


Abstract
Cytosine methylation in CpG dinucleotides is believed to be important in gene regulation, and is generally associated with reduced levels of transcription. Methylation-mediated gene silencing involves a series of DNA–protein and protein–protein interactions that begins with the binding of methyl-CpG binding proteins (MBPs) followed by the recruitment of histone-modifying enzymes that together promote chromatin condensation and inactivation. It is widely known that alterations in methylation patterns, and associated gene activities, are often found in human tumors. However, the mechanisms by which methylation patterns are altered are not currently understood. In this paper, we investigate the impact of oxidative damage to a methyl-CpG site on MBP binding by the selective placement of 8-oxoguanine (8-oxoG) and 5-hydroxymethylcytosine (HmC) in a MBP recognition sequence. Duplexes containing these specific modifications were assayed for binding to the methyl-CpG binding domain (MBD) of one member of the MBP family, methyl-CpG binding protein 2 (MeCP2). Our results reveal that oxidation of either a single guanine to 8-oxoG or of a single 5mC to HmC, significantly inhibits binding of the MBD to the oligonucleotide duplex, reducing the binding affinity by at least an order of magnitude. Oxidative damage to DNA could therefore result in heritable, epigenetic changes in chromatin organization.
INTRODUCTION
Cytosine methylation patterns in higher eukaryotes are important in gene regulation. Following DNA replication, methylation occurs enzymatically at the C5 position of cytosine residues, preferentially in hemimethylated CpG dinucleotides (1–3). Methylation in promoter regions is generally associated with transcription repression (4–6). The proposed mechanism by which CpG methylation represses gene expression is through the binding of specific proteins, the methyl-CpG binding proteins (MBPs). Methyl-CpG binding protein 2 (MeCP2) and other members of the MBP family contain a well-conserved 70–75 amino acids domain that discriminates between oligonucleotides (ODNs) containing methylated and unmethylated CpG dinucleotides (7–17). Upon binding to methylated DNA, the MBPs then recruit cytosine methyltransferases, histone deacetylases and other proteins involved in chromatin remodeling (7–24). The initial binding of the MBPs to methylated CpGs is a critical event in the epigenetic regulation of gene activity.
Cytosine methylation patterns are frequently altered in human tumors (25–29). It has long been recognized that the hydrolytic deamination of 5-methylcytosine (5mC) residues to thymine could account for some mutations observed in human tumors. More recently however, it has been recognized that epigenetic silencing of tumor suppressor genes, or the aberrant loss of methylation of promoter regions of transforming genes are frequently observed in human tumors, in the absence of mutation. The mechanisms by which other forms of DNA damage might result in the alteration of methylation patterns and transcriptional activity have not been thoroughly studied.
The precise nature of the high affinity of MBPs for methylated CpG dinucleotides is not as yet fully understood, however, specific contacts with guanine functional groups as well as a hydrophobic patch interacting with the symmetric 5-methyl groups through the well-conserved methyl-CpG binding domain (MBD) have been implicated (8,9,20,30–34). Upon the basis of these previous findings, we predicted that oxidation of guanine or 5mC would disrupt MBP interactions with DNA. Our study focuses on the effects of two endogenous oxidative damage products derived from reactive oxygen species (ROS) on MBP binding.
The guanine oxidation damage product, 8-oxoguanine (8-oxoG), is a major form of DNA damage (35–40). The oxidation of guanine to 8-oxoG would convert the N7 position of guanine from a hydrogen bond acceptor into a hydrogen bond donor, as well as replace the 8-proton with an oxygen atom (Figure 1A). Either modification could potentially interfere with the recognition of the methyl-CpG dinucleotide by MBPs. It is known that conversion of the N7 hydrogen-bond acceptor of guanine into the hydrogen-bond donor of 8-oxoG is exploited by the 8-oxoG repair enzyme hOGG1 glycosylase to discriminate between undamaged and oxidized guanine residues (41,42), suggesting that the purine N7 position may be a generally important landmark for the specificity of some DNA–protein interactions.
Figure 1.
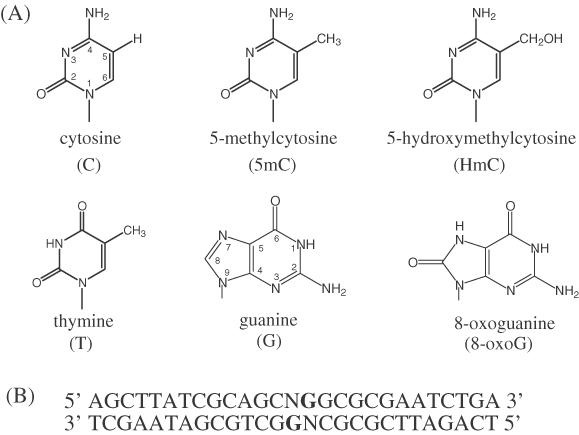
(A) Structures of cytosine, 5-methylcytosine, 5-hydroxymethylcytosine, thymine, guanine and 8-oxoG. (B) Sequence of ODN duplex used in EMSA. Replacement of position N with cytosine, 5mC or HmC in both strands yields duplexes C/C, 5mC/5mC and HmC/HmC, respectively. The duplexes are named based upon the modification at the N position, with the slash denoting that the modification is on different strands within the central CpG. Modification in the upper strand is indicated before the slash, whereas modification to the lower strand is indicated after the slash. Four duplexes containing 8-oxoG were used and are named in a manner similar to, e.g. the 5mC/5mC8oxoG duplex contains 5mC at position N in both the upper and lower strands with 8-oxoG replacing the boldface G in the lower strand. The duplex containing 5mC at position N in the upper strand and T at position N in the lower strand was given the name 5mC/T.
In the human genome, only ∼4% of cytosine residues are methylated (43). Because 5mC is a minor base, substantially less work has been done on damage products derived from 5mC. In addition, some 5mC reaction products deaminate readily to the corresponding thymine derivatives and are therefore lost in the background generated by the more predominant thymine (44). The methyl groups of both thymine and 5mC are susceptible to oxidation with 5mC being slightly more reactive (44,45). The oxidation of 5mC can generate 5-hydroxymethylcytosine (HmC), 5-formylcytosine and 5-carboxycytosine (44,46). HmC (Figure 1A) has been detected in mammalian cells and a specific HmC glycosylase activity has been partially purified from mammalian tissues (47–50).
It is well recognized that the thymine methyl group is an important landmark for sequence-specific DNA–protein interactions (51,52). The oxidation of the methyl group of thymine generates 5-hydroxymethyluracil (HmU) recently shown to interfere with transcription factor binding (53–55). We therefore suspected that oxidation of the hydrophobic methyl group of 5mC to the hydrophilic hydroxymethyl group could also interfere with MBP binding.
To investigate the effects of guanine and 5mC oxidation on MBP binding, a series of ODN substrates were prepared that contain 5mC, HmC and 8-oxoG residues at selected positions (Figure 1B). The binding of the MBD of MeCP2 to this series of ODN duplexes was then studied using a gel electrophoretic mobility shift assay (EMSA). Our results reveal that the methylation in either strand of a CpG-containing ODN increases the binding of the MBP by an order of magnitude, and symmetric methylation increases the binding affinity by two orders of magnitude. The apparent methylation-induced increased binding affinity is near the theoretical maximum predicted for the contribution of a methyl group (56). The replacement of a 5mC residue with HmC, or the replacement of guanine by 8-oxoG is observed to effectively reverse the increase in binding affinity afforded by a 5mC residue. Therefore, oxidation of 5mC to HmC or guanine to 8-oxoG in vivo could inhibit the initial binding of MBPs, interfering with subsequent steps in the chromatin condensation cascade, resulting in potentially heritable epigenetic alterations.
MATERIALS AND METHODS
ODN synthesis
The seven ODN 27mers (Figure 1B) containing normal bases and the modified base HmC were prepared by standard solid-phase synthesis using either the Gene Assembler Plus (Pharmacia) or Expedite Nucleic Acid Synthesis System (Applied Biosystems) automated DNA synthesizers (57,58). The sequence (Figure 1B, top strand) and its complement (Figure 1B, bottom strand) were chosen based upon binding experiments conducted by Free et al. (9) and Hendrich et al. (32). The HmC phosphoramidite was prepared according to the method developed by this laboratory (59). All other phosphoramidites used were obtained from Glen Research. The HmC-containing ODNs were removed from the solid support and deprotected in aqueous ammonia (Aldrich) at 65°C for 3 days. All other ODNs were removed from the solid support and deprotected in aqueous ammonia (Aldrich) at 60°C overnight. The deprotected ODNs were purified with Poly-Pak II cartridges (Glen Research). The sequence composition of the ODNs was confirmed via high-performance liquid chromatography (HPLC) analysis following digestion of the ODNs with nuclease P1 (Sigma) at 37°C for 1 h and bacterial alkaline phosphatase (Sigma) at 37°C overnight. ODNs containing 8-oxoG at the central guanine position (Figure 1B, boldface) were synthesized by TriLink Biotechnologies (San Diego, CA).
Protein expression and purification
The pAFB105 construct encoding 6× His-tagged MBD of mouse MeCP2, residues 77–165 (8,9), in a pET6H vector, was overexpressed in Escherichia coli BL21 (DE3)/pLysS. The expression and purification was carried out essentially according to the protocol of Free et al. (9). A total of 4 litres of LB was inoculated with an overnight culture of a transformant E.coli BL21 (DE3)/pLysS at a 1:20 dilution and grown to an A600 of ∼0.5–0.6 prior to induction with 0.5 mM isopropyl-β-d-thiogalactoside for 3 h. The cells were harvested, washed in 50 mM HEPES, pH 7.9, 0.1 M NaCl, and then resuspended in binding buffer (5 mM imidazole, 20 mM Tris–HCl, pH 8.0, 0.25 M NaCl, 10% glycerol, 0.1% Triton X-100 and 10 mM 2-mercaptoethanol) and sonicated. Cellular debris were pelleted at 16 000 r.p.m. at 4°C for 20 min. Cleared lysate was retained and mixed with 6 ml of 50% Ni-nitriloacetic acid Superflow agarose resin (Qiagen) in binding buffer. Binding was conducted at 4°C for 1 h with gentle shaking at ∼250 r.p.m. The mixture was packed onto a 20 ml column (BioRad) with a final resin volume of 3 ml. The column was washed 3 × 3 ml of binding buffer containing 30 mM imidazole, and eluted with 3 × 1.5 ml of binding buffer containing 0.5 M imidazole. All elution fractions were loaded onto a 3.5 ml Fractogel EMD SO32− 650(M) column (Merck), washed with 2 × 3.5 ml binding buffer, and step eluted with 4 × 3.5 ml binding buffer containing 0.25 M NaCl, 3 × 3.5 ml binding buffer containing 0.5 M NaCl and 2 × 3.5 ml binding buffer containing 1 M NaCl. The majority of pure protein eluted in the fractions containing 0.5 M NaCl. The eluates were pooled and dialyzed against 2 litres of 100 mM NaCl, 10% glycerol and 0.1% Triton X-100. The purified protein was judged as >95% pure by SDS–PAGE, and western-blot analysis showed that the protein reacted strongly with anti-6× His antibody (Santa Cruz Biotech).
Electrophoretic mobility shift assay
The 27mer ODNs (Figure 1B) were 5′-32P-end labeled by T4 polynucleotide kinase (New England Biolabs) with [γ-32P]ATP (ICN Life Sciences) under conditions recommended by the enzyme supplier. The labeled ODNs were then purified using G50 Sephadex columns (Boehringer Mannheim). The labeled complementary strand (Figure 1B, upper strand) was incubated with 1.5-fold excess of the unlabeled strand (Figure 1B, lower strand) in 20 mM HEPES, pH 7.3, 1 mM EDTA at 95°C for 10 min and then allowed to cool to room temperature slowly for duplex formation. Nine duplexes were annealed, with all possible combinations of C, 5mC and HmC at position N indicated in both strands (Figure 1B). To confirm duplex formation and cytosine modifications, the annealed ODNs were digested with MspI and HpaII (New England Biolabs) according to the conditions recommended by the enzyme supplier (59), and the products were sized on denaturing 20% (v/v) polyacrylamide gels. MspI was able to cleave duplexes in which the cytosine was modified, both symmetrically and asymmetrically in the CpG, with 5mC and HmC, whereas HpaII was only able to cleave the unmodified duplex (data not shown). These results are in accordance with the data presented by Tardy-Planchaud et al. (59) and confirm the presence of cytosine modifications in the duplexes. The four duplexes containing 8-oxoG were also labeled and annealed as described above. The presence of 8-oxoG within the duplexes was confirmed by treatment of the duplexes with hOGG1 glycosylase (New England Biolabs) according to the conditions recommended by the enzyme supplier. The reaction products were visualized on denaturing 20% (v/v) polyacrylamide gels.
Purified MBD at varying concentrations (0, 0.25, 0.5, 1, 2, 4, 8, 16, 32, 64, 128 and 256 nM) was incubated with 2 nM labeled duplex, 50 ng/μl of poly(dA-dT)·poly(dA-dT) (Sigma) in 20 mM HEPES, pH 7.3, 1 mM EDTA, 10 nM (NH4)2SO4, 1 mM DTT, 0.2% Tween-20 and 30 mM KCl for 15 min at room temperature in a 30 μl reaction volume, prior to the addition of 7.5 μl loading buffer (60% 0.25× TBE and 40% glycerol) (9). The binding reactions were then loaded onto 10% non-denaturing polyacrylamide (37.5:1, acrylamide/bis-acrylamide) gels that had been pre-run for 1.5 h at 200 V at 4°C. Samples were electrophoresed at 250 V for 2.5 h at 4°C. Visualization and quantification of the gels were carried out using a PhosphorImager (Molecular Dynamics) and the ImageQuant 5.0 software (Amersham Biosciences).
Binding model and data analysis
It is believed that the MBD of MeCP2 binds to its symmetrically methylated DNA substrate as a monomer (8); therefore, the following non-cooperative, single-site binding scheme describes the binding of MBD to its substrate:
![]()
where E is unbound MBD, O is the unbound ODN duplex and EO is the MBD–ODN duplex complex. From this scheme, the dissociation constant (Kd), or the concentration of MBD where half of the ODN duplex is bound, for the MBD–ODN duplex complex can be defined as follows:
![]()
Rearrangement of yields the following equation:
![]()
From the EMSA data, the radioactivity in each band was quantified using ImageQuant 5.0 software and the fraction bound was plotted as a function of protein concentration. The fraction of duplex bound was determined at each protein concentration as follows:
![]()
When [E] ≫ [O], then [E]total − [EO] = [E] ≈ [E]total, where [E]total is the total concentration of MBD, both bound and unbound, substitution of into yields the equation:
![]()
To determine the dissociation constants for 14 ODN duplexes used in this study, the average of a minimum of three data sets obtained for each duplex were fit to by non-linear regression using SigmaPlot software (SPSS Science).
The apparent-free energy change (ΔΔGap) in MBD binding to the fully methylated duplex compared with all other duplexes was calculated according to the following equation:
![]()
where R = 1.987 × 10−3 kcal mol−1 K−1 and T = 298 K. A positive ΔΔGap would indicate decreased binding of MBD to the duplex of interest as compared to the symmetrically methylated duplex, whereas a negative ΔΔGap indicates increased binding of MBD to the duplex of interest. Error propagation in the determination of the ΔΔGap from experimentally determined Kd values were calculated using the basic equation for error propagation given by Bevington (60).
RESULTS
The ODN duplexes with the sequences represented in Figure 1B were synthesized based upon a sequence that had been previously used to examine the binding properties of the MBD of MeCP2 (9,32). The 27mer contains a central CpG dinucleotide, which was systematically modified as described below. The nine duplexes synthesized, with all possible combinations of hemi- and symmetrical C, 5mC and HmC (Figure 1A) within the central CpG, were assayed for binding to the MBD of MeCP2 at different protein concentrations ranging from 0 to 256 nM (Figures 2 and 3). Four duplexes containing a single 8-oxoG (Figure 1A) either across from 5mC, next to 5mC, within a fully methylated CpG dinucleotide, or within an unmethylated CpG dinucleotide were also assayed for binding to the MBD of MeCP2 in the same manner (Figures 3C and 4). A last duplex containing 5mC in position N on the upper strand and T in position N in the lower-strand (Figure 1B), resulting in a T:G mispair next to a 5mC:G base pair, was also assayed (Figures 2 and 3A). The Kd (Table 1) was determined for each duplex using non-linear regression to fit the equation for simple, non-cooperative monomeric binding.
Figure 2.

Binding of C/C, C/5mC, 5mC/5mC and 5mC/T duplexes to varying concentrations of MBD from 0 to 256 nM assayed via EMSA.
Figure 3.
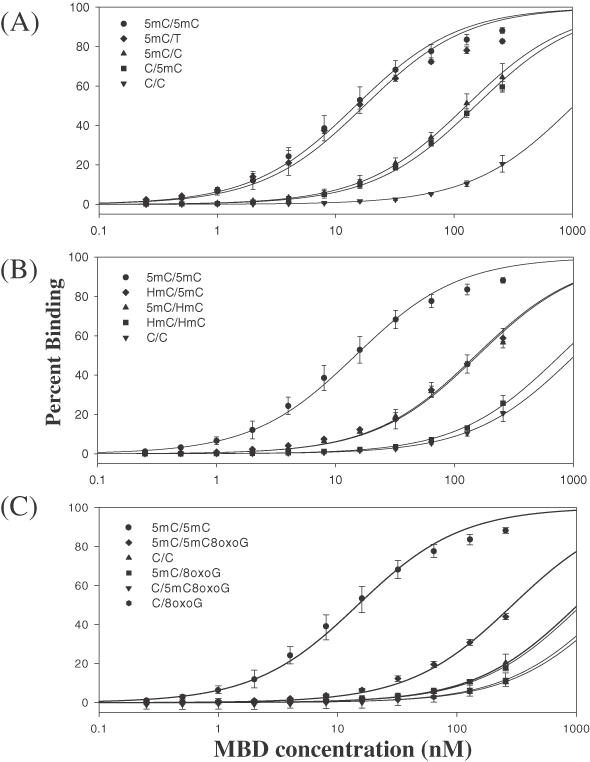
Non-linear regression of the plot of average percentage binding (determined from EMSA with three or more sets of titrations per duplex) and concentration of MBD for duplexes containing (A) normal purines within the CpG site, (B) HmC within the CpG site and (C) 8-oxoG within the CpG site.
Figure 4.
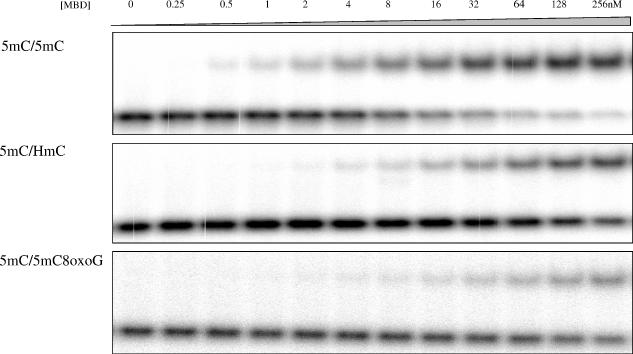
Binding of 5mC/5mC, 5mC/HmC and 5mC/5mC8-oxoG duplexes to varying concentrations of MBD from 0 to 256 nM assayed via EMSA.
Table 1. Dissociation constant and free energy change for MBD of MeCP2 binding to 27mer duplexes containing all possible combinations of hemi- and symmetrical C, 5mC and HmC in a CpG pair, containing 8-oxoG in the un-, hemi- and symmetrically methylated CpG dinucleotide, and containing a T:G mismatch within a methyl CpG sequence context.
| Duplex | Kd (nM) | Correlation coefficient (R2) | ΔΔG (kcal mol−1) |
|---|---|---|---|
| 5mC/5mC | 14.7 ± 1.0 | 0.99 | N/A |
| C/C | 1030 ± 20 | 0.99 | 2.52 ± 0.04 |
| HmC/HmC | 777 ± 15 | 0.99 | 2.35 ± 0.04 |
| C/HmC | 1100 ± 10 | 0.99 | 2.55 ± 0.04 |
| HmC/C | 953 ± 11 | 0.99 | 2.47 ± 0.04 |
| C/5mC | 152 ± 4 | 0.99 | 1.38 ± 0.04 |
| 5mC/C | 127 ± 3 | 0.99 | 1.28 ± 0.04 |
| 5mC/HmC | 157 ± 8 | 0.98 | 1.40 ± 0.05 |
| HmC/5mC | 151 ± 7 | 0.98 | 1.38 ± 0.05 |
| 5mC/5mC8oxoG | 292 ± 11 | 0.99 | 1.77 ± 0.05 |
| 5mC/8oxoG | 1110 ± 41 | 0.98 | 2.56 ± 0.05 |
| C/5mC8oxoG | 1930 ± 130 | 0.96 | 2.89 ± 0.06 |
| C/8oxoG | 2130 ± 49 | 0.99 | 2.95 ± 0.04 |
| 5mC/T | 17.5 ± 2.0 | 0.97 | 0.01 ± 0.08 |
Using SigmaPlot, the Kd for MBD binding to each of the duplexes was determined from non-linear regression of the three or more sets of data obtained from EMSA. The plot of the average percentage binding of each duplex against the concentration of MBD was fit to (see Materials and Methods). The ranges seen in the Kd values are the standard error for the regression analysis. ΔΔGap values compare the free energy change of MBD binding to the symmetrically methylated duplex versus binding to all other duplexes. In representing the duplexes, the top 32P-labeled strand (Figure 1B) is written before the slash, and the unlabeled bottom strand is written after the slash (Figure 1B).
It has been shown that the binding of the MBD of MeCP2 requires only one methylated CpG dinucleotide, and that the MBD of MeCP2 is necessary and sufficient for binding to its target duplex (8). Previously, the Kd for the binding of MBD of MeCP2 to a single methyl CpG pair was reported by Nan et al. (8) to be ∼10−9 M. The Kd for a symmetrically methylated CpG pair obtained from our binding studies, though determined with a different duplex, is comparable to data reported by both Nan et al. (8) and Free et al. (9), 14.7 ± 1.0 nM (Table 1). Nan et al. (8) also reported reduced binding of MBD to DNA containing either hemimethylated CpG or unmethylated CpG pairs. Our data confirm that duplexes with hemi- or unmethylated CpG dinucleotide sequences do not bind MBD as well as the symmetric methyl-CpG duplex, however, we note that a significant quantitative difference between the binding of the hemi- and unmethylated duplexes is observed (Figure 2). The Kd for a hemimethylated CpG duplex is 10-fold higher than that of a fully methylated CpG pair, whereas the Kd for an unmethylated duplex is ∼100-fold higher (Table 1 and Figure 3A). The ΔΔGap for the hemimethylated duplexes compared to the fully methylated duplex are 1.28 ± 0.04 and 1.38 ± 0.04 kcal mol−1, and the ΔΔGap for an unmethylated duplex is 2.52 ± 0.04 kcal mol−1 (Table 1). These data reveal that each 5mC residue contributes equally to the binding of MBD, as each residue lowers the Kd by a factor of 10 when compared to the unmethylated CpG pair. Also, these data indicate that the apparent-free energy change of removing two 5mC residues from the methyl CpG dinucleotide is additive (Table 1).
Furthermore, these data are consistent with MBD binding to its target duplex as a monomer (8) that recognizes both methylated cytosines in a fully methylated duplex (33) much like the MBD of MBD1 (34). The binding curve for the fully methylated duplex reveals only one inflection point (Figure 3A; 5mC/5mC), suggesting that one MBD binds both 5mC residues in a methyl CpG pair. Two inflection points would be expected if binding entailed one MBD per 5mC in a symmetrically methylated CpG dinuclotide.
Our data from the assay of the 5mC/T duplex for binding to the MBD of MeCP2 are also consistent with the data reported by Hendrich et al. (30). Indeed the MBD of MeCP2, homologous to the MBD of MBD4, also recognizes a T:G mismatch in a methyl-CpG dinucleotide. Our data show that the Kd obtained for the 5mC/T duplex is nearly identical to the Kd obtained for the 5mC/5mC duplex (Table 1 and Figure 3A), with no significant difference in the apparent-free energy change of MBD binding between the two duplexes (Table 1).
The binding constants for duplexes containing the 5mC oxidation damage product HmC were also determined. Replacing one of the 5mC residues in a symmetrically methylated CpG dinucleotide with HmC, regardless of which 5mC is replaced, decreases binding (Figures 3B and 4), increasing the Kd 10-fold (Figure 3B and Table 1), with ΔΔGap values of 1.38 ± 0.04 and 1.40 ± 0.05 kcal mol−1 (Table 1), thereby indicating that the binding is equivalent to that of the hemimethylated duplexes. Replacement of both methylcytosines with HmC further reduces the affinity of the duplex, placing its Kd and free energy change in the same range as that of an unmodified CpG pair (Figure 3B and Table 1). Our results suggest that, in the context of MBD binding, oxidizing the methyl group of a 5mC in a methyl CpG dinucleotide is functionally equivalent to demethylating that same 5mC residue.
In addition, the effect of 8-oxoG within a fully methylated CpG was determined. Our results show a dramatic reduction in the binding of MBD to a fully methylated CpG as compared to a fully methylated CpG that had one oxidized guanine (Figures 3C and 4). The Kd increased >10-fold from 14.7 ± 1.0 nM to 292 ± 11 (Figure 3C and Table 1). The presence of 8-oxoG within an unmethylated CpG also adversely affects MBD binding, resulting in a 2-fold increase in the Kd as compared to an undamaged unmethylated CpG (Table 1 and Figure 3C). The presence of 8-oxoG across from 5mC in a hemimethylated CpG also reduces the binding of MBD to the level of an unmethylated duplex (Figure 3C). The substitution of guanine by 8-oxoG in the hemimethylated duplexes reduces binding substantially when the 8-oxoG is adjacent to or paired with the 5mC residue, however, the effect of the 8-oxoG-substitution is greater when adjacent to the 5mC residue (Figure 3C and Table 1). These data confirm that multiple sites of the methylated CpG dinucleotide are needed for strong binding by the MBD (Figure 5), and that oxidative damage to any one site can dramatically interfere with the interaction.
Figure 5.

Molecular model of the sequence 5′-GCmCGGC-3′. The methyl groups of 5mC are depicted in green, and the N7 position of guanines within the methyl-CpG dinucleotide are depicted in dark blue. Multiple sites within the methylated CpG dinucleotide are needed for strong binding by the MBD. All four major sites of contact, two methyl groups and two hydrogen bond accepting nitrogens, are in the major groove of the DNA within close proximity of one another. Disruption of MBD binding results from oxidative damage to any one of these four sites.
DISCUSSION AND CONCLUSION
In higher eukaryotes, cytosine methylation is crucial for gene regulation during development (61,62), in genomic imprinting (63,64) and X chromosome inactivation (65,66). Underscoring the importance of methylation in normal development and differentiation, aberrant methylation patterns have been observed in many human cancers (25–29). The mechanisms by which these perturbations arise are an important and as yet relatively unexplored aspect of cancer development.
Very little is known currently about potential mechanisms by which DNA damage could result in epigenetic changes. Previously Turk et al. (67) examined the impact of 8-oxoG on the capacity of the human DNA methyltransferase to methylate the CpG dinucleotide. They observed that the replacement of guanine by 8-oxoG inhibits methylation of a cytosine residue adjacent to that of 8-oxoG residue, but had little effect on methylation of the CpG dinucleotide when the target cytosine was paired opposite to 8-oxoG residue (67). We observe that, at a hemimethylated CpG dinucleotide, the replacement of either guanine residue with 8-oxoG substantially diminishes MBD binding, with a greater effect seen when an 8-oxoG residue is placed adjacent to the 5mC residue (Table 1 and Figure 3C). Currently, it is unknown if guanine residues in specific sequences are more prone to oxidation.
Previously, Watanabe et al. (68) demonstrated that the replacement of guanine by 7-methylguanine inhibited MBD binding. The inhibition in binding resulted from N7 methylation could be explained on the basis of an unfavorable steric clash with the N7 methyl group, or by the loss of the N7 position as a hydrogen bond acceptor. The predominant keto tautomeric form of 8-oxoG places a proton at the N7 position, resulting in the conversion of a hydrogen bond acceptor into a donor (42). Our results with 8-oxoG discussed above are in accordance with these methylation studies, and provide additional evidence that the N7 position of guanine acts as a hydrogen bond acceptor in the formation of the MBD–DNA complex.
The methyl group of 5mC is the most likely site for oxidation damage to the pyrimidine components of the methylated CpG dinucleotide, resulting in the formation of HmC (Figure 1A). Previously, we predicted that ∼20 5mC residues would be oxidized to HmC per human cell per day under normal metabolic conditions, with even greater numbers during oxidative stress (44,59). The conversion of 5mC to HmC would not be predicted to be mutagenic or to substantially perturb DNA structure (59). Indeed, the genomes of T-even phages contain HmC residues instead of cytosine residues (69). Although not a miscoding lesion, mammalian genomes have a specific glycosylase to remove HmC, and Teebor and co-workers (49,50) have argued that the primary role of HmU-glycosylases is to repair products derived from 5mC oxidation.
The methyl groups of both thymine and 5mC are important for sequence-specific DNA–protein interactions. The proximity of the methyl groups in the major groove of a B-form DNA helix for a symmetrically methylated CpG sequence is illustrated in Figure 5. The contributions of both Van der Waals interactions and solvation effects are believed to contribute to the favorable binding affinity resulting from the presence of the methyl group (56). The contribution of the Van der Waals interaction can be as small as 0.2 kcal mol−1, whereas the solvation effect can be as large as 1.5 kcal mol−1. When thymine is replaced by either uracil or HmU within the binding site of the AP1 transcription factor, the binding energy decreases by a modest 0.2–0.4 kcal mol−1 (55). In contrast, the contribution of the methyl group of 5mC to MBD binding is estimated here to be ∼1.4 kcal mol−1, representing the upper limit for the effect of a methyl group on the specificity of a DNA–protein interaction (56).
The 5-methyl group of 5mC is critical for the discrimination of MBPs between methylated and unmethylated DNA. We predicted, and confirmed here experimentally, that the oxidation of 5mC to HmC impedes binding of proteins that recognize the methyl group. In accordance with similar studies with thymine oxidation damage products and sequence-specific DNA-binding proteins that recognize the thymine methyl group (55,70), our results suggest that pyrimidine methyl group oxidation generally can substantially interfere with DNA–protein interactions.
Epigenetic programming in eukaryotic cells relies upon the specific placement of methyl groups in selected CpG dinucleotides. The mechanisms by which these methylation patterns are initially established are not well understood (6,7,71). Once established, symmetrically methylated CpG dinucleotides are converted into hemimethylated dinucleotides as a result of semiconservative DNA replication. The hemimethylated parental strand then directs methylation to the progeny strand by the maintenance methylase, regenerating the symmetrically methylated site (1–3). As with the MBP binding examined here, the preferential methylation of hemimethylated sites is based upon the specific recognition of the methyl group of the 5mC residue in the parental strand by the maintenance methylase (72).
Following DNA replication and methylation, MBPs including MeCP2 studied here, selectively bind to the methylated CpG sites (7–17). Our results indicate that symmetric methylation increases the affinity for MBP binding, relative to the unmethylated sequence by a factor of 100. The additional affinity afforded by methylation is near the theoretical maximum possible for a methyl substituent, indicating the importance of cytosine methylation in the selective binding of the MBPs. Additional proteins with affinity for the MBPs then accumulate and covalently modify histone proteins in the immediate vicinity, leading to condensed, less transcriptionally active chromatin regions (7–24). Through this sequence of events, the methylation signal on the DNA can be used to re-establish a condensed regional chromatin structure following cell division.
The DNA of all organisms is constantly damaged. Although substantial work has been reported previously on an array of lesions that can either miscode or block DNA replication, significantly less work has been done on the role of DNA damage in altering epigenetic programming. Existing data suggest that the binding of MBPs to methylated DNA is a critical link in the cascade of events that maintain heritable gene silencing. The results of the studies reported here, in accordance with previous studies, indicate that contact points on both the guanine and 5mC residues of a methylated CpG dinucleotide are required for the high-affinity binding of the MBPs, and that interaction with the 5mC methyl group provides the high level of discrimination between methylated and unmethylated DNA sequences. The oxidation of either guanine or 5mC is shown here to eliminate discrimination. If unrepaired, these oxidative lesions would break the normal chain of events that allow propagation of epigenetic signals, perhaps explaining in part the disregulation of transcriptional activity observed in some tumors.
Acknowledgments
ACKNOWLEDGEMENTS
We gratefully acknowledge the assistance of Dr Jonathan Neidigh in the preparation of graphics. This study was supported in part by the National Institutes of Health (GM50351, CA84487). V.V. is supported in part by the Loma Linda University School of Medicine Medical Scientist training program.
REFERENCES
- 1.Ehrlich M. and Wang,F.Y.-H. (1981) 5-Methylcytosine in eukaryotic DNA. Science, 212, 1350–1357. [DOI] [PubMed] [Google Scholar]
- 2.Riggs A.D. and Jones,P.A. (1983) 5-Methylcytosine, gene regulation and cancer. Adv. Cancer Res., 40, 1–30. [DOI] [PubMed] [Google Scholar]
- 3.Doerfler W. (1983) DNA methylation and gene activity. Ann. Rev. Biochem., 52, 93–124. [DOI] [PubMed] [Google Scholar]
- 4.Antequera F., Boyes,J. and Bird,A. (1990) High levels of de novo methylation and altered chromatin structure at CpG islands in cell lines. Cell, 62, 503–514. [DOI] [PubMed] [Google Scholar]
- 5.Jaenisch R. and Bird,A. (2003) Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nature Genet., 33, 245–254. [DOI] [PubMed] [Google Scholar]
- 6.Jones P.L. and Wolffe,A.P. (1999) Relationships between chromatin organization and DNA methylation in determining gene expression. Semin. Cancer Biol., 9, 339–347. [DOI] [PubMed] [Google Scholar]
- 7.Bird A.P. and Wolffe,A.P. (1999) Methylation-induced repression-belts, braces, and chromatin. Cell, 99, 451–454. [DOI] [PubMed] [Google Scholar]
- 8.Nan X., Meehan,R.R. and Bird,A.P. (1993) Dissection of the methyl-CpG binding domain from the chromosomal protein MeCP2. Nucleic Acids Res., 21, 4886–4892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Free A., Wakefield,R.I., Smith,B.O., Dryden,D.T., Barlow,P.N. and Bird,A.P. (2001) DNA recognition by the methyl-CpG binding domain of MeCP2. J. Biol. Chem., 276, 3353–3360. [DOI] [PubMed] [Google Scholar]
- 10.Meehan R.R., Lewis,J.D. and Bird,A.P. (1992) Characterization of MeCP2, a vertebrate DNA binding protein with affinity for methylated DNA. Nucleic Acids Res., 20, 5085–5092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lewis J.D., Meehan,R.R., Henzel,W.J., Maurer-Fogy,I., Jeppesen,P., Klein,F. and Bird,A. (1992) Purification, sequence, and cellular localization of a novel chromosomal protein that binds to methylated DNA. Cell, 69, 905–914. [DOI] [PubMed] [Google Scholar]
- 12.Magdinier F. and Wolffe,A.P. (2001) Selective association of the methyl-CpG binding protein MBD2 with the silent p14/p16 locus in human neoplasia. Proc. Natl Acad. Sci. USA, 98, 4990–4995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nguyen C.T., Gonzales,F.A. and Jones,P.A. (2001) Altered chromatin structure associated with methylation-induced gene silencing in cancer cells: correlation of accessibility, methylation, MeCP2 binding and acetylation. Nucleic Acids Res., 29, 4598–4606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nan X. and Bird,A. (2001) The biological functions of the methyl-CpG-binding protein MeCP2 and its implication in Rett syndrome. Brain Dev., 23, S32–S37. [DOI] [PubMed] [Google Scholar]
- 15.Nan X., Tate,P., Li,E. and Bird,A. (1996) DNA methylation specifies chromosomal localization of MeCP2. Mol. Cell. Biol., 16, 414–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Holliday R. and Ho,T. (2002) DNA methylation and epigenetic inheritance. Methods, 27, 179–183. [DOI] [PubMed] [Google Scholar]
- 17.Bhakat K.K. and Mitra,S. (2003) CpG methylation-dependent repression of the human O6-methylguanine DNA methyltransferase gene linked to chromatin structure alteration. Carcinogenesis, 24, 1337–1345. [DOI] [PubMed] [Google Scholar]
- 18.Kimura H. and Shiota,K. (2003) Methyl-CpG-binding protein, MeCP2, is a target molecule for maintenance DNA methyltransferase, Dnmt1. J. Biol. Chem., 278, 4806–4812. [DOI] [PubMed] [Google Scholar]
- 19.Fuks F., Hurd,P.J., Wolf,D., Nan,X., Bird,A.P. and Kouzarides,T. (2003) The methyl-CpG-binding protein MeCP2 links DNA methylation to histone methylation. J. Biol. Chem., 278, 4035–4040. [DOI] [PubMed] [Google Scholar]
- 20.Nan X., Ng,H.H., Johnson,C.A., Laherty,C.D., Turner,B.M., Eisenmann,R.N. and Bird,A. (1998) Transcriptional repression by the methyl-GpG-binding protein MeCP2 involved a histone deacetylase complex. Nature, 393, 386–389. [DOI] [PubMed] [Google Scholar]
- 21.Knoepfler P.S. and Eisenman,R.N. (1999) Sin meets NuRD and other tails of repression. Cell, 99, 447–450. [DOI] [PubMed] [Google Scholar]
- 22.Jones P.L., Veenstra,G.J., Wade,P.A., Vermaak,D., Kass,S.U., Landsberger,N., Strouboulis,J. and Wolffe,A.P. (1998) Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nature Genet., 19, 187–191. [DOI] [PubMed] [Google Scholar]
- 23.Ng H.-H., Zhang,Y., Hendrich,B., Johnson,C.A., Turner,B.M., Erdjument-Bromage,H., Tempst,P., Reinberg,D. and Bird,A. (1999) MBD2 is a transcriptional repressor belonging to the MeCP1 histone deacetylase complex. Nature Genet., 23, 58–61. [DOI] [PubMed] [Google Scholar]
- 24.Ng H.-H., Jeppesen,P. and Bird,A. (2000) Active repression of methylated genes by the chromosomal protein MBD1. Mol. Cell. Biol., 20, 1394–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jones P.A. and Buckley,J.D. (1990) The role of DNA methylation in cancer. Adv. Cancer Res., 54, 1–23. [DOI] [PubMed] [Google Scholar]
- 26.Jones P.A., Rideout,W.M.,III, Shen,J.-C., Spruck,C.H. and Tsai,Y.C. (1992) Methylation, mutation and cancer. BioEssays, 14, 33–36. [DOI] [PubMed] [Google Scholar]
- 27.Schmutte C. and Jones,P.A. (1998) Involvement of DNA methylation in human carcinogenesis. Biol. Chem., 379, 377–388. [DOI] [PubMed] [Google Scholar]
- 28.Santini V., Kantarjian,H.M. and Issa,J.-P. (2001) Changes in DNA methylation in neoplasia: pathophysiology and therapeutic implications. Ann. Int. Med., 134, 573–586. [DOI] [PubMed] [Google Scholar]
- 29.Schmutte C., Yang,A.S., Nguyen,T.T., Beart,R.W. and Jones,P.A. (1996) Mechanisms for the involvement of DNA methylation in colon carcinogenesis. Cancer Res., 56, 2375–2381. [PubMed] [Google Scholar]
- 30.Hendrich B., Hardeland,U., Ng,H.-H., Jiricny,J. and Bird,A. (1999) The thymine glycosylase MBD4 can bind to the product of deamination at methylated CpG sites. Nature, 401, 301–304. [DOI] [PubMed] [Google Scholar]
- 31.Nan X., Campoy,J.F. and Bird,A. (1997) MeCP2 is a transcriptional repressor with abundant binding sites in genomic chromatin. Cell, 88, 471–481. [DOI] [PubMed] [Google Scholar]
- 32.Hendrich B. and Bird,A.P. (1998) Identification and characterization of a family of mammalian methyl-CpG binding protiens. Mol. Cell. Biol., 18, 6538–6547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wakefield R.I., Smith,B.O., Nan,X., Free,A., Soteriou,A., Uhrin,D., Bird,A.P. and Barlow,P.N. (1999) The solution structure of the domain from MeCP2 that binds to methylated DNA. J. Mol. Biol., 291, 1055–1065. [DOI] [PubMed] [Google Scholar]
- 34.Ohki I., Shimotake,N., Fujita,N., Jee,J., Ikegami,T., Nakao,M. and Shirakawa,M. (2001) Solution structure of the methyl-CpG binding domain of human MBD1 in complex with methylated DNA. Cell, 105, 487–497. [DOI] [PubMed] [Google Scholar]
- 35.Kasai H. and Nishimura,S. (1984) Hydroxylation of deoxyguanosine at the C-8 position by ascorbic acid and other reducing agents. Nucleic Acids Res., 12, 2137–2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dizdaroglu M. (1985) Formation of an 8-hydroxyguanine moiety in deoxyribonucleic acid on gamma-irradiation in aqueous solution. Biochemistry, 24, 4476–4481. [DOI] [PubMed] [Google Scholar]
- 37.Grollman A.P. and Moriya,M. (1993) Mutagensis by 8-oxoguanine: an enemy within. Trends Genet., 9, 246–249. [DOI] [PubMed] [Google Scholar]
- 38.Shibutani S., Takeshita,M. and Grollman,A.P. (1991) Insertion of specific bases during DNA synthesis past the oxidation-damaged base 8-oxodG. Nature, 349, 431–434. [DOI] [PubMed] [Google Scholar]
- 39.Kuchino Y., Mori,F., Kasai,H., Inoue,H., Iwai,S., Miura,K., Ohtsuka,E. and Nishimura,S. (1987) Misreading of DNA templates containing 8-hydroxydeoxyguanosine at the modified base and at adjacent residues. Nature, 327, 77–79. [DOI] [PubMed] [Google Scholar]
- 40.Cheng K.C., Cahill,D.S., Hiroshi,K., Nishimura,S. and Loeb,L.A. (1992) 8-Hydroxyguanine, an abundant form of oxidative DNA damage, causes G→T and A→C substitutions. J. Biol. Chem., 267, 166–172. [PubMed] [Google Scholar]
- 41.Bruner S.D., Norman,D.P.G. and Verdine,G.L. (2000) Structural basis for recognition and repair of the endogenous mutagen 8-oxoguanine in DNA. Nature, 403, 859–866. [DOI] [PubMed] [Google Scholar]
- 42.Jang Y.H., Goddard,W.A.,III, Noyes,K.T., Sowers,L.C., Hwang,S. and Chung,D.S. (2002) First principles calculations of the tautomers and pKa values of 8-oxoguanine: implications for mutagenicity and repair. Chem. Res. Toxicol., 15, 1023–1035. [DOI] [PubMed] [Google Scholar]
- 43.Ehrlich M., Gama-Sosa,M.A., Huang,L.H., Midgett,R.M., Kuo,K.C., McCune,R.A. and Gehrke,C. (1982) Amount and distribution of 5-methylcytosine in human DNA from different types of tissues of cells. Nucleic Acids Res., 10, 2709–2721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Burdzy A., Noyes,K.T., Valinluck,V. and Sowers,L.C. (2002) Synthesis of stable-isotope enriched 5-methylpyrimidines and their use as probes of base reactivity in DNA. Nucleic Acids Res., 30, 4068–4074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Masuda T., Shinoara,H. and Kondo,M. (1975) Reactions of hydroxyl radicals with nucleic acid bases and the related compounds in gamma-irradiated aqueous solution. J. Radiat. Res., 16, 153–161. [DOI] [PubMed] [Google Scholar]
- 46.Privat E. and Sowers,L.C. (1996) Photochemical deamination and demethylation of 5-methylcytosine. Chem. Res. Toxicol., 9, 745–750. [DOI] [PubMed] [Google Scholar]
- 47.Penn N.W. (1976) Modification of brain deoxyribonucleic acid base content with maturation in normal and malnourished rats. Biochem. J., 155, 709–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Penn N.W., Suwalski,R., O'Riley,C., Bojanowski,K. and Yura,R. (1972) The presence of 5-hydroxymethylcytosine in animal deoxyribonucleic acid. Biochem. J., 126, 781–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Boorstein R.J., Chiu,L.N. and Teebor,G.W. (1989) Phylogenetic evidence of a role for 5-hydroxymethyluracil-DNA glycosylase in the maintenance of 5-methylcytosine in DNA. Nucleic Acids Res., 17, 7653–7661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cannon S.V., Cummings,A. and Teebor,G.W. (1988) 5-Hydroxymethylcytosine DNA glycosylase activity in mammalian tissue. Biochem. Biophys. Res. Commun., 151, 1173–1179. [DOI] [PubMed] [Google Scholar]
- 51.Brennan C.A., Van Cleve,M.D. and Gumport,R.I. (1986) The effects of base analogue substitutions on the methylation by the EcoRI modification methylase of octadeoxyribonucleotides containing modified EcoRI recognition sequences. J. Biol. Chem., 261, 7279–7286. [PubMed] [Google Scholar]
- 52.Brennan C.A., Van Cleve,M.D. and Gumport,R.I. (1986) The effects of base analogue substitutions on the cleavage by the EcoRI restriction endonuclease of octadeoxyribonucleotides containing modified EcoRI recognition sequences. J. Biol. Chem., 261, 7270–7278. [PubMed] [Google Scholar]
- 53.Frenkel K., Zhong,Z.J., Wei,H.C., Karkoszka,J., Patel,U., Rashid,K., Georgescu,M. and Solomon,J.J. (1991) Quantitative high-performance liquid chromatography analysis of DNA oxidized in vitro and in vivo. Anal. Biochem., 196, 126–136. [DOI] [PubMed] [Google Scholar]
- 54.Cadet J., Incardona,M.F., Odin,F., Molko,D., Mouret,J.F., Polverelli,M., Faure,H., Ducros,V., Tripier,M. and Favier,A. (1993) Measurement of oxidative base damage to DNA by using HPLC-32P-postlabelling and GC/MS-selective ion monitoring assays. IARC Sci. Publ., 124, 271–276. [PubMed] [Google Scholar]
- 55.Rogstad D.K., Liu,P., Burdzy,A., Lin,S.S. and Sowers,L.C. (2002) Endogenous DNA lesions can inhibit the binding of the AP-1 (c-Jun) transcription factor. Biochemistry, 41, 8093–8102. [DOI] [PubMed] [Google Scholar]
- 56.Plaxco K.W. and Goddard,W.A.,III (1994) Contributions of the thymine methyl group to the specific recognition of poly- and mononucleotides: an analysis of the relative free energies of solvation of thymine and uracil. Biochemistry, 33, 3050–3054. [DOI] [PubMed] [Google Scholar]
- 57.Nielsen J., Taafaard,M., Marugg,J.E., van Boom,J.H. and Dahl,O. (1986) Application of 2-cyanoethyl-N,N,N′,N′-tetraisopropylphosphoramidite for in situ preparation of 2′-deoxyribonucleoside phosphoramidites and their use in polymer supported synthesis of oligodeoxyribonucleosides. Nucleic Acids Res., 14, 7391–7403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gait M.J. (1984) Oligonucleotide Synthesis: A Practical Approach. IRL Press, Oxford, UK, pp. 35–81. [Google Scholar]
- 59.Tardy-Planechaud S., Fujimoto,J., Lin,S.S. and Sowers,L.C. (1997) Solid phase synthesis and restriction endonuclease cleavage of oligodeoxynucleotides containing 5-(hydroxymethyl)-cytosine. Nucleic Acids Res., 25, 553–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bevington P.R. (1969) Data Reduction and Error Analysis for the Physical Sciences. McGraw-Hill, NY, pp. 56–65. [Google Scholar]
- 61.Li E., Bestor,T.H. and Jaenisch,R. (1992) Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell, 69, 915–926. [DOI] [PubMed] [Google Scholar]
- 62.Shiota K. and Yanagimachi,R. (2002) Epigenetics by DNA methylation for development of normal and cloned animals. Differentiation, 69, 162–166. [DOI] [PubMed] [Google Scholar]
- 63.Tucker K.L., Beard,C., Dausmann,J., Jackson-Grusby,L., Laird,P.W., Lei,H., Li,E. and Jaenisch,R. (1996) Germ-line passage is required for establishment of methylation and expression patterns of imprinted but not of nonimprinted genes. Genes Dev., 10, 1008–1020. [DOI] [PubMed] [Google Scholar]
- 64.Amir R.E., Van den Veyver,I.B., Wan,M., Tran,C.Q., Francke,U. and Zoghbi,H.Y. (1999) Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nature Genet., 23, 185–188. [DOI] [PubMed] [Google Scholar]
- 65.Li E. (2002) Chromatin modification and epigenetic reprogramming in mammalian development. Nature Rev. Genet., 3, 662–673. [DOI] [PubMed] [Google Scholar]
- 66.Bird A. (2002) DNA methylation patterns and epigenetic memory. Genes Dev., 16, 6–21. [DOI] [PubMed] [Google Scholar]
- 67.Turk P.W., Laayoun,A., Smith,S.S. and Weitzman,S.A. (1995) DNA adduct 8-hydroxyl-2′-deoxyguanosine (8-hydroxyguanine) affects function of human DNA methyltransferase. Carcinogenesis, 16, 1253–1255. [DOI] [PubMed] [Google Scholar]
- 68.Watanabe S., Ichimura,T., Naoyuki,F., Tsuruzoe,S., Ohki,I., Shirakawa,M., Kawasuji,M. and Nakao,M. (2003) Methylated DNA-binding domain 1 and methylpurine-DNA glycosylase link transcriptional repression and DNA repair in chromatin. Proc. Natl Acad. Sci. USA, 100, 12859–12864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wyatt G.R. and Cohen,S.S. (1953) The bases of the nucleic acids of some bacterial and animal viruses: the occurrence of 5-hydroxymethylcytosine. Biochem. J., 55, 774–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rogstad D.K., Heo,J., Vaidehi,N., Goddard,W.A.,III, Burdzy,A. and Sowers,L.C. (2004) 5-Formyluracil-induced perturbations of DNA function. Biochemistry, 43, 5688–5697. [DOI] [PubMed] [Google Scholar]
- 71.Baylin S. and Bestor,T.H. (2002) Altered methylation patterns in cancer cell genomes: cause or consequence? Cancer Cell, 1, 299–305. [DOI] [PubMed] [Google Scholar]
- 72.Hardy T.A., Baker,D.J., Newman,E.M., Sowers,L.C., Goodman,M.F. and Smith,S.S. (1987) Size of the directing moiety at carbon 5 of cytosine and the activity of human DNA(cytosine-5) methyltransferase. Biochem. Biophys. Res. Commun., 145, 146–152. [DOI] [PubMed] [Google Scholar]