


Abstract
Protein–DNA interactions facilitate the fundamental functions of living cells and are universal in all living organisms. Several investigations have been carried out, essentially identifying pairs of interactions between the amino acid residues in proteins and the bases in DNA. In the present study, we have detected the recognition motifs that may constitute a cluster of spatially interacting residues in proteins, which interact with the bases of DNA. Graph spectral algorithm has been used to detect side chain clusters comprising Arg, Lys, Asn, Gln and aromatic residues from proteins interacting with DNA. We find that the interaction of proteins with DNA is through clusters in about half of the proteins in the dataset and through individual residues in the rest. Furthermore, inspection of the clusters has revealed additional interactions in a few cases, which have not been reported earlier. The geometry of the interaction between the DNA base and the protein residue is quantified by the distance d and the angle θ. These parameters have been identified for the cation–π/H-bond stair motif that was reported earlier. Among the Arg, Lys, Asn and Gln residues, the range of (d, θ) values of the interacting Arg clearly falls into the cation–π and the hydrogen bond interactions of the ‘cation–π/H-bond’ stair motif. Analysis of the cluster composition reveals that the Arg residue is predominant than the Lys, Asn and Gln residues. The clusters are classified into Type I and Type II based on the presence or absence of aromatic residues (Phe, Tyr) in them. Residue conservation in these clusters has been examined. Apart from the conserved residues identified previously, a few more residues mainly Phe, Tyr and Arg have also been identified as conserved and interactive with the DNA. Interestingly, a few residues that are parts of interacting clusters and do not interact directly with the DNA have also been conserved. This emphasizes the importance of recognizing the protein side chain cluster motifs interacting with the DNA, which could serve as signatures of protein–DNA recognition in the families of DNA binding proteins.
INTRODUCTION
Recognition of DNA by proteins, and the specific interactions of these proteins with the nucleic acid bases of DNA are inevitable for carrying out the basic processes of living cells such as growth, differentiation, maturation, etc. The phenomenon of biological molecular recognition is mediated by the complex interplay of non-covalent interactions. These include the canonical hydrogen bonds, electrostatic and van der Waals interactions, as well as the non-canonical interactions such as cation–π interactions, CH–π interactions, π–π stacking, etc. The recognition principles of protein–protein and protein–DNA interfaces are guided by many such non-covalent interactions.
The cation–π interactions, among the non-canonical interactions, are found to be operative between a side chain carrying a positive charge such as Arg, Lys or a side chain carrying a partial charge such as Asn or Gln and a π system. The π system can be an aromatic ring of Phe, Tyr, Trp, His (1) or of a nucleic acid base. The cation–π interactions are the source of recognition of many ligand–receptor interactions in proteins (2). A recent study has found cation–π interactions along with π–π stacking and hydrogen bonds as molecular determinants of ATP binding in proteins (3). In DNA, the presence of the cation–π interaction between a divalent cation such as Mg2+ and nucleic acid bases [both DNA and RNA (tRNA)] has been observed earlier (4). The contribution of the cation–π interaction to the specificity and stability of protein–DNA interface has been recognized only recently (5,6).
A review (7) of the structural basis of macromolecular recognition outlines various efforts to characterize the protein–protein and protein–DNA interfaces. Buried surface area (8,9) and shape complementarity information upon complexation are used as measures to characterize the protein–protein (10) as well as protein–DNA interactions (11). In general, although the determinants of protein–protein recognition are reasonably well characterized (8,10,12,13), there exists no simple code for protein–DNA recognition (14). Attempts have been made to characterize the protein–DNA interfaces to get better DNA binding signatures (15,16) and therefore to bring in more insight into the principles of protein–DNA recognition. Recently, the role of cation–π interactions in the stability and specificity of protein–DNA complexes has been studied in detail (5). A motif involving the cation–π interaction called the cation–π/H-bond stair motif has been reported by Rooman and coworkers (6). This motif is composed of a cation–π interaction formed by a protein residue such as Arg, Lys, Asn or Gln with a base of DNA, and an associated hydrogen bond also formed by the same amino acid residue with an adjacent DNA base. In this motif, a preferential recognition of Guanine (G) by Arg and adenine (A) by Lys and Asn is observed especially in the major groove of B-DNA (6).
In all the analyses of the protein–DNA interfaces described above, specific interactions are addressed at the pair-wise level. Macromolecular recognitions, however, are not necessarily confined to pair-wise interactions. In many instances, sets of amino acid residues, which are close to one another in sequence or space, determine the specificity of such interactions. In other words, a cluster of amino acids in spatial proximity is involved in recognition. Such clusters at the protein–protein interfaces have been analyzed earlier (17). Here, we describe a study of the protein–DNA interface, essentially capturing multiple interactions of protein residues with the DNA, as a cluster of interacting residues. This is carried out by the graph spectral method which has been developed in the context of identifying side chain clusters of interacting residues in protein structures (18) and has been further applied to the identification of protein–protein interface clusters (17).
The aim of this paper is to identify clusters of amino acid side chains in proteins, which are engaged in the formation of the cation–π/H-bond stair motifs, at the protein–DNA interface. In the present study, we have considered the cationic (Arg, Lys), neutral, charge-delocalized (Asn, Gln) and aromatic residue (Phe, Tyr and Trp) side chains of proteins. Clusters of those residues that interact with the DNA have been identified. Our study has revealed that some proteins from the dataset recognize the DNA bases as a cluster of residues while some do not. In many cases, aromatic residues or the additional Arg, Lys, Asn or Gln residues, which are part of the cluster, support the residues that are directly involved in the interaction with the DNA. The amino acid residues constituting such clusters are also highly conserved within the family. Such an approach using side chain clustering has enabled us to identify a network of protein–DNA interactions as well as identify functionally important amino acid residue clusters in the vicinity of the protein–DNA interaction site.
MATERIALS AND METHODS
In the present study, the protein chains from the crystal structures of protein–DNA complexes have been taken from the Protein Data Bank (PDB) (19) and the clustering algorithm has been applied to detect the side chain clusters in the proteins from these protein–DNA complexes.
Dataset
The positively charged amino acid residues such as Arg and Lys, and the neutral, charge-delocalized side chains of Asn and Gln are considered as cations and the nucleic acid bases of DNA as π systems for the detection of cation–π/H-bond stair motifs (6). On this basis, a dataset consisting of 52 protein–DNA complexes (Table 1) has been considered by Rooman and coworkers (6) for characterizing the cation–π/H-bond stair motif interactions at the protein–DNA interface. A total of 77 cation–π/H-bond stair motif interactions of the protein residues with the bases of DNA are reported in their study. We have also considered the same dataset with a difference in the analysis procedure. We have obtained clusters of amino acid side chains from the proteins of these protein–DNA complexes of the dataset, using the graph spectral algorithm (18). In our study, we have clustered all the cationic (Arg, Lys), the neutral charge-delocalized (Asn, Gln) and aromatic (Phe, Tyr, Trp) residues in the proteins from the dataset and examined, those clusters that interact with the bases of DNA.
Table 1. A list of protein–DNA complexes considered in the present study.
| ETS domain | 1awcA, 1bc8C, 1pueE |
| Helix–loop–helix | 1am9A |
| Cro and repressor | 1lmb3, 1lmb4, 1rpeL, 3croL |
| Trp repressor | 1troA |
| Integration host factor | 1ihfA |
| DNA repair protein | 1e3mA |
| Recombinase DNA binding domain | 1hcrA |
| Homeodomain | 1akhB, 1au7A, 1b72A, 1b72B, 1fjlA, 1mnmC, 1mnmD, 2hddA, 9antA |
| RAP1 | 1ignA |
| TC3 transposase | 1tc3C |
| Interferon regulatory factor like | 2irfL |
| Paired domain | 1pdnC |
| DNA polymerase | 1bpyA |
| REL homology | 1a3qA, 1a3qB, 1tsrB, 2ramA |
| STAT family | 1bg1A |
| Single-strand DNA binding domain | 1jmcA |
| Transcription factor T domain | 1xbrA |
| Lambda integrase like N terminal domain | 1crxA |
| Transcription factor DNA binding domain | 1sknP |
| Transcription factor IIB | 1aisA |
| SRF like | 1egwA, 1mnmA |
| TATA box binding domain | 1ytbA |
| Histone like protein | 1azpA |
| DNA repair glycosylase | 1ebmA |
| Endonuclease | 1bgbA, 1bhmA, 3pviA, 1a73A |
| DNAQ Like 3′–5′ Exonuclease | 2kfnA, 4bdpA |
| Methyl transferase | 6mhtA |
| Zinc finger | 1a1gA, 1meyC, 1ubdC |
| Zn6/Cys6 DNA binding domain | 1zmeD |
| Leucine Zipper domain | 1gd2E, 2dgcA |
| Hormone receptor | 1hcqA, 1latA, 2nllB |
Methods
Cluster detection by the graph spectral method
The side chain clusters of amino acids participating in the cation–π interactions are detected using an in-house side chain clustering algorithm developed based on the graph theory (18). A brief description of the procedure is given here.
A protein graph is constructed with amino acid residues (specifically the Cβ atoms) of the side chains of proteins as nodes and non-covalent interactions existing between them as edges. An edge is defined between two nodes based on the extent of side chain interaction existing between the nodes. It is quantified by the use of a contact criterion called the percentage contact criterion or the overlap criterion, which specifies the number of side chain atom pairs that comes within a distance of 4.5 Å (18). This criterion allows users to specify the extent of interaction in the side chains involved, which reflects in the nature of the clusters obtained. An edge is defined between the residues i and j, if and only if the overlap criterion evaluated between the side chains is greater than the contact criterion specified by the user. The graph, thus constructed, can be mathematically represented in the form of a connectivity matrix [Aij]. A degree matrix [Dii] is then constructed by summing the elements of each row in the connectivity matrix [Aij]. A Laplacian matrix [Lij] of order n × n is constructed from [Aij] and [Dii] as follows: [Lij] = [Dii] − [Aij].
The Laplacian matrix is diagonalized using LAPACK (version 3.0.3) (20) to get the eigen values and eigen vectors. The cluster information is then derived from the eigen vectors of the second lowest eigen value. The cluster-forming nodes have degenerate eigen vector components for the second lowest eigen value (21).
Identification of cation–π clusters
The side chain clusters from the proteins of the dataset are obtained with an overlap criterion ranging from 2 to 8%. A low cutoff of 2% results in more number of loosely connected clusters. However, a high contact criterion yields lesser number of clusters in which the side chain interactions are stronger (18). Most protein families in the dataset produce side chain clusters at 6% contact cutoff and detailed analysis is therefore reported for the clusters obtained with this contact cutoff.
As mentioned earlier, only the side chains of Arg, Lys, Asn, Gln and the aromatic residues from the proteins are considered for cluster detection. The clusters comprising at least one of the residues (Arg, Lys, Asn, Gln) that participate in the cation–π/H-bond stair motif interaction with the bases of DNA are selected for detailed analysis and are referred to as ‘interacting clusters’. The other clusters from the proteins that do not have any residues interacting with the DNA are ignored in the present study. The amino acid residues (Arg, Lys, Asn and Gln) that participate in the cation–π stair motif interaction with bases of DNA (6) and also found as a component of clusters are called ‘reported interacting residues’. The other Arg, Lys, Asn and Gln residues obtained from the same cluster are denoted as ‘additional residues’. The interactions of these additional residues, if any, with the DNA, as well as the other interactions by the reported residues that do not necessarily fall into the cation–π/H-bond definition are defined as the ‘additional interactions’. An ‘isolated residue’ is defined as an amino acid residue interacting with the DNA base through the cation–π stair motif, which is not part of any cluster. It, however, belongs to a protein, which already contains an interacting cluster. The interactions through such residues are called as ‘isolated interactions’. A ‘non-cluster-forming’ residue is the same as an ‘isolated residue’ except for the fact that the residue is present in a protein that has no interacting cluster. The above definitions are followed throughout the paper.
Geometrical analysis of clusters
The cation–π interaction can be quantified by the geometrical parameters d and θ as given in Figure 1. The parameter d is the distance between the cation and the centroid of the aromatic ring, while θ is the angle between the line joining the cation, the centroid and the normal to the aromatic plane at the centroid.
Figure 1.
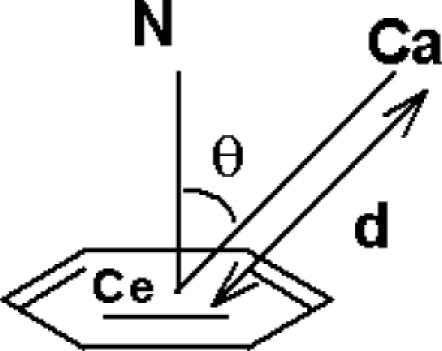
Definition of geometrical parameters for cation–π interaction. Ca, the atom from the protein residue considered as the cation; Ce, nucleic acid ring centroid; N, ring normal at the centroid. d is the distance between Ca and Ce. θ is the angle Ca–Ce–N.
The positive charge centers of Arg (NE, CZ, NH1, NH2), Lys (CE, NZ) and the atoms carrying partial charge in Asn (CG, ND1), Gln (CD, NE2) are considered as cations in the present study. The aromatic rings of the nucleic acid bases A, G, C and T and the amino acid side chains of Phe, Tyr and Trp are considered as π systems. The least square plane of the aromatic ring and the unit normal to the plane have been calculated using the algorithm given by Blow (22). Both the five- and the six-member rings of the A and G bases and Trp are treated separately for finding the normal from their respective centroid.
The protein side chains Arg, Lys, Asn, Gln present in a cluster are considered interactive with the nucleic acid base aromatic ring if d ≤ 6.0 Å. All the interactions in this d limit and 0 ≤ θ ≤ 90° are identified. This d limit has been used to identify the amino–aromatic interactions (pair wise) in proteins previously (23) and the same is used in our present study. McFail-Isom and coworkers (4) have used the same (d, θ) geometry to detect the cation–π interaction of inorganic cations such as Mg2+ with the DNA and RNA bases. The interaction of protein aromatic residues, with the bases of DNA is also evaluated. Such aromatic residues are considered to interact with the bases, if the distance between any side chain atom of the protein aromatic ring and the nucleic acid base is ≤4.5 Å. Thus two different distances are evaluated, one for characterizing the interaction of cationic residue side chains with the bases of the DNA (d) and the other for evaluating the general side chain contacts existing between the protein aromatic rings, if any, and the bases in an interacting cluster.
Analysis of accessible surface area of clusters
The accessible surface area is calculated using NACCESS (24), which implements Lee and Richards (9) algorithm. The percentage relative accessibility (ASA) of the residues in the protein–DNA complex as well as in the uncomplexed protein is obtained using NACCESS. The ASA of the uncomplexed protein is calculated assuming it to be similar in conformation to its DNA bound state. The difference, δASA (Å2) is the percentage accessibility lost by the protein residues upon complexation with the DNA. The δASA is calculated for clusters from the protein–DNA complexes. The average loss in percentage accessibility, δASAavg (Å2) is also calculated for these clusters.
RESULTS AND DISCUSSION
Interface cluster as a function of overlap criterion
By employing the formalism outlined in the previous section, the protein side chain clusters containing the reported interacting residues at the protein–nucleic acid interface have been identified and analyzed. Clusters consisting of Arg, Lys, Asn, Gln, the aromatic residues Phe, Tyr, Trp are detected from the PDB files using the overlap criterion ranging from 2 to 8%. As expected, the number of interacting amino acid residues in a cluster decreases as the overlap criterion is increased (details presented in Figure S1). Large numbers of clusters are obtained with the reported Arg residues at all the cut-off levels. This is due to a greater number of contacts that Arg makes with other residues in the cluster. On the other hand, the participation of the reported Lys, Gln and Asn is small in the 8% overlap case. The participation of the Lys, Asn and Gln residues is predominant at lower cutoffs. This is because of the fact that lesser number of contacts also qualifies to be connected and become a part of the cluster. Thus, the Arg residues seem to interact very strongly with other residues in the cluster when compared to Lys, Asn and Gln.
The reported interacting clusters from proteins in different cut-off regions have been analyzed. Out of the 77 reported residues, 38 interacting with the DNA could form clusters at the 6% overlap (6). They belong to 11 different families and the interacting residues in these clusters are mainly Arg and Lys residues. A reduction in overlap criterion to 4% resulted in adding only three more proteins to this list. A list of proteins forming interacting clusters at various overlap criteria is given in Table S1.
A complete list of clusters formed by the reported residues in proteins is given in Table 2 with the reported residues represented in bold face. It is evident from Table 2 that in many proteins of the dataset, more than one reported residue has been found as a single interacting cluster. In many instances, both the reported residues are detected in the same cluster as in the ETS domain proteins (1bc8C, 1pueE), PBX1 homeodomain (1b72B), homing endonuclease (1a73A) and zinc fingers. Apart from this, in RAP1 protein (1ignA) and in PAP1 Leucine Zipper (1gd2E), the reported residues are found as part of different interacting clusters. Furthermore, there are proteins (1au7A, 1ignA, 1a3qA, 1a1gA and 1ubdC) in which one of the reported residues is in a cluster whereas the others do not form a cluster. Such isolated interactions are represented in bold italic in Table 2.
Table 2. Interactiona of the bases of DNA with the cluster-forming residues.
| PDB | Cluster no. | Protein cluster compositionb | δASAavg (Å2)c | Interacting DNA based |
|---|---|---|---|---|
| 1awcA | 1 | Arg 376t | 25.22 | G 8(D) G 9(D) |
| Arg 379t | C 6(D),C 7(D),G 8(D) | |||
| Tyr 380p | A 10(D),A 11(D),C 32(E),T 33(E) | |||
| Tyr 382p | ||||
| Lys 389t | ||||
| Tyr 397h | ||||
| 1bc8C | 1 | Arg 61t | 25.12 | G 5(A) G 6(A) |
| Arg 64t | C 4(A) G 5(A),C 3(A) | |||
| Tyr 65p | G 6(A),A 7(A),A 8(A),C13(B),T14(B) | |||
| Tyr 67p | ||||
| Lys 74t | ||||
| Tyr 82h | ||||
| 1pueE | 1 | ARG 232t | 40.25 | G 8(A) G 9(A),A 10(A) |
| ARG 235t | G 7(A) G 8(A),G 6(A) | |||
| 1lmb3 | 1 | GLN 44p | 27.1 | T 3(1) A 4(1) |
| GLN 33p | ||||
| 1lmb4 | 1 | ASN 55− | 30.95 | G 13(1) G 14(1) |
| LYS 4− | G 13(1),G 14(1) | |||
| 1rpeL | 1 | GLN 28p | 17.87 | A 24(A) A 25(A) |
| GLN 17h | ||||
| ARG 10h | ||||
| 3croL | 1 | GLN 28p | 19.58 | T 3(B) A 4(B) |
| GLN 17h | ||||
| ARG 10h | ||||
| GLN 32− | A 4(B), C 5(B) | |||
| LYS 7p | ||||
| 1akhA | 1 | ASN 120t | 31.7 | G 25(C) A 26(C) |
| ARG 124m | T 24(C),G 25(C),A 26(C) | |||
| 1akhB | 1 | ARG 185t | 39.55 | T 5(C) G 6(C),T 7(C) |
| ASN 182t | T 37(C),A 38(C) | |||
| 1b72B | 1 | ASN 286t | 40.95 | G 8(D) A 9(D) |
| ARG 290m | T 7(D) G 8(D),A 9(D),T 33(E) | |||
| 1au7A | 1 | GLN 44t | 20.57 | T 459(C) A 460(C) |
| GLN 27t | ||||
| ARG 20t | ||||
| Isolated | ARG 49t | T 483(D) G 484(D) | ||
| 1ignAe | 1 | ARG 404 | 11.68 | G 30(D) G 31(D),G 32(D) |
| PHE 407 | ||||
| TYR 388 | ||||
| ARG 408 | G 31(D), G 32(D) | |||
| 2 | ARG 542 | 18.23 | T 22(D) G 23(D) | |
| ARG 546 | G 23(D) G 24(D), T 25(D) | |||
| PHE 545 | ||||
| PHE 526 | ||||
| Isolated | ASN 401 | A 7(C) C 8(C) | ||
| 1a3qA | 1 | ARG 52h | 27.6 | G606(D)G607(D),G608(D),C512(C),C513(C) |
| LYS 221h | G 607(D),G 608(D) | |||
| Isolated | ARG 54t | G 605(D) G 606(D) | ||
| 1a3qB | 1 | ARG 52h | 30.15 | G 506(C),G 507(C),C 612(D) |
| LYS 221h | G 507(C) G 508(C),T 611(D) | |||
| 2ramA | 1 | ARG 33h | 15.8 | G 6(D) A 7(D) |
| ARG 187t | ||||
| 1a73Ae | 1 | ARG 74 | 29.97 | A 17(D) G 18(D) |
| GLN 63 | G 16(D) A 17(D) | |||
| ASN 57 | A 15(D), G 16(D) | |||
| 6mhtA | 1 | ARG 240p | 16.7 | A 425(D) G 426(D) |
| ARG 228p | A 425(D) | |||
| TYR 242− | ||||
| 1a1gAf | 1 | ARG 124 | 27.43 | G 7(B) G 8 (B),C 9(B) |
| ASN 121 | G 8(B) C 9 (B) | |||
| ARG 146 | G 6(B) G 7 (B),C 55(C) | |||
| Isolated | ARG 174 | C 3(B) G 4 (B),T 5(B) | ||
| Isolated | ARG 180 | A 1(B) G 2 (B),C 3(B) | ||
| 1meyCf | 1 | ASN 19 | 35.45 | G 9(A) A 10 (A) |
| GLN 16 | A 10(A) A 11 (A) | |||
| 2 | LYS 22 | 27.85 | A 8(A) G 9 (A) | |
| GLN 44 | C 7(A) A 8 (A) | |||
| 3 | ARG 72 | 25.1 | A 4(A) G 5 (A) | |
| LYS 50 | G 5(A), G 6(A),T 7(B),G 8(B) | |||
| 1ubdCf | 1 | LYS 339 | 30.4 | G 32(B) G 33(B) |
| ARG 342 | T 31(B) G 32(B), C 8(A) | |||
| Isolated | ASN 369 | A 29(B) A 30(B) | ||
| Isolated | GLN 396 | A 27(B) A 28(B) | ||
| 1gd2Ee | 1 | ARG 82 | 38.6 | G −6(B) G −5(B),A 4(A) |
| ASN 86 | T 2(A) A 3(A) | |||
| GLN 85 | G −6(B) | |||
| 2 | ARG 94 | 33.96 | C −1(A) G 1(A) | |
| GLN 90 | T −3(B), T −4(B),G 1(A),T 2(A) | |||
| 1tc3C | 1 | ARG 236p | 24.65 | G 7(A) G 8(A) |
| ARG 240h | G 8(A), T 9(A) | |||
| 2nllB | 1 | ARG 328m | 16.78 | G 514(C) G 515(C),T 516(C),G523(D) |
| TYR 315y | ||||
| PHE 327t | ||||
| GLN 332p |
aThe reported cation–π/H-bond stair motif interactions of the protein residue (column 2) with the corresponding DNA bases (column 4) are given in bold face. The additional interactions detected by the present study are given in normal font. The other cation–π/H-bond stair motif interactions which belong to the same protein but do not form clusters are indicated as ‘isolated’ and are given in bold, italics.
bThe conservation information obtained from HOMSTRAD (26). The extent of conservation is given as superscript. t, totally conserved (>99% of list from the alignment); h, highly conserved(>90%); p, partially conserved(50–90%); m, conserved mutation; and dashes, non-conserved.
cδASAavg is the average loss of accessible surface area (Å2) upon complexation with DNA, averaged over all residues in a given cluster.
dNucleic acid base, the residue number, chain identifier (in parenthesis).
eSingle member family(26).
fMultispecific family of zinc fingers.
Thus, there are instances with the occurrence of one or more reported residues interacting with the bases of DNA either in the same or in different interacting clusters. In addition to this, there are additional Arg, Lys, Asn or Gln and aromatic residues present in these clusters that may or may not interact with the bases of the DNA.
In contrast to the above set, the reported residues from the proteins in eight families of the dataset do not form any cluster even with the least overlap criterion (2%). Such reported interactions are called the ‘non-cluster-forming’ interactions. There are 28 such non-cluster-forming and isolated interactions present in the dataset. In order to verify whether these non-cluster-forming interactions form clusters with any other amino acid residue in the protein, a separate analysis was carried out with all the 20 amino acid residues. It was found that even in the presence of all the amino acid residues these 28 reported residues do not form clusters (data not shown). Thus, it appears that some proteins interact with the DNA as a cluster of residues while others prefer to interact at the individual residue level.
The clusters detected at the 6% overlap are used for further analyzes, as it gave a good trade-off between the strength of the interaction existing between the side chains and the number of the reported interactions detected as clusters. It is interesting to note that in most cases if there is an interacting cluster, it occurs at the 6% overlap cutoff. Only in a few cases, the interacting clusters appear at the 4 or the 2% cut-off and are not seen at the 6% overlap criterion. This emphasizes the fact that the presence of an interacting cluster is a property of the given protein.
Geometry of protein–DNA interface interaction
The amino acid residues interacting with the DNA through the cation–π/H-bond stair motif can be classified as (1) those that are part of the cluster which contains other cationic and/or aromatic residues of proteins and (2) those that are not part of any cluster. The geometrical details of both types of residues interacting with the DNA are quantified in terms of the parameters (d, θ) described in the Methods. The frequency distribution of the parameters d and θ are analyzed for the cluster-forming and the non-cluster-forming categories. The plots are shown in figures S2a and S2b, respectively, for the two cases.
The statistical significance of the difference in the d and θ distributions between the cluster-forming and the non-cluster-forming cases were analyzed the using the Mann–Whitney –U-test and the Kolmogorov–Smirnov test. Both the tests consistently agreed on the fact that there is a statistically significant difference in d distribution between the cluster-forming and the non-cluster-forming cases (U-test: U = 5776, P = 0.001, Kolmogorov–Smirnov test: D = 0.21, P = 0.03). On the other hand, the θ distributions are not statistically significant (U-test: U = 4840, P = 0.43, Kolmogorov–Smirnov test: D = 0.151, P = 0.22). The significant difference in d distribution is due to the presence of the additional interactions in the cluster-forming cases.
As reported earlier (6), Arg residues dominate the cation–π/H-bond stair motifs. Our observation also shows that Arg dominates in its interaction with the DNA, both as cluster and as isolated interactions. The θ of Arg falls into two distinct groups, in the range of 20–45° and 75–90° which correspond, respectively, to the cation–π and the H-bond interaction of the cation–π/H-bond stair motif.
Detection of additional interactions
It is interesting to note that several additional interactions of Arg, Lys, Asn and Gln residues in the interacting clusters, not detected by earlier studies, are emerging from our analysis. A large number of these newly identified interactions are in the distance range d (5.0 Å–6.0 Å) and in θ range of (50–90°). These additional interactions are not as strong as the cation–π or it is associated H-bonded interaction in the motif. However, these newly detected additional residues may play a supportive role in strengthening the interaction of the reported residues with DNA in the interacting clusters.
Types of clusters
A complete list of the interacting clusters (consisting of the reported residues) from all the proteins obtained at the 6% overlap criterion is given in Table 2. An examination of the list shows the presence of two different types of interacting clusters classified based on the presence or the absence of the aromatic residues in a cluster. The Type I clusters consist of the reported interaction, along with the additional Lys or Arg as well as the aromatic residues like Phe and Tyr in a cluster. Unlike the case of the protein–protein interfaces (10,17), Trp is not detected as part of interacting cluster from the present dataset. This might be because of the presence of a bulky side chain of Trp that is less preferred at the DNA interfacial regions.
Apart from the reported cationic residues and the aromatic residues, the Type I clusters contain additional Arg, Lys, Asn and Gln residues. These additional residues in the cluster may or may not interact directly with the DNA base. For example, there is a Type I cluster observed in the SAP1 Transcription factor from Homo sapiens (1bc8C). In this cluster, Arg 61 and Arg 64 (shown as yellow vdw spheres, Figure 2a) are the two reported residues involved in the cation–π/H-bond stair motif formation with the bases of DNA. Arg 61 simultaneously forms a cation–π interaction with G5 of DNA and also a H-bond with G6 in the cation–π stair motif. Similarly, Arg 64 forms cation–π stair motif with the bases C4 and G5. Thus, Arg 61 and Arg 64 are involved in the specific recognition of G (Figure 2a). Apart from these two residues, the clustering method has picked Tyr 65, Tyr 67, Lys 74, and Tyr 82 as part of the same cluster. Tyr 65 (brown, Figure 2a) forms a hydrogen bond with A7 of DNA and a cation–π interaction with Arg 61. This is an instance of a cation–π interaction formed exclusively between the protein side chains (Arg 61–Tyr 65) with (d, θ) – (3.83 Å, 17.69°). The orientation of the Arg 61 is held intact by its interaction with Tyr 65 in the cluster and thereby imparts specific recognition of G of DNA. Lys 74 forms a cation–π interaction with Tyr 82 (both in blue, Figure 2a). Tyr 67 (also brown, Figure 2a) interacts non-specifically with the backbone of the DNA. Hence, there are the two subgroups of residues in the cluster, one comprising Arg 61, 64 and Tyr 65, involved in specific base recognition, and the second one consisting of Lys 74 and Tyr 82, not involved in direct interaction with the bases. Tyr 67 links the two subgroups of residues by interacting with Arg 64 and Lys 74. Tyr 67 is also found to interact with the DNA backbone. A similar trend of interacting residues is also observed in the other GA-binding protein of Mus musculus (1awcA). Apart from the ETS proteins, the residues reported from RAP1 (1ignA), methyltransferase (6mhtA) and the thyroid hormone receptor protein (2nllB) form the Type I cluster. An examination of the size of the interacting clusters in ETS proteins (1awcA, 1bc8C) and thyroid hormone receptor (2nllB) as a function of the overlap criterion has shown that the number of residues in the clusters increases (cluster expands) with decrease in the overlap criterion. This trend of cluster expansion as a function of the overlap criterion has been observed earlier in the case of the active site clusters (18). Hence these expanding clusters can be considered as a profile characteristic of the particular family of DNA binding proteins especially of those forming Type I clusters.
Figure 2.
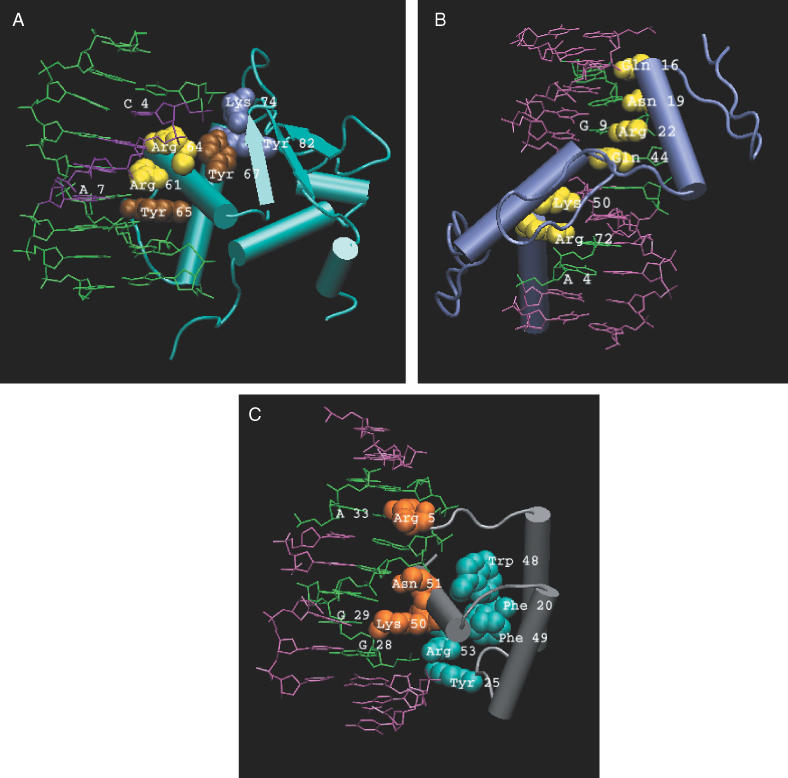
(A) Interface cluster (Type I) in SAP1 ETS transcription factor (1bc8C) of H.sapiens. The residues constituting the cluster are represented as vdw spheres. The sequence of DNA that directly interacts with the amino acids in the cluster is shown in purple. The residues Arg 61 and 64 interacting with DNA through cation–π interaction are shown in yellow, Tyr 65 and Tyr 67 in brown vdw spheres. Lys 74 and Tyr 82 are colored blue and are not involved in base recognition. Tyr 67 interacts with the backbone of DNA. The Figures 2 and 3 are prepared using VMD (28). (B) Interface clusters (Type II) in a synthetic zinc finger construct (1meyC). The residues Gln 16, Asn 19, Lys 22, Gln 44, Lys 50, Arg 72 are directly involved in base recognition and are represented as yellow vdw spheres. The bases of DNA that are specifically recognized by these residues are coloured green. (C) Interface and non-interface interactions in Engrailed Homeodomain of D.melanogaster (2hddA). The non-cluster-forming residues involved in specific base recognition are represented as orange vdw spheres. The cluster that is highly conserved and close to the interacting site is colored blue. Regions of DNA interacting with the proteins are colored green.
Type II clusters consist only of Arg, Lys, Asn, or Gln residues and aromatic residues are completely absent from these clusters. Most of the DNA binding protein families from the dataset, such as repressors, Homeodomains, REL homology domains, zinc fingers and Leucine Zipper proteins yield clusters of this category. In many families, there is more than one reported cationic residue in the cluster, and a strong van der Waals contact is observed amongst both these reported interacting protein side chains. In a synthetic zinc finger construct (1meyC) a series of residues, namely Gln 16 and Asn 19, Lys 22 and Gln 44, Lys 50 and Arg 72 (shown as yellow van der Waals spheres, Figure 2b), interact with specific DNA bases as separate clusters. On examining the clusters, it is evident that, not only do the protein residues recognize specific DNA bases, but also undergo tight interaction with the other residue side chains in the cluster. The strength of these tight interactions of the residues forming Type II clusters is established by their tendency to form clusters even at the high overlap criteria of 10 and 12%. It is seen that the size of the Type II clusters from yeast mating protein alpha (1akhA) and PBX1 homeodomain (1b72B), REL homology proteins, zinc fingers from the dataset is the same from 6% to a very high overlap criterion of 12%. In other words, these clusters do not expand upon decreasing the overlap criterion and a tight interaction of the side chains is observed. Not only the van der Waals contacts, but also π–π interaction of the non-aromatic (delocalized π orbital of Asn, Gln and Arg) residues could be instrumental in imparting greater rigidity to these side chains so as to bind the bases of DNA more strongly and specifically.
It can be seen from Table 2 that in RAP1 DNA binding protein (1ignA), PAP1 Leucine Zipper (1gd2E) and in zinc finger (1meyC), more than one interacting cluster is found. In such cases, both these interacting clusters are of the same type. Both the interacting clusters are Type I in RAP1 DNA binding protein and Type II in zinc finger and the PAP1 Leucine Zipper (Table 2). In some proteins along with the interacting clusters, isolated interactions of the cation–π/H-bond type are also present. These isolated interactions are indicated in bold italics in Table 2.
ASA loss upon complexation
In order to distinguish the clusters that interact with the DNA from all the other clusters in a protein, we have evaluated the percentage relative accessibility of the residues from the protein–DNA complexes as well as from the proteins in isolation. The loss of accessible surface area [δASA (Å2)] of the residues upon complexation with the DNA and the average loss in percentage accessibility per cluster [δASAavg (Å2)] are calculated.
There are a total of 141 clusters obtained from the proteins of the dataset, whose reported interacting residues formed clusters. Amongst these, 71 clusters show zero δASA and are not interacting with the DNA. About 48 clusters have lost significant ASA (δASAavg > 10%) upon complexation, while 22 show negligible loss of ASA (δASAavg ≤ 10%). Amongst the 48 interacting clusters, 27 contain residues interacting specifically with the bases of the DNA through the cation–π/H-bond stair motif. The δASAavg for all these 27 clusters is listed in Table 2. The details of the other clusters that show interaction with either the DNA backbone or the bases (not through cation–π/H-bond motif) are provided in Table S2.
It is interesting to note that among the interacting clusters the loss in ASA is more for Type II clusters (δASAavg ≥ 25% per cluster) than in Type I cluster (δASAavg ≤ 25%). This may be because of lesser number of residues present in the Type II cluster. Two or three residues mediate the protein–DNA interaction in Type II cluster whereas in the Type I cluster, it is shared by a larger number of residues.
In summary, the loss of ASA can be used as an indicator to distinguish between interacting and non-interacting clusters as well as the Type I and Type II clusters.
Conservation of the residues in the clusters
The conserved residues in proteins play an important role in protein–DNA interactions. It is known from earlier studies that the amino acid residues, which interact with the DNA bases, are better conserved compared to other residues present at the surface of the protein (25). Although the amino acid residues interacting with the DNA backbone are highly conserved, the extent of conservation of residues interacting directly with the bases vary widely across families. Luscombe and Thornton (25) have classified three types of families: non-specific, multi-specific and highly specific families, based on the DNA binding specificities of the amino acid residues. ETS domain and REL Homology domains belong to a highly specific family while the Homeodomains, zinc fingers and hormone receptors belong to multi-specific family.
In order to extract conservation information for the cluster-forming residues, we have made use of the structure-based alignments provided by HOMSTRAD for homologous protein families (26). The conservation information extracted this way is reported for all residues of the interacting clusters as superscript in Table 2. The structure-based sequence comparison has also revealed a greater degree of structural alignment of α helices in ETS domains, Homeodomains, TC3 transposase (1tc3C) and Thyroid hormone receptor (2nllB). It also emphasizes the importance of a particular secondary structural conformation of a protein for its interaction with the DNA.
All the amino acid residues involved in the cation–π stair motif interaction with the nucleic acid bases are totally conserved across the family. This includes both the cluster-forming and the non-cluster-forming residues. Additionally, we find complete or partial conservation of several additional residues, which are part of the interacting clusters. These residues may or may not directly interact with the DNA bases. In the case of ETS domains forming Type I clusters, e.g. in SAP1 transcription factor (1bc8C), the Arg residues (Arg 61, Arg 64) are in direct contact with the base of DNA and are totally conserved across all the family members. Among the other residues of the cluster, Tyr 65 is in direct interaction with the DNA base and is partially conserved amongst the family. Lys 74 and Tyr 82 are part of the interacting cluster and are highly conserved. Both of these residues do not directly interact with the DNA base. However, they are involved in a cation–π type of interaction among themselves. Thus, it can be seen that the entire cluster comprises residues that are well conserved, thereby making the cluster as a whole, significant from a conservation point of view. A very similar situation prevails in GA-binding protein alpha (1awcA) of M.musculus (Table 2). In addition to this, a clear preference is seen for i, i + 3 Arg, i + 4 Tyr as interacting residues in ETS proteins (6), forming a conserved cluster at the interface.
In Type II clusters from Homeodomains, REL homology proteins and TC3 transposase, the residues that are directly interacting with the DNA bases are highly conserved across the family. The residues Arg 232, Arg 235 in PU1 transcription factor of H.sapiens (1pueE), Asn 286, Arg 290 in PBX1 homeodomain (1b72B), Arg 236, Arg 240 in TC3 transposase (1tc3C) recognize specific DNA bases as clusters in each of the proteins. All these residues are well conserved, yielding conserved clusters in the family. A high likelihood of i Asn and i + 3 or i + 4 Arg in an interacting cluster is seen in Mat α2 and PBX1 homeodomains of the Homeodomain family. Though the pair-wise interactions of the above mentioned residues in ETS as well as in Homeodomain proteins are reported earlier (6), the fact that they manifest as conserved clusters of spatially interacting amino acid residues at the interface is evident only from the present analysis.
Zinc fingers show no characteristic residue-type conservation within the family. This is because of the divergent mutations/substitutions that these proteins have undergone for carrying out different functions. From the alignment it is evident that the conservation information provided for clusters in zinc finger proteins concern mainly the conservation of the residue positions than the actual type of residues. Here the preference of i Asn, i + 3 Arg/Lys or i Lys and i + 3 Arg to form a single interacting cluster is widely exhibited among subfamilies. Also, in the case of phage repressors and methyltransferase (6mhtA) a characteristic conservation of a particular amino acid is not observed. However, a preference for i Gln, i + 11 Gln in the same cluster is seen in the case of phage repressors. More structural information is indeed needed to bring out clear preferences of amino acids in these proteins.
To summarize, as can be seen from Table 2, the reported cluster-forming amino acid residues recognizing specific bases of the DNA are highly conserved. In many cases, more than one reported residue is found to form a single completely conserved cluster. Furthermore, in several proteins the additional residues found in the same cluster are also conserved whether these residues interact directly with the DNA bases or not. This makes the cluster as a whole, conserved across the family. The presence of such conserved clusters in a family is a characteristic feature of that family and can be used as a specific signature motif for recognizing specific bases of DNA. These results emphasize the fact that the recognition of the DNA bases by amino acid residues goes beyond pair-wise interactions.
As mentioned earlier, 28 out of the 77 reported residues could not become part of clusters even at an overlap criterion as low as 2%. In such cases, we investigated other clusters with conserved residues in the vicinity of the residues interacting with the DNA. In the case of some Homeodomains, we found non-interacting clusters in close proximity to the interacting site. Such proximal clusters are found in Engrailed (2hddA) Antennapedia (9antA), Paired protein (1fjlA) homeodomains. It must be noted that several residues in these non-interacting clusters are highly conserved across the family. These conserved clusters in Engrailed homeodomain (2hddA) are depicted in cyan and the reported interacting residues as orange vdw spheres in Figure 2c. It is interesting to see that a totally conserved Trp residue (Trp 48 in 2hddA) is a part of the non-interacting cluster though no Trp is found to interact directly with any base of the DNA or as a part of any interacting cluster. These non-interacting clusters, which are proximal to the interacting residues may play a role in stabilizing the orientation of the helices and provide a suitable orientation of the long loop region that harbors the reported interacting residue (Figure 2c). The conserved Phe and Trp in all the non-interacting clusters from the above-mentioned proteins are also part of the hydrophobic core of the protein (27).
Prediction of plausible DNA binding clusters from uncomplexed protein
The clusters interacting with the DNA have been identified from the protein–DNA complexes. In an attempt to identify plausible clusters that could bind DNA from the uncomplexed structures, we scanned the PDB for unbound crystal structures, equivalent to the DNA-bound structures selected for analysis in the present case. However, we were not able to obtain suitable unbound structures for comparison. In many instances, the uncomplexed structures were either not homologous to the DNA-bound ones or not equivalent in terms of their structural domains. A solution structure of ETS domain from murine ETS-1 (1etd) and its minimized average model (1etc) were available. This protein is homologous to the ETS domain, GA-binding protein (1awcA) and was chosen for analysis.
The details of the clusters obtained at 6% for the DNA-bound protein (1awcA) is given in Table 2. In case of the minimized average NMR structure of murine ETS-1, the composition of the corresponding cluster is not the same as that of the bound protein and is fragmented into smaller clusters although they comprise equivalent aromatic residues Tyr 412 (Tyr 397 in 1awcA), Tyr 410 (Phe 395 in 1awcA) and Tyr 395(Tyr 380 in 1awcA). In the NMR model-1, the composition is slightly different, with Tyr 412 and Lys 404 (Lys 389 in 1awcA) as part of one cluster and Tyr 395 as part of another cluster. In the bound structure, the reported interacting residues, Arg 376 and Arg 379 are part of a large aromatic cluster. Curiously, the interacting residues Arg 391, Arg 394 are not part of any of the small clusters obtained in the minimized as well as the NMR model-1. It is very clear from Figure 3b, that the orientation of these Arg side chains in the bound and the NMR model-1 are completely different. This is not surprising since NMR experiments scan a range of conformations and the interacting Arg residues have adopted different conformations as shown in Figure 3a. The availability of more crystal structures will enable us to probe further into the nature of DNA binding signatures and validate our present findings.
Figure 3.

(A) Orientation of the Arg side chains in the NMR models of murine ETS-1 (1etd). The NMR model-1 is represented as the cyan Cα trace. The orientation of the Arg residue from all the models is shown as orange lines. (B) Comparison of the side chain orientation of Arg in the DNA bound GA-binding protein (1awc) with the minimized average NMR structure (1etc) of murine ETS-1 The DNA bound protein (1awc) is shown as cyan Cα trace and the minimized average structure (1etc) in orange. Arg residues are shown as lines in 1etc and as CPK in 1awc.
In summary, the recognition of DNA by proteins can either be at the individual residue level or as a sequence motif, which is a part of secondary structure such as a helix. In addition to this, the recognitions can also be at the tertiary structure level where the residues are spatial neighbors and are not necessarily sequence neighbors. The present analysis based on graph spectra has aided in identifying clusters of spatially interacting residues, which are important in protein–DNA recognition. The reported interacting residues being a part of spatially interacting clusters impart greater significance to the specific protein–DNA interaction. Studies of this kind can aid in increasing the understanding of the recognition observed at the protein–DNA interface and would help us gain new insights about the protein–DNA recognition code.
CONCLUSIONS
In the present study, we have tested the ability of those protein residues, which are already engaged in the cation–π/H-bond stair motif interaction with specific bases of DNA (6) to form clusters. The previous study had revealed that out of the 52 protein–DNA complexes considered, 37 form the cation–π stair motif interaction with the DNA bases. Out of the 77 reported amino acid residues that interact with the bases of the DNA through the cation–π/H-bond stair motif, 38 form side chain clusters with other residues in the protein at 6% overlap and the others do not. Thus about half of the amino acid residues that form cation–π stair motif in the current dataset, present themselves as clustered motifs to the DNA while the others seem to prefer pair-wise interaction. Nevertheless, the ability to form clusters is not a phenomenon restricted to any particular DNA binding protein class or family. There seems to be no rule pertaining to the fold, family, or function of proteins that guide the ability of proteins at the protein–DNA interface to form clusters.
A greater number of Arg residues are part of clusters owing to the fact that Arg could form significant number of contacts with other residues in its vicinity. This might impart greater specificity to the Arg residues to recognize particular nucleic acid bases. The Asn and Gln residues form clusters preferably at a lower criterion (<6%), as they do not form many side chain contacts with other residues in the clusters.
The analysis of the geometry of the protein residues interacting with the DNA through cation–π stair motif has revealed that the average (d, θ) values fall into distinct ranges corresponding to the cation–π (4.16 Å, 34.19°) and the H-bond (4.18 Å, 85.16°) interactions. There is a significant statistical difference in the distribution of d between the cluster-forming and the non-cluster-forming categories. There is no such difference in the θ distribution.
Two types of clusters are found to interact with the DNA. Type I clusters consist of Arg, Lys, Asn and Gln residues along with the aromatic residues like Phe and Tyr in a cluster. Trp is however completely absent from the protein–DNA interface region in the present study and is not detected as a part of any interacting cluster. The second type of cluster (Type II) consists only of Arg, Lys, Asn and Gln residues and is devoid of any aromatic residue. The presence of strong van der Waals overlap existing among the side chains, suggests tight interaction of the residues occurring at the protein–DNA interface. This is revealed by the consistent presence of these clusters even at a very high overlap criterion. In many proteins, when there is more than one reported interaction, both the residues are part of a single cluster. The Type I clusters also show smaller (δASAavg) compared to the Type II clusters. The protein residues forming Cation–π/H-bond stair motif interactions and detected as clusters are completely conserved within the family. Additionally, most of the additional residues comprising the cluster also show a great degree of conservation. This makes the clusters as a whole, obtained at the interface, conserved and significant from an evolutionary perspective. The presence of such conserved clusters at the interface imparts more structural and functional significance to these protein–DNA interactions. This highlights the importance of side chain – side chain contacts prevailing at the protein–DNA interface. Also, these conserved interacting clusters within families can act as specific signatures or profiles characteristic of the particular family of DNA binding proteins.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at NAR Online.
Acknowledgments
ACKNOWLEDGEMENTS
Support from the computational genomics grant from the Department of Biotechnology, India is acknowledged. Authors thank Prof. N.V. Joshi, CES, IISc, for the valuable discussions on the statistical analysis carried out in the paper.
REFERENCES
- 1.Dougherty D.A. (1996) Cation–π interactions in chemistry and biology: a new view of benzene, Phe, Tyr and Trp. Science, 271, 163–168. [DOI] [PubMed] [Google Scholar]
- 2.Zacharias N. and Dougherty,D.A. (2002) Cation-π interactions in ligand recognition and catalysis. Trends Pharmacol. Sci., 23, 281–287. [DOI] [PubMed] [Google Scholar]
- 3.Mao L., Wang,Y., Liu,Y. and Hu,X. (2004) Molecular determinants for ATP binding in proteins: a data mining and quantum chemical analysis. J. Mol. Biol., 336, 787–807. [DOI] [PubMed] [Google Scholar]
- 4.McFail-Isom L., Shui,X. and Williams,L.D. (1998) Divalent cations stabilize unstacked conformation of DNA and RNA by interacting with base π systems. Biochemistry, 37, 17105–17111. [DOI] [PubMed] [Google Scholar]
- 5.Wintjens R., Lievin,J., Rooman,M. and Buisine,E. (2000) Contribution of cation–π interactions to the stability of protein-DNA complexes. J. Mol. Biol., 302, 395–410. [DOI] [PubMed] [Google Scholar]
- 6.Rooman M., Lievin,J., Buisine,E. and Wintjens,R. (2002) Cation–π/H bond stair motifs at protein–DNA interfaces. J. Mol. Biol., 319, 67–76. [DOI] [PubMed] [Google Scholar]
- 7.Wodak S.J. and Janin,J. (2003) Structural basis of molecular recognition. Adv. Protein Chem., 61, 7–73. [DOI] [PubMed] [Google Scholar]
- 8.Chothia C. and Janin,J. (1975) Principles of protein–protein recognition. Nature, 256, 705–708. [DOI] [PubMed] [Google Scholar]
- 9.Lee B. and Richards,F.M. (1971) The interpretation of protein structures: estimation of static accessibility. J. Mol. Biol., 55, 379–400. [DOI] [PubMed] [Google Scholar]
- 10.Jones S. and Thornton,J.M. (1996) Principles of protein–protein interactions. Proc. Natl Acad. Sci. USA, 93, 13–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jones S., van Heyningen,P., Berman,H.M. and Thornton,J.M. (1999) Protein–DNA interactions: a structural analysis. J. Mol. Biol., 287, 877–896. [DOI] [PubMed] [Google Scholar]
- 12.Janin J., Miller,S. and Chothia,C. (1988) Surface, subunit interfaces and interior of oligomeric proteins. J. Mol. Biol., 204, 155–164. [DOI] [PubMed] [Google Scholar]
- 13.Fabian G., Stenberg,D.M., Vakser,I.A. and Ben-Tal,N. (2001) Residue pairing preferences at protein–protein interfaces. Prot. Struct. Funct. Genet., 43, 89–102. [PubMed] [Google Scholar]
- 14.Pabo C.O. and Nekludova,L. (2000) Geometric analysis and comparison of protein–DNA interfaces: why is there no simple code for recognition. J. Mol. Biol., 301, 597–624. [DOI] [PubMed] [Google Scholar]
- 15.Nadassy K., Wodak,S.J. and Janin,J. (1999) Structural features of protein–nucleic acid recognition sites. Biochemistry, 38, 1999–2017. [DOI] [PubMed] [Google Scholar]
- 16.Luscombe N.M., Laskowski,R.A. and Thornton,J.M. (2001) Amino acid-base interactions: a three dimensional analysis of protein–DNA interactions at an atomic level. Nucleic Acids Res., 29, 13, 260–2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brinda K.V., Kannan,N. and Vishveshwara,S. (2002) Analysis of homodimeric interface by graph spectral methods. Protein Eng., 15, 265–277. [DOI] [PubMed] [Google Scholar]
- 18.Kannan N. and Vishveshwara,S. (1999) Identification of side chain clusters in protein structures by graph spectral method. J. Mol. Biol., 292, 441–464. [DOI] [PubMed] [Google Scholar]
- 19.Berman H.M., Westbrook,J., Feng,Z., Gilliland,G., Bhat,T.N., Weissig,H., Shindyalov,I.N. and Bourne,P.E. (2000) The protein data bank. Nucleic Acids Res., 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anderson E., Bai,Z., Bischof,C., Blackford,S., Demmel,J., Dongarra,J., Du Croz,J., Greenbaum,A., Hammarling,S., McKenney,A. and Sorensen,D. (1999) Lapack User's Guide, 3rd Edn. Society for Industrial and Applied Mathematics, Philadelphia, PA. [Google Scholar]
- 21.Hall K.M. (1970) An r-dimensional quadratic placement algorithm. Manag. Sci., 17, 219–229. [Google Scholar]
- 22.Blow D.M. (1959) To fit a set of points by least squares. Acta Cryst., 13,168. [Google Scholar]
- 23.Burley S.K. and Petsko,G.A. (1986) Amino-aromatic contacts in proteins. FEBS Lett., 203, 139–143. [DOI] [PubMed] [Google Scholar]
- 24.Hubbard S.J. (1996) 'NACCESS vZ.2.1.1. Computer Program, Biomolecular Structure and Modelling Unit, University College, London, UK.
- 25.Luscombe N.M. and Thornton,J.M. (2002) Protein–DNA interactions: amino acid conservation and the effects of mutations on binding specificity. J. Mol. Biol, 320, 991–1009. [DOI] [PubMed] [Google Scholar]
- 26.Mizuguchi K., Deane,C.M., Blundell,T.L. and Overington,J.P. (1998) HOMSTRAD: a database of protein structure alignments for homologous families. Protein Sci., 7, 2469–2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mcknight, Keith R.Yamamoto (1992) Transcriptional regulation. Cold Spring Harbour Laboratory Press, Cold Spring Harbour, NY, pp. 535–575. [Google Scholar]
- 28.Humphrey W., Dalke,A. and Schulten,K. (1996) VMD—visual molecular dynamics. J. Mol. Graphics, 14, 33–38. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.