pH 8.0, 40% (w/v) sucrose, 0.6% SDS, 0.25% bromophenol blue and 0.25% xylene cyanol]. The reaction products were subjected to electrophoresis for 0.5 h at 150 V through a 12% polyacrylamide gel containing 0.1% SDS in 0.5× TBE (45 mM Tris base, 45 mM boric acid and 1 mM EDTA). The gel was dried on DEAE-cellulose paper and autoradiographed. Labeled DNA products were quantified with the use of a PhosphorImager. E.coli SSB and all 8 NTPs used for ATPase and helicase assays were from Amersham Biosciences and Roche Applied Science, respectively. ATP analogs such as adenosine-5′-(γ-thio)triphosphate (ATPγS) and adenosine-5′-(β,γ-imido)triphosphate [App(NH)p] were obtained from Sigma–Aldrich.
Gel mobility shift assay
DNA-binding activity was measured in reaction mixtures (20 μl) containing 50 mM Tris–HCl (pH 8.5), 100 mM NaCl, 2 mM DTT, 2 mM MgCl2, 0.25 mg/ml BSA and the 5′-32P-labeled partial duplex DNA substrate (15 fmol) in the presence and absence of 2 mM ATP. Reactions were incubated at 30°C for 10 min. After addition of 4 μl of 6× loading dye (50% glycerol and 0.25% bromophenol blue), aliquots of reaction mixtures were electrophoresed for 1 h at 100 V through a 5% polyacrylamide gel in 0.5× TBE. The gel was dried on DEAE-cellulose paper and autoradiographed. Labeled DNA products were quantified with the use of a PhosphorImager.
RESULTS
Pfh1 contains DNA-dependent ATPase and helicase activities
Difficulties in isolating an intact Pfh1 protein hampered the precise characterization of its helicase and ATPase activities. For example, a substantial (>90%) fraction of the NusA-Pfh1 protein eluted in the void volume during the gel filtration step in our previous purification efforts (17), and the helicase and ATPase activities in this fraction were much lower than expected from the amount of protein present (data not shown). Even the Pfh1 protein that was included in this step displayed variable levels of helicase and ATPase activities from preparation to preparation (data not shown). In order to increase the yield of active enzyme, we improved the purification procedure by adding a FLAG epitope to the C-terminus of NusA-Pfh1 to produce NusA-Pfh1-F. The expression of this recombinant protein in S.pombe complemented the deletion of the pfh1+ gene (data not shown). The NusA-Pfh1-F protein was purified using Ni2+-nitriloacetic acid (NTA) agarose, anti-FLAG M2 Ab-agarose, and two successive rounds of Superdex 200 HR columns as described in Materials and Methods. As shown in Figure 1, the elution of the ATPase and helicase activities were not precisely coincidental with the amount of protein present in the fractions obtained during the second gel filtration step. Although the peak activities of ATPase and helicase were coincidental with the protein peak at fractions 18, 19 and 20 (5.1, 12.1 and 4.7 μg/ml, respectively), fractions eluted earlier (fractions 14–16) were devoid of detectable helicase and ATPase activities despite the presence of substantial amount of protein (compare fractions 18–20 with fractions 14–17). To unambiguously demonstrate that the ATPase and helicase activities detected in the peak fractions (fractions 18–20) were intrinsic to Pfh1, we examined whether these activities in the pooled peak fractions could be simultaneously depleted by anti-FLAG M2 agarose beads (Figure 2). Prior to this experiment, we examined whether the presence of the NusA protein tag inhibited the enzymatic activities associated with Pfh1. To this end, the NusA-Pfh1-F enzyme was digested with thrombin (Figure 2A). Thrombin cleavage resulted in a small decrease in ATPase and helicase activities (Figure 2B and C), indicating that the NusA tag does not interfere with the enzymatic activities of Pfh1 in vitro. This is in keeping with the observation that the NusA-Pfh1-F complemented the deletion of pfh1+ in vivo. We used the proteolyzed enzyme preparation for subsequent immunodepletion experiments. The addition of anti-FLAG M2 agarose beads depleted both ATPase (Figure 2D) and helicase (Figure 2E) activities efficiently and to the same extent, whereas the addition of buffer alone or unrelated protein G resin depleted neither of them (Figure 2D and E). These results demonstrate that both activities are intrinsic to the purified Pfh1 protein. Since the NusA moiety fused to Pfh1 did not affect any of its activities in vitro or its in vivo function, we used the fusion protein to further characterize this enzyme.
Figure 1.
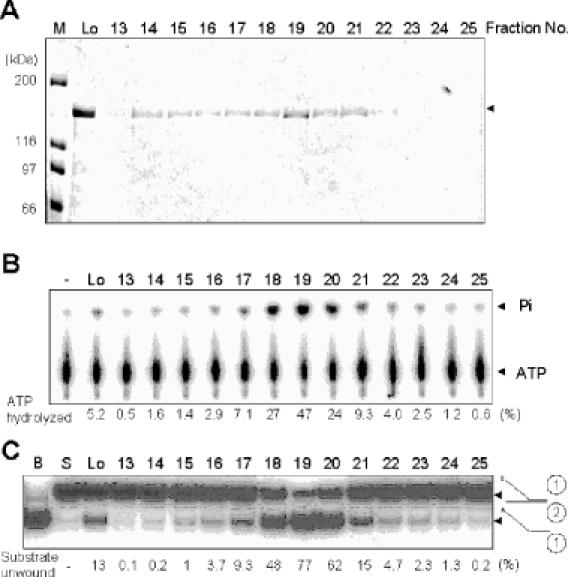
Co-migration of the ATPase and helicase activities with the NusA-Pfh1-F protein. Peak fractions obtained from the first Superdex 200 column were subjected to a second round of Superdex 200 column chromatography. The resulting fractions were analyzed for proteins and enzymatic activities. Fractions analyzed are indicated at the top of each figure. (A) Aliquots (40 μl) of each fraction as indicated were subjected to 8% SDS–PAGE and the gel was stained with Coomassie blue. Lo, load-on of the second round of Superdex 200 column chromatography. M denoted the size (in kDa) of molecular weight marker proteins. The NusA-Pfh1-F protein eluted is as indicated by an arrowhead. (B) The ssDNA-dependent ATPase activity of NusA-Pfh1-F was measured using the standard reaction mixtures as described in Materials and Methods. An aliquot (2 μl) of each fraction was used to measure ATPase activity. The reaction products were analyzed by thin layer chromatography as described in Materials and Methods. The extents of ATP hydrolysis were measured by determining the relative ratios of radioactivities of inorganic 32P-phosphate to uncleaved [γ-32P]ATP and were indicated (as percentage of total ATP) at the bottom of the figure. (C) Helicase activity of NusA-Pfh1-F was measured using the reaction mixtures (20 μl) containing 30 fmol of the indicated DNA substrate as described in Materials and Methods. An aliquot (0.2 μl) of each fraction was used for helicase assays. B denotes boiled substrate. The schematic structure of the substrate used is shown at the right-hand side of the figure. The asterisk in the substrate indicates the position of the 32P-labeled end.
Figure 2.
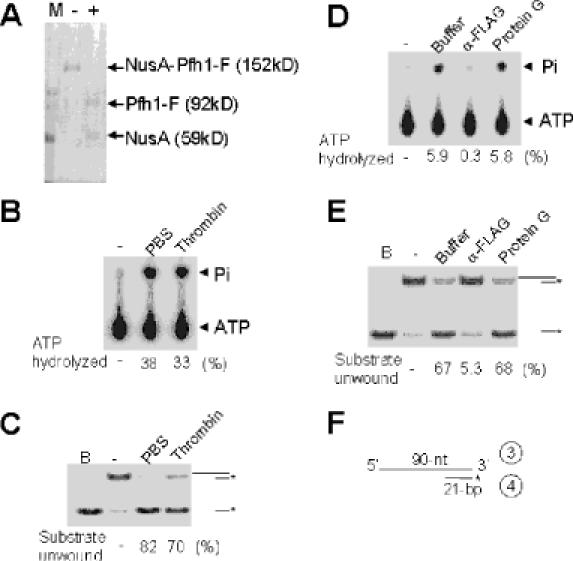
The helicase and ATPase activities are intrinsic to Pfh1. (A) NusA-Pfh1-F (10 μg, 200 μl of the peak fraction obtained from the first Superdex 200 column) was incubated with PBS (−) or 5 U of thrombin (+) at 4°C for 10 h. The input protein and digested products were as indicated. (B) DNA-dependent ATPase activities of PBS-treated (PBS) and thrombin-digested (Thrombin) NusA-Pfh1-F proteins (2 μl, 100 ng each) were measured using reaction conditions described in Materials and Methods. (C) Helicase activities of PBS-treated and thrombin-digested NusA-Pfh1-F proteins [0.2 μl each from (A)] were measured using 15 fmol of the indicated DNA substrate as described in Materials and Methods. The products displaced were analyzed on a 10% polyacrylamide gel. The schematic structures of the DNA substrates used are shown at the right-hand side of the figure. The asterisk in the substrate indicates the position of the 32P-labeled end. B denotes the boiled substrate control. (D) and (E) Aliquots (50 μl, 2.5 μg total) of thrombin-digested NusA-Pfh1-F were mixed with 50 μl containing either 10 μl of either anti-FLAG M2 agarose resin (α-FLAG) or protein G agarose resin (protein G) and the mixtures were incubated at 4°C for 4 h to allow formation of immunocomplexes. Buffer indicates immunodepletion without resin. After the resins were spun down, an aliquot (1 μl, 25 ng) of the supernatant was examined for ATPase (D) or helicase (E) activities. B denotes boiled substrate. For each experiment, the amount of ATP hydrolyzed or substrate unwound are indicated as percentage of input substrate at the bottom of each figure. (F) The schematic structure of the DNA substrate used is shown.
Hydrodynamic properties of NusA-Pfh1-F
Exclusion chromatography and glycerol gradient sedimentation were used to determine the hydrodynamic properties of NusA-Pfh1-F. These experiments were carried out with the pooled fractions (fractions 18–20) obtained from the second sizing column. NusA-Pfh1-F eluted before ferritin (440 kDa, 61 Å) during the gel filtration step and yielded a Stokes radius of 63.5 Å (Figure 3A). As shown in Figure 3B, NusA-Pfh1-F sedimented with a sedimentation coefficient of 6.2S during glycerol gradient centrifugation. Assuming a partial specific volume of 0.725 ml/g, NusA-Pfh1-F was calculated to have a native molecular mass of 163 kDa and frictional ratio of 1.76 (Figure 3C) by the method of Siegel and Monty (28). Since the calculated molecular weight of NusA-Pfh1-F from its amino acid sequence is 152 kDa, its hydrodynamic properties indicate that NusA-Pfh1-F is a monomer and possesses a highly prolate shape. The hydrodynamic properties also suggest that our preparation of NusA-Pfh1-F was properly folded.
Figure 3.
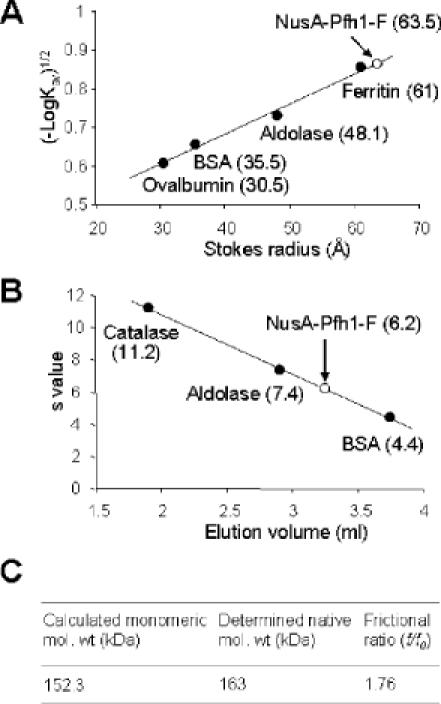
NusA-Pfh1-F is active as a monomer. NusA-Pfh1-F was subjected to gel filtration column chromatography [(A), Superdex 200] and glycerol gradient centrifugation [(B) 15–35% glycerol in 5 ml of buffer T500, 45 000 r.p.m. in a Beckman SW55 rotor]. The size markers used for gel filtration were ferritin (440 kDa, 61 Å), aldolase (158 kDa, 48.1 Å), BSA (66 kDa, 35.5 Å) and ovalbumin (43 kDa, 30.5 Å). The marker proteins used for glycerol gradient were catalase (11.2S), aldolase (7.4S) and BSA (4.4S). The determined Stokes radius and sedimentation values of NusA-Pfh1-F determined were shown in (A) and (B), respectively. (C) By using the sedimentation coefficient and stokes radius, the native molecular weight (163 kDa) and the frictional ratio (f/f0, 1.76) of NusA-Pfh1-F are calculated as described by Siegel and Monty (28).
Examination of the substrate specificity of Pfh1 helicase activity
We examined whether fork-like structures stimulated Pfh1 helicase activity using two different substrates: one contained a 5′ overhang with a 5′ 25 nt ssDNA region, while the other was a Y-structured substrate possessing both 5′ and 3′ 25 nt ssDNA overhangs (Figure 4A). The unwinding of the Y-structured substrate in the presence of 2.4 and 7.2 fmol of enzyme was substantially more efficient (20- and 13-fold, respectively) than that observed with the 5′ overhang substrate (Figure 4A and C). Next, we examined whether the enzyme could unwind a duplex DNA devoid of an ssDNA 5′ tail. For this purpose, two additional substrates were prepared; one with a 5′ 60 nt ssDNA overhang and a 30 bp duplex at the 3′ end, and the other a 30 nt ssDNA flanked by 30 bp duplexes at both ends (Figure 4B). Pfh1 utilized both substrates with almost identical efficiency (Figure 4B and D). This result indicates that Pfh1 can enter an internal ssDNA region and does not depend on a 5′ free end as an entry site, provided that the size of the internal ssDNA is longer than 30 nt. We further confirmed our observation that a fork-like structure stimulated the Pfh1 helicase activity using several ΦX174-based substrates; all substrates contained the same partial duplex but varied in the length of their 5′ tail. The unwinding efficiency of Pfh1 increased in proportion to the 5′ tail length (data not shown); when blunt or short (5 nt) 5′-tailed substrates were used, unwinding was marginal. However, the levels of unwound product formed increased greatly when the length of the 5′-ssDNA tail was >10 nt. Unwinding was stimulated further by increasing the tail length to >21 nt. Further increase in the 5′ tail length up to 40 nt did not stimulate the unwinding activity of Pfh1 (data not shown). These results are consistent with the result described above that unwinding is increased by the presence of a 5′ fork structure and indicate that a 5′ ssDNA region of at least 10 nt is required to stimulate Pfh1 helicase activity.
Figure 4.
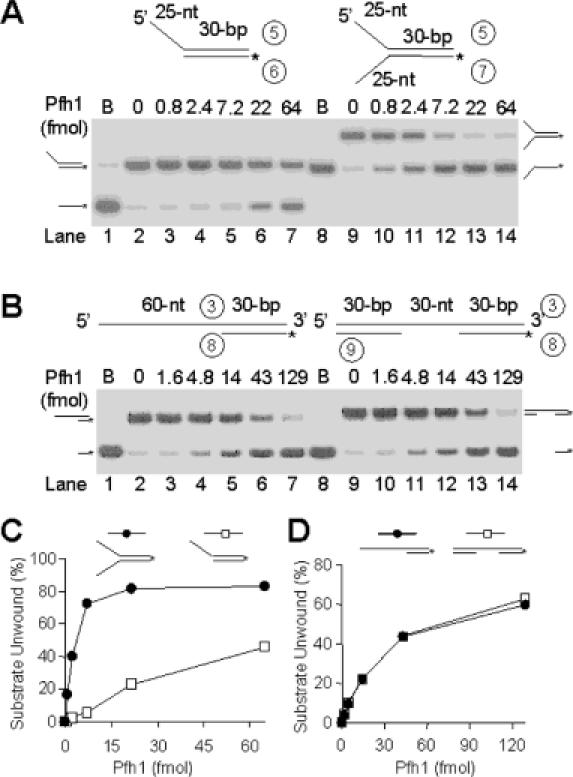
The substrate specificity of Pfh1 helicase. The schematic structures of DNA substrates used are shown at the top of the figure. The asterisks indicate the position of 32P-labels. (A) Pfh1 helicase activity is stimulated by a fork structure in the substrate. The 5′ overhang and Y-structure partial duplex DNA substrates were as indicated. Both substrates contained 30 bp duplex with an identical nucleotide sequence. Increasing levels of NusA-Pfh1-F (0, 0.8, 2.4, 7.2, 22 and 64 fmol) were incubated with 15 fmol of either the 5′ overhang or fork DNA substrate in standard reaction mixtures (see Materials and Methods) at 30°C for 10 min, and the products were analyzed on 10% polyacrylamide gel. B denotes the boiled substrate control. The positions where the substrate and unwound products migrated are as indicated. (B) Pfh1 helicase activity is not dependent on a 5′ free ssDNA end as an entry site. The increasing amounts of NusA-Pfh1-F (0, 1.6, 4.8, 14, 43 and 129 fmol) were incubated with 15 fmol of each DNA substrate in a 20 μl standard reaction mixture at 30°C for 20 min. (C) and (D) Quantification of results obtained in (A) and (B), respectively. The amounts of unwound products (as a percentage of total substrate) were plotted against the amount of enzyme used.
Since the helicase activity of Pfh1 is implicated in Okazaki fragment processing, we examined its ability to displace a flap structure containing an RNA segment in the unannealed 5′ tail, a likely structure formed during Okazaki fragment extension. Since Pfh1 cannot hydrolyze ATP in the presence of RNA as a cofactor, one possible prediction was that the 5′ end of an ssRNA would inhibit the unwinding activity of Pfh1. To test this prediction, we prepared two substrates; one with an RNA–DNA chimeric flap that contained a 12 nt RNA and 15 nt DNA segment, and the other a flap that consisted solely of DNA (Figure 5A). Contrary to the prediction, the RNA–DNA chimeric flap substrate was unwound as efficiently as the DNA only substrate (Figure 5A and B). This finding suggests that when the primer RNA–DNA of Okazaki fragments is converted into a ssRNA–DNA flap by the displacement DNA synthesis by pol δ, it would not interfere with the helicase activity of Pfh1.
Figure 5.
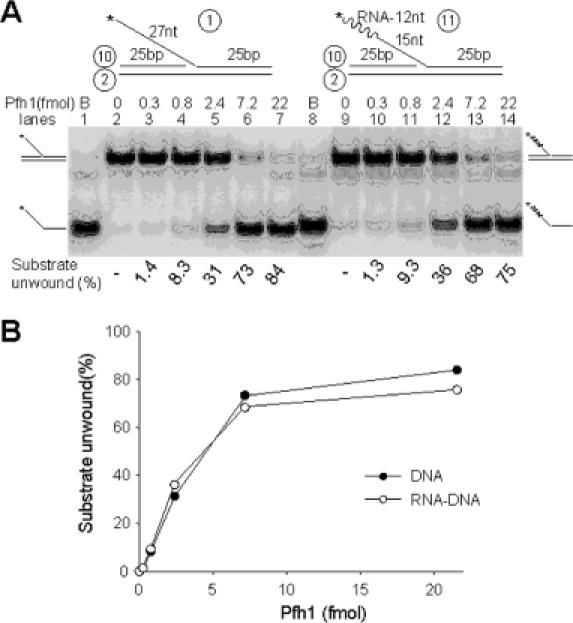
The terminal RNA moiety present in a 5′ flap does not interfere with Pfh1 helicase activity. (A) The two flap-structured substrates are shown at the top of the figure. The circled numbers indicate the oligonucleotides listed in Table 1. The asterisks indicate the position of 32P-labels. Both substrates contained a 5′ flap identical in length and nucleotide sequence except that one consisted of a 12 nt RNA (wavy lines) and a 15 nt DNA segment (thin lines), while the other consisted solely of DNA. Increasing levels (0, 0.3, 0.8, 2.4, 7.2 and 22 fmol) of NusA-Pfh1-F were incubated at 30°C for 5 min with 15 fmol of the indicated substrates in standard reaction mixtures (20 μl) containing 100 mM NaCl, and the products were analyzed on 10% polyacrylamide gel. B denotes boiled substrate control. The migration positions of the substrate and unwound products are as indicated. (B) The amount of each substrate unwound in (A) are plotted against the level of Pfh1 added. Closed and open circles denote results obtained with the substrate containing the DNA only or RNA–DNA chimeric flap, respectively.
Processivity of Pfh1 helicase activity
To determine the maximum length of duplex DNA that can be unwound by Pfh1, three different DNA substrates were prepared that contained 30, 40 and 52 bp partial duplexes, but the same 5′ ssDNA tail length (21 nt). As shown in Figure 6A, Pfh1 unwound all three substrates, but the rate of unwinding decreased dramatically as the length of duplex DNA increased up to 40 bp. During the 20 min incubation period, the 30 bp duplex DNA was displaced efficiently by Pfh1, while the 40 and 52 bp duplex DNAs were poorly unwound (Figure 6A). We also examined the processivity of the Pfh1 helicase activity by carrying out substrate-challenge experiments in which an excess of unlabeled DNA substrate was added at different time points (0, 2 and 5 min) after initiation of the unwinding reaction (Figure 6B). The unwinding of the helicase substrate was blocked immediately by the addition of excess (50-fold) cold competitor DNA at any time point after initiation (Figure 6B). The addition of increasing levels of competitor DNA also inhibited the unwinding reaction in a dose-dependent manner (data not shown). These results demonstrate that the Pfh1 helicase has the ability to unwind a limited duplex length in a highly distributive manner.
Figure 6.
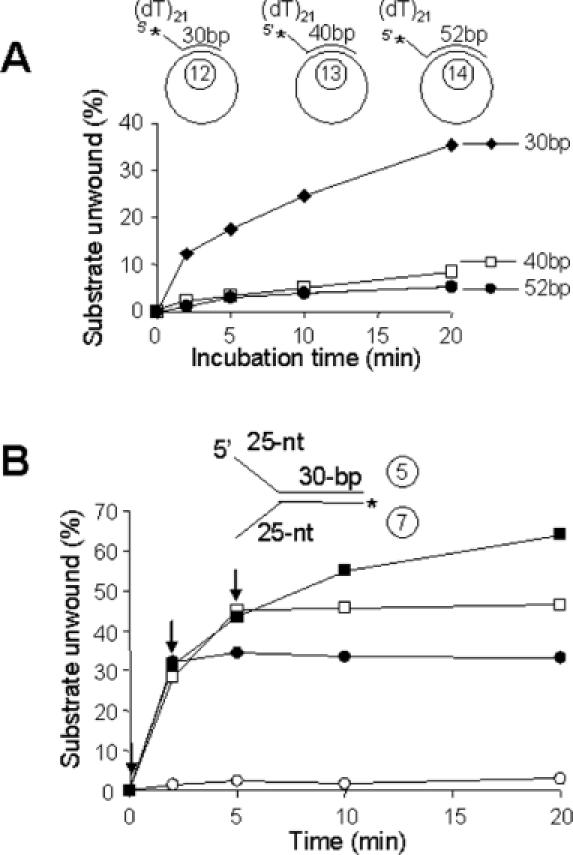
Pfh1 helicase unwinds short duplex DNA in a distributive manner. (A) The oligonucleotides indicated were annealed to ΦX174 sscDNA to construct the 30, 40 or 52 bp partial duplex substrates as shown. The reaction mixtures (200 μl) were pre-incubated at 30°C for 2 min with 150 fmol of each substrate, and the reactions were initiated by the addition of Pfh1 (105 fmol/20 μl reaction). Aliquots (20 μl) were withdrawn at each time points as indicated and the products were analyzed on a 10% polyacrylamide gel. Note that higher levels of Pfh1 were used in this experiment in order to overcome sequestration of Pfh1 by the lengthy ΦX174 ssDNA. (B) Substrate-challenge experiments. Four reaction mixtures (200 μl) containing NusA-Pfh1-F (23 ng, 150 fmol) and the Y-structured DNA substrate (150 fmol) as indicated were assembled on ice and preincubated at 30°C for 3 min in the absence of ATP. ATP (2 mM) was added to each reaction mixture to initiate the unwinding reaction (time 0). Unlabeled substrate DNA (50-fold molar excess, 7.5 pmol/200 μl reaction) was added to each reaction mixture at 0 min (open squares), 2 min (closed circles), 5 min (open squares) after the initiation of reaction (arrows). Aliquots (20 μl) were withdrawn at each time point, and the products were analyzed on a 10% polyacrylamide gel. Closed squares, the amount of substrate unwound in the absence of the competitor DNA.
Pfh1 efficiently can displace a secondary structure-containing flap from a template
Since the Pfh1 helicase is able to unwind a 5′-tailed substrate preferentially and is stimulated by a fork structure (Figures 4 and 5), we examined the influence of the nature of 3′ overhang strand. To this end, we investigated whether Pfh1 could unwind a 5′-tailed flap DNA that consists of 3′ duplex DNA, and found that the enzyme unwound both Y-structured DNA and flap DNA substrates with equal efficiency (data not shown). This finding coupled with the ability of Pfh1 to be loaded onto internal ssDNA regions (Figure 4B) prompted us to examine whether Pfh1 could unwind a flap DNA that contains a secondary structure in which the 5′ ssDNA end was not available to the Pfh1 enzyme. As shown in Figure 7A, the two 48 nt flap DNA substrates (one with random sequences and the other with 16 repeats of CTC) were displaced with almost identical efficiency at various levels of enzyme (Figure 7A, lanes 1–5 and 11–15, respectively; and Figure 7C). Interestingly, the substrate with a flap containing 16 repeats of CTG that can anneal to itself and form a hairpin (29) was also unwound efficiently using Pfh1 (Figure 7A, lanes 6–10 and B). Because this substrate is likely to contain an ssDNA gap varying in size between the hairpin and the duplex junction, unwinding can occur via loading onto the available ssDNA gap. Alternatively, Pfh1 could enter directly through the duplex hairpin. In order to distinguish between these possibilities, we prepared two additional substrates; one with a fixed 10 nt gap and the other lacking a gap (Figure 7B). The unwinding reaction occurred efficiently with the substrate containing the 10 nt gap (Figure 7B, lanes 8–14 and D), but poorly with the substrate devoid of a gap between the hairpin and the junction (Figure 7B, lanes 14–19 and D).
Figure 7.
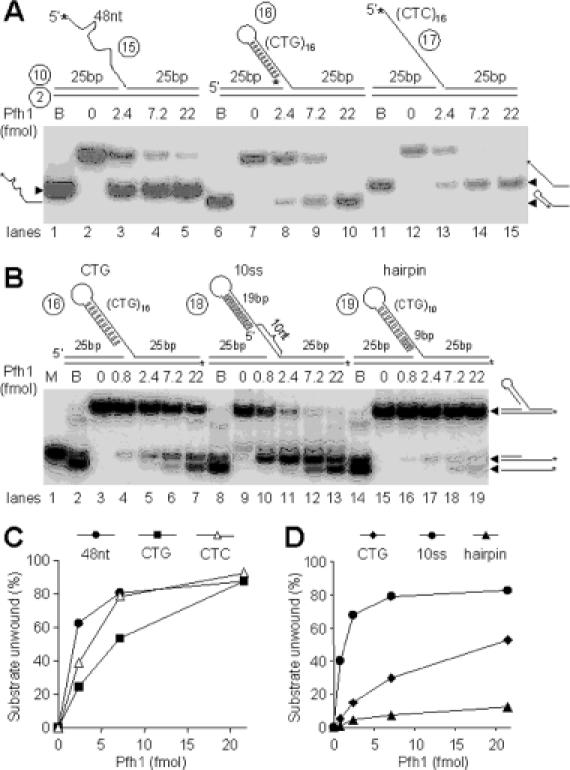
Pfh1 helicase can efficiently displace the hairpin-containing flap strand from DNA substrates. The schematic structures of substrates and oligonucleotides used are shown at the top of each figure. (A) The flap contains either 48 nt random sequence, (CTG)16, or (CTC)16 repeat at the 5′ tail. The asterisks indicate 32P-labeled ends. Increasing levels (0, 2.4, 7.2 and 22 fmol) of NusA-Pfh1-F in a 20 μl reaction mixture (see Materials and Methods) containing 100 mM NaCl were incubated with 15 fmol of each DNA substrate at 30°C for 10 min, and the products were analyzed on 10% polyacrylamide gel. B denotes boiled substrate controls. (B) The flap DNA substrates with a 10 nt or no gap between the hairpin and the junction are shown along with the (CTG)16 flap substrate at the top of the figure. The asterisks indicate 32P-labeled ends. Increasing amounts of Pfh1 (0, 0.8, 2.4, 7.2 and 22 fmol) in a 20 μl reaction mixture containing 100 mM NaCl were incubated at 30°C for 10 min with 15 fmol of each DNA substrate indicated at the top of figure, and the products were analyzed on 10% polyacrylamide gel. B and M denotes boiled substrate controls and size marker for intermediate substrate, respectively. (C) Quantification of the amount of substrate unwound in (A) as a function of Pfh1 added. (D) Quantification of the levels of the two products formed in (B) as a function of Pfh1 added.
Requirements of helicase and ATPase activities of Pfh1
Helicases use NTPs as their source of energy for translocation and unwinding. As expected, the Pfh1-catalyzed unwinding was absolutely dependent on ATP and Mg2+ and little or no activity was observed in the presence of non-hydrolyzable ATP analogs, such as ADP (<5%), ATPγS (<1%) and AppNp (<1%) (Table 2). The optimal salt concentration required for the reaction was 50–100 mM NaCl. Helicase activity was inhibited 85 and >99% upon addition of 0.2 and 0.3 M NaCl, respectively (Table 2). In the presence of optimal levels of Mg2+ (2 mM), unwinding occurred most efficiently in the presence of 0.8–2 mM ATP. Among the eight NTPs, ATP and dATP supported unwinding activity with equal efficiency (Table 2). CTP and dCTP supported unwinding activity to a limited extent (50 and 58% of ATP at 2 mM) (Table 2). However, this finding is not consistent with our observations that CTP and dCTP were not hydrolyzed efficiently by Pfh1 (Table 3), raising the possibility that the unwinding activity observed with CTP and dCTP may be due to the presence of contaminating levels of ATP or dATP. Therefore, Pfh1 appears to utilize ATP and dATP only as the energy source for unwinding duplex DNA. We also investigated the enfluence of ssDNA-binding proteins from various sources (S.pombe, human, E.coli and bacteriophage T4) on the unwinding activity of Pfh1. Pfh1 was significantly inhibited only at the highest levels (80 fmol) of S.pombe RPA. Human RPA, E.coli SSB or T4g32 hardly affected the unwinding activity of Pfh1 (Table 2).
Table 2. Requirements for helicase activity of Pfh1.
| Additions or omissions | Amount added (final concentration) | Relative activity (%)a |
|---|---|---|
| Completeb | 100 | |
| Add NaCl | 0.01, 0.05, 0.1, 0.2, 0.3 M | 24, 100, 117, 15, <1 |
| Add MgCl2 | 0, 0.5, 1, 2, 5,10 mM | <1, 31, 63, 100, 63, 30 |
| Add S.pombe RPA | 5, 20, 80 fmol | 91, 51, 19 |
| Add human RPA | 5, 20, 80 fmol | 99, 88, 39 |
| Add E.coli SSB | 5, 20, 80 fmol | 99, 98, 67 |
| Add T4g32 | 5, 20, 80 fmol | 101, 99, 102 |
| Omit ATP | <1 | |
| Add ATP | 0.08, 0.2, 0.8, 2, 4 mM | 45, 72, 113, 100, 62 |
| Add GTP | 0.2, 2 mM | <1, 2.2 |
| Add UTP | 0.2, 2 mM | <1, 8.8 |
| Add CTP | 0.2, 2 mM | 3.8, 50 |
| Add dATP | 0.2, 2 mM | 98, 112 |
| Add dGTP | 0.2, 2 mM | 1.5, 3.3 |
| Add dTTP | 0.2, 2 mM | 1.4, 5.7 |
| Add dCTP | 0.2, 2 mM | 3.9, 58 |
| Add ADP | 2 mM | 5 |
| Add GDP | 2 mM | 1 |
| Add ATPγS | 0.8, 4 mM | <1, <1 |
| Add App(NH)p | 0.8, 4 mM | <1, <1 |
aThe value of 100% in this experiment corresponded to 8.2 fmol of the DNA substrate unwound.
bThe complete reaction contained 15 fmol of the Y-structure DNA substrate used in Figure 4A and 50 fmol of NusA-Pfh1-F in the standard reaction mixture described in Materials and Methods.
Table 3. Requirements for ATPase activity of Pfh1.
| Additions or omissions | Amount added (final concentrations) | Relative activity (%)a |
|---|---|---|
| Completeb | 100 | |
| Add ATPγS | 0.1, 0.2, 0.4, 1 mM | 91, 91, 81, 62 |
| Add App(NH)p | 0.2, 0.4, 1, 2 mM | 95, 89, 89, 67 |
| Add NaCl | 0.01, 0.025, 0.05, 0.1, 0.2, 0.3 M | 82, 96, 100, 99, 49, 9 |
| Add MgCl2 | 0, 0.05, 0.2, 0.5, 1, 2, 5 mM | 0, 18, 74, 96, 101, 100, 84 |
| Omit MgCl2 | <1 | |
| Add MnCl2 | 0.05, 0.2, 2 mM | 31, 115, 11 |
| Add CaCl2 | 0.05, 0.2, 2 mM | 6, 27, 40 |
| Add CoCl2 | 0.05, 0.2, 2 mM | 10, 4, 6 |
| Omit ATP | ||
| Add GTP | 200 μM | 2.5 |
| Add UTP | 200 μM | 1.0 |
| Add CTP | 200 μM | 1.7 |
| Add dATP | 200 μM | 95 |
| Add dGTP | 200 μM | 3.0 |
| Add dTTP | 200 μM | 0.8 |
| Add dCTP | 200 μM | 2.0 |
| Omit M13ssDNA | <1 | |
| Add M13ssDNA | 1, 5, 50 ng | 41, 89, 100 |
| Add Φ×174ssDNA | 1, 5, 50 ng | 50, 89, 94 |
| Add Φ×174dsDNA | 1, 5, 50 ng | 8, 21, 53 |
| Add Poly(U) | 1, 5, 50 ng | <1 |
aThe value of 100% in this experiment corresponded to the hydrolysis of 2100 pmol of ATP.
bThe complete reaction contained 50 fmol of NusA-Pfh1-F in the standard reaction mixture described in Materials and Methods.
The DNA-dependent ATPase activity was further characterized by using the standard reaction conditions described in Materials and Methods. As observed in the DNA helicase assay, ATP hydrolysis required Mg2+ (Table 3), and 0.2 mM Mn2+ efficiently supported ATP hydrolysis while Ca2+ (>0.2 mM) partially supported ATP hydrolysis. In the presence of 0.2 mM ATP, ATP hydrolysis was not competitively inhibited by the addition of non-hydrolyzable ATP analogs, such as ATPγS (up to 0.4 mM) or App(NH)p (up to 1 mM) (Table 3). Consistent with the effects of NaCl on unwinding (Table 2), DNA-dependent ATPase activity was inhibited to a similar extent at high concentrations of NaCl (51% at 0.2 M and 91% at 0.3 M NaCl) (Table 3). The ssDNA-dependent ATPase activity was scarcely affected by the addition of S.pombe RPA and E.coli SSB (data not shown). We also examined the hydrolysis of other nucleotides by Pfh1. The helicase hydrolyzed only dATP efficiently (Table 3), in agreement with its ability to support the displacement reaction (Table 2). Several polynucleotides were also examined for their ability to support ATP hydrolysis under standard reaction conditions. Without DNA, the enzyme was virtually inactive (<1% ATP hydrolyzed). M13 and Φx174 sscDNA supported ATP hydrolysis most effectively. Φx174 dsDNA was active, but less efficient (53% of M13 sscDNA at 50 ng) (Table 3). Homopolymeric RNA such as poly(U) did not support ATPase activity.
Expression of wild-type pfh1+ abolishes the ability of pfh1-R20 or pfh1-R23 mutant alleles to suppress the growth defect of cdc24-M38 mutation
The pfh1-R20 and pfh1-R23 alleles contain a point mutation in conserved amino acids, but not within the conserved helicase motifs (17). The cdc24-M38 allele produces a C-terminally truncated 369 amino acid protein (14,15,17). Therefore, it is possible that these pfh1 and cdc24 mutant alleles result in a partial loss of function. If the loss-of-function of pfh1+ is essential to suppress the temperature-sensitive phenotype of cdc24-M38, the expression of wild-type pfh1+ is predicted to restore the temperature-sensitive phenotype of cdc24-M38 pfh1-R20 and cdc24-M38 pfh1-R23 mutants. To test this prediction, we introduced wild-type pfh1+ in a multicopy plasmid into cdc24-M38 pfh1-R20 or cdc24-M38 pfh1-R23 double mutant strains and examined the resulting phenotypes. As shown in Figure 8, the introduction of the control plasmid (pREP81x) did not affect the phenotypes of cdc24-M38 pfh1-R20 or cdc24-M38 pfh1-R23 double mutants. In contrast, expression of pfh1+ (pREP81x-pfh1wt) abolished the growth of the double mutant cells at 37°C. The cold-sensitive growth defect caused by both pfh1-R20 and pfh1-R23 mutations was suppressed by pREP81x-pfh1wt, indicating that pfh1+ complemented these mutations. Consistent with these results, genetic analysis indicates that both pfh1-R20 and pfh1-R23 are recessive alleles (unpublished data). In conclusion, our above results confirm that suppression of the temperature-sensitivity of cdc24-M38 requires the impairment of the in vivo function of pfh1+.
Figure 8.
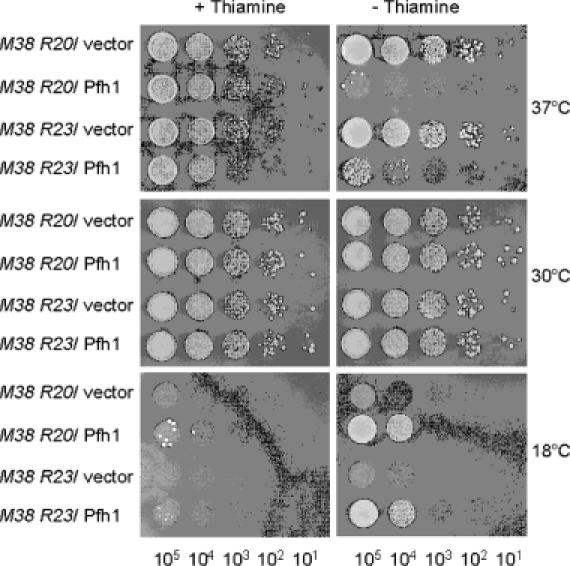
Expression of wild-type pfh1+ abolishes the ability of pfh1-R20 or pfh1-R23 to suppress the temperature-sensitive growth defect of cdc24-M38. The cdc24-M38 pfh1-R20 (M38 R20) or the cdc24-M38 pfh1-R23 (M38 R23) double mutant cells (strains 24R20 and 24R23, respectively) were transformed with pREP81x (vector) alone or pREP81x-pfh1 expressing wild-type pfh1+ (Pfh1) as denoted at the left-hand side of figure. Each transformant was grown in liquid media and the cells obtained were spotted in 10-fold dilutions (105, 104, 103, 102 and 101 cells) onto EMM plates or EMM plates containing thiamine (5 μg/ml). The plates were incubated for 3 days at 30°C and at 37°C or 7 days at 18°C as shown.
Pfh1-R20 has reduced helicase and ATPase activities
To support the above genetic observations, we isolated a mutant enzyme (NusA-Pfh1-R20-F) using the same purification strategy employed for the isolation of the wild-type enzyme and examined its helicase and ATPase activities. Since pfh1-R20 by itself is a cold-sensitive mutation and allows cdc24-M38 temperature-sensitive mutant cells to grow at 37°C, its ATPase activity was measured at 18, 30 and 37°C. The ssDNA-dependent ATPase activity of Pfh1-R20 was significantly reduced at all temperatures tested compared to that of the wild-type (Figure 9A). Although the Pfh-R20 mutation is not located within the conserved helicase domain, it may alter the conformation of the enzyme such that the enzymatic activities of Pfh1 are reduced. The amount of ATP hydrolyzed by the wild-type Pfh1 at 37°C was slightly lower than that at 30°C. The efficiency of ATP hydrolysis by the mutant enzyme, however, was dramatically reduced at 37°C, yielding only 10% of the wild-type level of ATP hydrolysis. The Km for ATP and kcat for ATPase activity of the wild-type and mutant enzymes were also determined: the Km for ATP and kcat of wild-type Pfh1 (0.378 mM and 41.1 s−1, respectively) were ∼6- and 10-fold higher than those (0.068 mM and 4.4 s−1, respectively) of Pfh1-R20. This result suggests that though the mutant protein has a higher affinity for ATP, it hydrolyzes ATP very slowly. In addition, we examined the helicase activity of the mutant protein. Pfh1-R20 displaced the 21 bp partial duplex DNA substrate very poorly at both 18 and 37°C compared to wild-type Pfh1 (Figure 9B). This suggests that the pfh1-R20 mutant allele is a loss-of-function mutation and that the enzyme produced by this allele is nearly inactive at 37°C. In the absence of ATP, the mutant enzyme was defective in binding the substrate DNA. However, in the presence of ATP its DNA binding activity was almost identical to the wild-type enzyme (Figure 9C). In summary, these results indicate that the ability of the mutant enzyme to hydrolyze ATP, unwind duplex DNA and bind substrate DNA in the absence of ATP was significantly lower than the wild-type. The fact that Pfh1-R20 is less active than the wild-type enzyme is consistent with the notion that suppression of the cdc24-M38 mutation requires the impaired function of pfh1.
Figure 9.
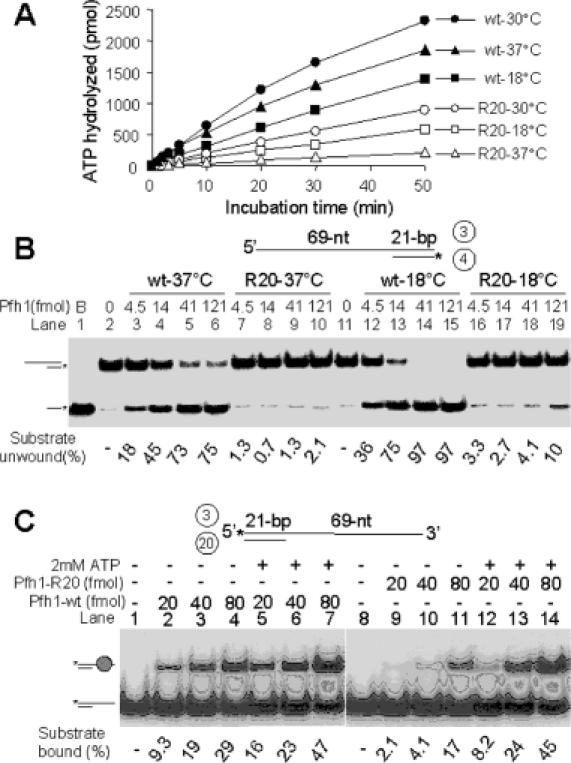
Pfh1-R20 mutant protein is impaired in its enzymatic activities. (A) ATP hydrolysis was measured at varying temperatures. The standard reaction mixtures (200 μl) for ATPase assays were pre-incubated at each temperature for 2 min with 0.2 mM of ATP, and the reaction was initiated by the addition (800 fmol each) of either NusA-Pfh1-F (wt) or NusA-Pfh1-R20-F (R20) mutant enzymes. Aliquots (20 μl) were withdrawn at each time point as indicated, and the amount of ATP hydrolyzed was measured as described in Materials and Methods. (B) Increasing levels (4.5, 13.5, 40.5 and 121 fmol) of either NusA-Pfh1-F (wt) or NusA-Pfh1-R20-F (R20) mutant enzymes were incubated with 15 fmol of DNA substrate (prepared from oligonucleotides 3 and 4) in a 20 μl standard reaction mixtures (see Materials and Methods) at 30°C for 20 min. The amounts of displaced products were analyzed on 10% polyacrylamide gel. B denotes boiled substrate control. (C) Oligonucleotides 3 and 20 were used to construct the 3′-overhang partial duplex DNA substrate. Increasing amounts (20, 40 and 80 fmol) of NusA-Pfh1-F (Pfh1-wt) and mutant NusA-Pfh1-R20-F (Pfh1-R20) were incubated with the substrate (15 fmol) in the presence (+) or absence (−) of 2 mM of ATP at 30°C for 10 min. The reaction products were subjected to electrophoresis for 1 h at 100 V through a 5% polyacrylamide gel in 0.5× TBE. The position of Pfh1–DNA complex is as indicated. The amount of substrate bound is presented at the bottom of the figure.
Genetic interactions between pfh1+ and dna2+
Cdc24 is known to function via a direct interaction with Dna2 (14,16). Because we have established a genetic interaction between cdc24 and pfh1 alleles (above) and hence a functional relationship between Cdc24 and Pfh1, we predicted that a functional relationship might exist between Dna2 and Pfh1. Such a relationship would indicate a role for Pfh1 in Okazaki fragment maturation. To test this possibility, we determined whether the pfh1-R20 allele could suppress the temperature-sensitive phenotype of a dna2-C2 mutant. The dna2-C2 mutation contains a single amino acid change (Leu to Ser) at position 1079, which is conserved throughout eukaryotes (16). dna2-C2 is a temperature-sensitive loss-of-function allele with a phenotype (at non-permissive temperature) indistinguishable from that of a dna2 null mutation (16).
dna2-C2 (HK10) and pfh1-R20 (GH101-2) strains were crossed. Tetrad dissection yielded dna2-C2 pfh1-R20 double mutants whose relevant genotypes were confirmed by PCR and DNA sequence analysis (data not shown). As predicted, dna2-C2 pfh1-R20 double mutant cells were able to grow at 37°C, indicating that pfh1-R20 suppresses the temperature-sensitive phenotype of dna2-C2 (Figure 10A and data not shown). Moreover, when a multicopy plasmid bearing the wild-type pfh1+ gene was introduced into dna2-C2 pfh1-R20 double mutants, suppression was lost (Figure 10A) and transformants grown at 37°C displayed a highly elongated cell shape similar to dna2-C2 mutant cells grown at non-permissive temperature (Figure 10B). In contrast, the same transformants displayed wild-type morphology when grown at 18°C (Figure 10B). Since cold-sensitive alleles of pfh1+ can suppress the temperature-sensitive phenotype of both cdc24-M38 and dna2-C2, this strongly suggests that pfh1+ plays a role in Okazaki fragment maturation.
Figure 10.

Expression of wild-type pfh1+ abolishes the ability of pfh1-R20 to suppress the temperature-sensitive growth defect of dna2-C2. (A) The dna2-C2 pfh1-R20 (C2 R20) double mutant cells (strain GH102-6) were transformed with pREP81x (vector) alone or pREP81x-pfh1 expressing wild-type pfh1+ (Pfh1) as denoted at the left-hand side of figure. Each transformant was grown in liquid media and the cells obtained were spotted and grown as described in Figure 8. (B) Photographs (×200) of cells obtained from transformation of pREP81x (vector) alone or pREP81x-pfh1 (Pfh1) into the GH102-6 cells.
In order to further support the significance of the genetic interactions between Pfh1 and Dna2 or Cdc24, we examined whether there were physical interactions between Pfh1 and Dna2 or Cdc24 using the yeast two-hybrid assays. These assays failed to detect any positive results (data not shown). Moreover, we found that Pfh1 did not interact with the two subunits (Pol 3 and Cdc27, the catalytic and PCNA-interacting subunits, respectively) of pol δ, PCNA, Fen1 and DNA ligase I in the yeast two-hybrid assays (data not shown). Instead, we detected synthetic sick or lethal interactions between pfh1-R20 and both pol 3-R18 and cdc27-R22, two cold-sensitive mutant alleles of pol δ that were isolated, like pfh1-R20, as suppressors of the temperature-sensitive growth defect of cdc24-M38 (H. Tanaka and S. MacNeill, unpublished data). When we analyzed tetrads from crosses of pfh1-R20 X pol3-R18 and pfh1-R20 X cdc27-R22, we found that the pfh1 pol3 and pfh1 cdc27 double mutants were poorly viable, even though single mutants are viable. In addition, the number of double mutant colonies detected among viable spores was far less than the expected number as shown in Table 4. This suggests that mutants bearing defective pol δ subunits cannot tolerate the loss of Pfh1 function. This genetic result is consistent with the notion that both Pfh1 and pol δ participate in the same process during lagging strand synthesis. In addition, our data suggest that the interactions between Pfh1 and other proteins involved in Okazaki fragment processing may only be functional and not physical.
Table 4. Synthetic lethal interactions between pfh1-R20 and pol3-R18 or cdc27-R22.
| pfh1-R20 x pol3-R18 | pfh1-R20 x cdc27-R22 | ||||
|---|---|---|---|---|---|
| Genotype | Observed | Expected | Genotype | Observed | Expected |
| pol3+ pfh1+ | 34 | 38 | cdc27+ pfh1+ | 11 | 12 |
| pol3-R18 pfh1+ | 20 | 38 | cdc27-R22 pfh1+ | 9 | 12 |
| pol3+ pfh1-R20 | 31 | 38 | cdc27+ pfh1-R20 | 9 | 12 |
| pol3-R18 pfh1-R20 | 1a | 38 | cdc27-R22 pfh1-R20 | 2 | 12 |
| Inviable spores | 66 | Inviable spores | 17 | ||
| Total spores | 152 | 152 | Total spores | 48 | 48 |
The pfh1-R20 mutant strain (GH101-2) was crossed with the pol3-R18 or cdc27-R22 mutant strains (H231-14 or H369-5, respectively), and resulting tetrads were dissected and grown at 30°C for 6 days. The genotype of each spore was confirmed with sequencing of the PCR-amplified fragment comprising the mutation.
aThe spore displayed a very severe growth defect.
DISCUSSION
In this study, we isolated the full-length Pfh1 enzyme and characterized its helicase and ATPase activities. Fusion of the NusA and FLAG epitopes to the N- and C-termini of Pfh1, respectively, allowed us to purify the enzyme to near homogeneity. Two rounds of successive gel filtration chromatographic steps yielded an active Pfh1 enzyme preparation, which contained ATPase and DNA helicase activities. Furthermore, the activities associated with the NusA-Pfh1-F were unaltered by removal of the NusA tag by thrombin digestion and both activities were simultaneously depleted by immunoprecipitation with anti-FLAG M2 Ab-coupled agarose beads. These results demonstrate unambiguously that both activities are intrinsic to the Pfh1 enzyme and that the presence of the NusA moiety fused to its N-terminus did not hinder its enzymatic activities. In contrast to these findings, the fusion of a GST moiety to an N- and C-terminal truncated version (255–789 of the 805 amino acids) of Pfh1 (GST-Pfh1-h) resulted in an inactive recombinant enzyme, and the removal of GST moiety was necessary to render it active (20). We noted that the specific activity of the ATPase activity of our NusA-Pfh1-F preparations is ∼10-fold higher than that reported with GST-Pfh1-h from which the GST moiety was removed (20). This suggests that either N-terminal or C-terminal deletions affect the ATPase activity of Pfh1 and probably its helicase activity as well. Whether the GST-Pfh1-h protein could functionally substitute for the wild-type Pfh1 in vivo was not determined. This experiment would provide information whether the N-terminal 255 amino acids are required for the cellular function of Pfh1.
Using the NusA-Pfh1-F enzyme that is active in vivo (data not shown), we determined a number of Pfh1's biochemical properties. The Pfh1 helicase of S.pombe shares several biochemical properties in common with Dna2 of S.cerevisiae (10,22): (i) both unwind duplex DNA in the 5′ to 3′ direction, preferring a 5′-tailed DNA substrate; (ii) both act in a highly distributive manner; (iii) both are active as a monomer; (iv) both preferentially utilize ATP and dATP as the energy sources; and, most significantly; and (v) both enzymes can unwind efficiently the DNA substrate containing a flap with secondary structure (22). However, it should be noted that Pfh1, with its 5′ to 3′ helicase activity, cannot itself resolve the secondary structure present at the flap, as this requires translocation to occur in the 3′ to 5′ direction. Instead, we have shown that the presence of the secondary structure in the flap does not interfere with the flap displacement ability of Pfh1 as long as a short internal ssDNA region is present. This property is unique to the Pfh1 enzyme, since S.cerevisiae or C.elegans Dna2 helicase by itself cannot efficiently displace a structured flap (unpublished data).
Based on the properties of Pfh1, it is possible to infer a role for Pfh1 in assisting the removal of long DNA flaps that contain secondary structure. Our previous observations indicated that in vitro displacement DNA synthesis by S.pombe pol δ is efficient since the removal of the primer RNA–DNA of the preceding Okazaki fragment was markedly stimulated by RPA (11,30). This finding reflects the generation of flaps long enough to support RPA binding. If this were the case in vivo, a large proportion of flaps generated by pol δ would have the potential to form secondary structures. The generation of long flaps with secondary structure would make S.pombe cells more dependent on helicase activity before the flap cleavage by the processing enzymes, Dna2 or Fen1. Since Dna2 of S.pombe lacks detectable ATPase/DNA helicase activity (unpublished data), it is likely that Pfh1 can function to facilitate removal of secondary structure containing flaps that are resistant to the cleavage by Dna2 and Fen1 (17). Since Pfh1 is unable to directly resolve the secondary structure in the flap, it is more likely to displace the entire hairpin-containing flap from the lagging template, resulting in a gap equivalent in size to an Okazaki fragment, which can be filled in by the S.pombe pol δ holoenzyme. We noted that Pfh1 is not stimulated by S.pombe RPA, although S.cerevisiae RPA stimulated both helicase and endonuclease activities of S.cerevisiae Dna2. At present, it is not clear why S.pombe RPA fails to stimulate Pfh1 helicase activity. It may be the case, however, that Pfh1 acts in vivo before the 5′ flap grows long enough (20–30 nt ssDNA) to stably bind RPA.
In addition to studying the enzymatic properties of Pfh1, we have characterized genetic interactions between cdc24-M38 and pfh1-R20 and between pfh1-R20 and dna2-C2 in order to establish a functional relationship between pfh1+ and dna2+ and cdc24+. Previous genetic studies demonstrated that cdc24+ is likely to function via direct interactions with dna2+ (14,16). Overexpression of wild-type pfh1+ interfered with the suppression by pfh1-R20 or pfh1-R23 of the temperature-sensitive phenotype of either cdc24-M38 or dna2-C2. These results indicate that when the function of dna2+ or cdc24+ is compromised by mutation, inactivation of pfh1+ is required to restore growth. The established roles of Cdc24 and Dna2 in Okazaki fragment maturation, the fact that mutant alleles of pfh1 can suppress the temperature-sensitive phenotype of both cdc24-M38 and dna2-C2, and the observation that overexpression of wild-type pfh1+ abolishes this suppression strongly suggest that Pfh1 plays a role in Okazaki fragment maturation in concert with Cdc24 and Dna2.
At present, the precise mechanism of Pfh1, pol δ and Dna2 function during the maturation of Okazaki fragments in S.pombe is unclear. In light of the helicase activity described in this study, Phf1 is likely to be involved in the generation of flaps from the 5′ end region of Okazaki fragments together with pol δ. This may be accomplished by the potential for Pfh1 to facilitate pol δ-mediated displacement DNA synthesis. Because S.cerevisiae Dna2 has been shown to support the processing of long flaps that can bind RPA (11), the inactivation of Pfh1 in S.pombe, leading to decreased production of long flaps, would relieve the requirement for Dna2-mediated long flap processing and suppress the dna2-C2 growth defect. Consistent with this model, we identified synthetic lethal or sick interactions, a close genetic link between pfh1+ and genes encoding two of the three essential subunits of pol δ in S.pombe. These findings support the idea that pfh1+ and pol δ carry out related functions during Okazaki fragment metabolism. To further define the functional interplay of Pfh1, pol δ, Dna2 and Cdc24 during the processing of Okazaki fragments in S.pombe, a rigorous biochemical analysis of these purified proteins will be required.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Dr Jerard Hurwitz (Sloan-Kettering Institute, USA) and Dr Peter Sherwood (Cold Spring Harbor Laboratory, New York) for critical reading of the manuscript. This work was supported by a grant from the Creative Research Initiatives of the Korean Ministry of Science and Technology given to Y.-S.S. Support for S.A.M. and H.T. came from the Wellcome Trust and JSPS.
REFERENCES
- 1.Hubscher U. and Seo,Y.S. (2001) Replication of the lagging strand: a concert of at least 23 polypeptides. Mol. Cells, 12, 149–157. [PubMed] [Google Scholar]
- 2.MacNeill S.A. (2001) DNA replication: partners in the Okazaki two-step. Curr. Biol., 11, R842–R844. [DOI] [PubMed] [Google Scholar]
- 3.Kornberg A. and Baker,T.A. (1992) DNA replication, 2nd edn. W. H. Freeman & Co, NY. [Google Scholar]
- 4.Bambara R.A., Murante,R.S. and Henricksen,L.A. (1997) Enzymes and reactions at the eukaryotic DNA replication fork. J. Biol. Chem., 272, 4647–4650. [DOI] [PubMed] [Google Scholar]
- 5.Waga S. and Stillman,B. (1998) The DNA replication fork in eukaryotic cells. Annu. Rev. Biochem., 67, 721–751. [DOI] [PubMed] [Google Scholar]
- 6.Waga S. and Stillman,B. (1994) Anatomy of a DNA replication fork revealed by reconstitution of SV40 DNA replication in vitro. Nature, 369, 207–212. [DOI] [PubMed] [Google Scholar]
- 7.Turchi J.J., Huang,L., Murante,R.S., Kim,Y. and Bambara,R.A. (1994) Enzymatic completion of mammalian lagging-strand DNA replication. Proc. Natl Acad. Sci. USA, 91, 9803–9807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bullock P.A., Seo,Y.S. and Hurwitz,J. (1991) Initiation of simian virus 40 DNA synthesis in vitro. Mol. Cell. Biol., 11, 2350–2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nethanel T. and Kaufmann,G. (1990) Two DNA polymerases may be required for synthesis of the lagging DNA strand of simian virus 40. J. Virol., 64, 5912–5918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bae S.H. and Seo,Y.S. (2000) Characterization of the enzymatic properties of the yeast dna2 Helicase/endonuclease suggests a new model for Okazaki fragment processing. J. Biol. Chem., 275, 38022–38031. [DOI] [PubMed] [Google Scholar]
- 11.Bae S.H., Bae,K.H., Kim,J.A. and Seo,Y.S. (2001) RPA governs endonuclease switching during processing of Okazaki fragments in eukaryotes. Nature, 412, 456–461. [DOI] [PubMed] [Google Scholar]
- 12.Brosh R.M. Jr, Driscoll,H.C., Dianov,G.L. and Sommers,J.A. (2002) Biochemical characterization of the WRN-FEN-1 functional interaction. Biochemistry, 41, 12204–12216. [DOI] [PubMed] [Google Scholar]
- 13.Brosh R.M. Jr, von Kobbe,C., Sommers,J.A., Karmakar,P., Opresko,P.L., Piotrowski,J., Dianova,I., Dianov,G.L. and Bohr,V.A. (2001) Werner syndrome protein interacts with human flap endonuclease 1 and stimulates its cleavage activity. EMBO J., 20, 5791–5801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gould K.L., Burns,C.G., Feoktistova,A., Hu,C.P., Pasion,S.G. and Forsburg,S.L. (1998) Fission yeast cdc24+ encodes a novel replication factor required for chromosome integrity. Genetics, 149, 1221–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tanaka H., Tanaka,K., Murakami,H. and Okayama,H. (1999) Fission yeast cdc24 is a replication factor C- and proliferating cell nuclear antigen-interacting factor essential for S-phase completion. Mol. Cell. Biol., 19, 1038–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kang H.Y., Choi,E., Bae,S.H., Lee,K.H., Gim,B.S., Kim,H.D., Park,C., MacNeill,S.A. and Seo,Y.S. (2000) Genetic analyses of Schizosaccharomyces pombe dna2+ reveal that dna2 plays an essential role in Okazaki fragment metabolism. Genetics, 155, 1055–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tanaka H., Ryu,G.H., Seo,Y.S., Tanaka,K., Okayama,H., MacNeill,S.A. and Yuasa,Y. (2002) The fission yeast pfh1+ gene encodes an essential 5′ to 3′ DNA helicase required for the completion of S-phase. Nucleic Acids Res., 30, 4728–4739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Foury F. and Kolodynski,J. (1983) pif mutation blocks recombination between mitochondrial rho+ and rho− genomes having tandemly arrayed repeat units in Saccharomyces cerevisiae. Proc. Natl Acad. Sci. USA, 80, 5345–5349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schulz V.P. and Zakian,V.A. (1994) The Saccharomyces PIF1 DNA helicase inhibits telomere elongation and de novo telomere formation. Cell, 76, 145–155. [DOI] [PubMed] [Google Scholar]
- 20.Zhou J.Q., Qi,H., Schulz,V.P., Mateyak,M.K., Monson,E.K. and Zakian,V.A. (2002) Schizosaccharomyces pombe pfh1+ encodes an essential 5′ to 3′ DNA helicase that is a member of the PIF1 subfamily of DNA helicases. Mol. Biol. Cell, 13, 2180–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu Q., Choe,W. and Campbell,J.L. (2000) Identification of the Xenopus laevis homolog of Saccharomyces cerevisiae DNA2 and its role in DNA replication. J. Biol. Chem., 275, 1615–1624. [DOI] [PubMed] [Google Scholar]
- 22.Bae S.H., Kim,D.W., Kim,J., Kim,J.H., Kim,D.H., Kim,H.D., Kang,H.Y. and Seo,Y.S. (2002) Coupling of DNA helicase and endonuclease activities of yeast Dna2 facilitates Okazaki fragment processing. J. Biol. Chem., 277, 26632–26641. [DOI] [PubMed] [Google Scholar]
- 23.Alfa C., Fantes,P.A., Hyams,L., McLeod,M. and Warbrick,E. (1993) Experiments with Fission Yeast. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 24.Okazaki K., Okazaki,N., Kume,K., Jinno,S., Tanaka,K. and Okayama,H. (1990) High-frequency transformation method and library transducing vectors for cloning mammalian cDNAs by trans-complementation of Schizosaccharomyces pombe. Nucleic Acids Res., 18, 6485–6489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moreno S., Klar,A. and Nurse,P. (1991) Molecular genetic analysis of fission yeast Schizosaccharomyces pombe. Methods Enzymol., 194, 795–823. [DOI] [PubMed] [Google Scholar]
- 26.Basi G., Schmid,E. and Maundrell,K. (1993) TATA box mutations in the Schizosaccharomyces pombe nmt1 promoter affect transcription efficiency but not the transcription start point or thiamine repressibility. Gene, 123, 131–136. [DOI] [PubMed] [Google Scholar]
- 27.Park J.S., Choi,E., Lee,S.H., Lee,C. and Seo,Y.S. (1997) A DNA helicase from Schizosaccharomyces pombe stimulated by single-stranded DNA-binding protein at low ATP concentration. J. Biol. Chem., 272, 18910–18919. [DOI] [PubMed] [Google Scholar]
- 28.Siegel L.M. and Monty,K.J. (1966) Determination of molecular weights and frictional ratios of proteins in impure systems by use of gel filtration and density gradient centrifugation. Application to crude preparations of sulfite and hydroxylamine reductases. Biochim. Biophys. Acta, 112, 346–362. [DOI] [PubMed] [Google Scholar]
- 29.Spiro C., Pelletier,R., Rolfsmeier,M.L., Dixon,M.J., Lahue,R.S., Gupta,G., Park,M.S., Chen,X., Mariappan,S.V. and McMurray,C.T. (1999) Inhibition of FEN-1 processing by DNA secondary structure at trinucleotide repeats. Mol. Cell, 4, 1079–1085. [DOI] [PubMed] [Google Scholar]
- 30.Bae S.H., Kim,J.A., Choi,E., Lee,K.H., Kang,H.Y., Kim,H.D., Kim,J.H., Bae,K.H., Cho,Y., Park,C. and Seo,Y.S. (2001) Tripartite structure of Saccharomyces cerevisiae Dna2 helicase/endonuclease. Nucleic Acids Res., 29, 3069–3079. [DOI] [PMC free article] [PubMed] [Google Scholar]