


Abstract
Ets2 is a member of the Ets family of transcription factors that in humans comprise 25 distinct members. Various Ets-domain transcription factors have been implicated in cancer development. Ets2 is expressed in prostate and breast cancer cells and is thought to have a role in promoting growth and survival in these cell types. However, a definitive role and the mechanisms whereby Ets2 acts in cancer cells are still unclear. Structural and functional similarities as well as overlapping DNA binding specificities complicate the identification of the specific roles of the various Ets factors. In this study, we used a triplex-forming oligonucleotide (TFO) to selectively inhibit Ets2 transcription in prostate cancer cells. We had previously shown that the Ets2-targeting TFO, which was directed to a unique purine-rich sequence critical for Ets2 promoter activity, acted with a high degree of sequence-specificity and target selectivity. TFO-mediated downregulation of Ets2 in prostate cancer cells induced important phenotypic changes, including inhibition of anchorage-dependent and anchorage -independent growth, cell cycle alterations and induction of apoptotic cell death. Expression of Ets2 under the control of a heterologous promoter abolished the anti-proliferative effects of the TFO in both short- and long-term assays, suggesting that these effects were a direct result of downregulation of Ets2 transcription and confirming target selectivity of the TFO. Furthermore, normal human fibroblasts, which expressed low levels of Ets2, were not affected by the Ets2-targeting TFO. Downregulation of Ets2 in prostate cancer cells was associated with reduced levels of the anti-apoptotic protein bcl-xL and growth regulatory factors cyclin D1 and c-myc. These data revealed a specific role of this transcription factor in promoting growth and survival of prostate cancer cells. Furthermore, the activity and selectivity of the Ets2-targeting TFO suggest that it might represent a valid approach to prostate cancer therapy.
INTRODUCTION
The ability to selectively modulate gene expression in mammalian cells can have far-reaching implications in biotechnology and medicine. Oligonucleotides appear to be ideal molecules for this purpose because of their intrinsic ability to bind nucleic acids in a sequence-specific manner. Antisense oligonucleotides (AOs) and small-interfering RNAs (siRNAs) bind to and induce degradation of the targeted RNA, thereby blocking the synthesis of the corresponding protein (1,2). An alternative gene targeting approach is based on the ability of single-stranded oligonucleotides to bind double-stranded DNA and form triple helices. The triplex-DNA-based or antigene approach can provide an effective way to target specific sequences in genomic DNA and modulate gene function via mutagenesis, recombination and transcriptional repression or activation (3–5). The general principles underlying formation of intra- and inter-molecular DNA triple helices have been extensively reviewed (3,4,6). Triplex-forming oligonucleotides (TFOs) bind to duplex DNA by forming base triplets, in which each base of the oligonucleotide recognizes a base pair in the duplex. Hydrogen bonds of the Hoogsteen or reverse-Hoogsteen type are formed between the bases of the oligonucleotide and purine bases of the duplex. The possible base combinations are limited by structural constrains creating a triplex binding code distinct from the binding code of duplex DNA. Purine-rich (GA) and mixed purine/pyrimidine (GT) TFOs bind preferentially antiparallel to the purine-rich strand of the duplex forming G:GC, A:AT and T:AT base triplets. Pyrimidine-rich TFOs bind parallel to the purine-rich target strand forming C+:GC and T:AT triplets (3,4). Binding of TFOs requires the presence of long and possibly uninterrupted homopurine sequences in the target DNA to ensure optimal stability and sequence specificity (3,4). Such homopurine sequences are common in gene regulatory regions and frequently overlap transcription factors binding sites, supporting the view that purine-rich elements may be relevant for control of gene expression and may be relatively easy targets of TFOs (7). Indeed, TFOs directed to triplex target sites within gene regulatory regions can be very effective in blocking transcription factor binding and transcription initiation in cell-free systems (4,8). TFOs have been shown to inhibit transcription from promoter–reporter constructs and expression of endogenous genes, indicating that they could be used as selective gene repressors in cells (4,8). This strategy has now proven to be successful in various experimental models and may provide also the means for design of new gene-targeted therapeutics (5,9).
Our group has investigated the triplex-DNA-based approach to block transcription of cancer-related genes (10–12). We have recently designed a TFO directed to a homopurine sequence in the promoter of the Ets2 gene (13). The triplex target site was located 40 bp upstream of the transcription initiation site and overlapped a putative Sp1 binding site (Figure 1). Proteins of the Sp1 family bound to this site in vitro and a promoter–reporter construct with a mutated Sp1 site had reduced activity in cells (13). The Ets2-targeting TFO bound with very high affinity and specificity to the target sequence, prevented binding of Sp1 and Sp3 in vitro and was an effective repressor of Ets2 transcription in cells (13). Experiments using control oligonucleotides with mismatched sequences as well as double-stranded oligonucleotides and promoter–reporter constructs with mutated triplex target sites demonstrated that the Ets2-TFO acted with a high degree of sequence-specificity and target selectivity both in vitro and in cells (13). These results confirmed that the anti-transcriptional activity of the Ets2-TFO was due to a triplex-DNA-mediated mechanism and was selective for the Ets2 gene.
Figure 1.
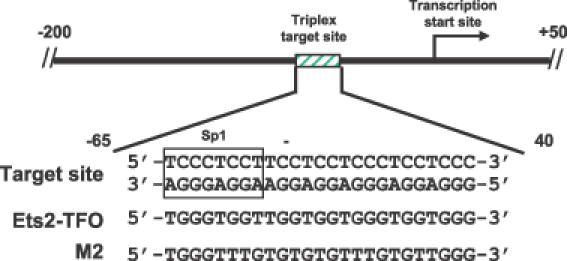
Sequences of the Ets2-TFO, M2 control oligonucleotide and target site in the Ets2 promoter. The positions of the triplex target site and transcription start site in the Ets2 promoter are indicated. Numbers are relative to the transcription start site.
The human Ets gene family includes 25 genes that code for transcription factors involved in the control of various aspects of cell proliferation, differentiation and development (14–16). Ets-domain transcription factors have been implicated in development of various forms of leukemias and solid tumors (14–16). Recent investigations suggest that Ets2 might have an important role in the pathogenesis of prostate cancer. Ets2 is expressed in basal cells of normal prostatic epithelium and is overexpressed in tissue specimens of primary and metastatic prostate cancers, particularly those with high Gleason grade (17,18). Prostate cancer cell lines DU145 and PC3, which are androgen independent, highly tumorigenic and metastatic in nude mice, have a higher level of Ets2 than LNCaP cells, which are androgen sensitive and exhibit a less aggressive behavior in vivo (19). Transfection of DU145 and PC3 cells with an Ets2 antisense construct or a dominant negative mutant reduces their ability to grow in an anchorage-independent manner compared to parental cells (19). Using similar approaches, Ets2 has been shown to have a role in breast and thyroid carcinogenesis (20,21). The oncogenic activity of Ets2 may be related to the ability of this transcription factor to activate expression of genes that promote cell proliferation and/or prevent apoptotic cell death (22). It has been difficult, however, to assess the specific functions and pathways regulated by Ets2 because of sequence and structural similarities between the various members of the Ets gene family and the absence of truly selective inhibitors of Ets2.
In the present study, we used the Ets2-targeting TFO to investigate the consequences of selective downregulation of Ets2 expression in prostate cancer cells. We show that downregulation of Ets2 expression in prostate cancer cells resulted in growth inhibition, reduced clonogenic potential and anchorage-independent growth, delayed cell cycle progression and induction of apoptotic cell death. The effects of the Ets2-TFO occurred with a high degree of specificity and target selectivity. Expression of Ets2 under the control of a heterologous promoter prevented the growth inhibition induced by the TFO, indicating that its effects depended on downregulation of the target gene. Furthermore, the Ets2-TFO did not affect growth of normal human fibroblasts, which expressed very low levels of Ets2. Downregulation of Ets2 was associated with reduced expression of bcl-xL, c-myc and cyclin D1, which have previously been shown to be regulated by Ets2 in other cell types and are critical for cell proliferation and viability. Thus, downregulation of Ets2 expression by the Ets2-TFO had profound effects on proliferation, apoptosis and clonogenic potential of prostate cancer cells. This strategy may permit an accurate analysis of the role of this transcription factor and the identification of specific Ets2 target genes in prostate cancer cells. New insights into the pathogenesis and progression of prostate cancer, thus, could be gained using this approach. Ultimately, a selective Ets2 inhibitor might be of therapeutic interest for the treatment of prostate cancer.
MATERIALS AND METHODS
Oligonucleotides
Source of phosphodiester (PO) and phosphorothioate (PS) oligonucleotides and preparation of oligonucleotide solutions have been described previously (13). The sequences of the triplex target site, Ets2-TFO and control oligonucleotide are shown in Figure 1.
Cell lines, plasmids and transfection
Human prostate cancer DU145 and PC3 cells, normal human fibroblasts and breast cancer MCF-7 cells were grown in RPMI 1640 supplemented with 10% heat-inactivated fetal bovine serum (Life Technologies). Normal human fibroblasts were kindly provided by M. Trojanowska (MUSC). The Ets2 expression plasmid (pSGK-Ets2) and empty vector (pSGK) were described previously (19). The pEGFP plasmid was purchased from Clontech. The pGL3-Ets2 promoter luciferase reporter construct was generated as described previously (13). Cells were plated either in 6-, 48- or 96-well plates and transfected with either Ets2-TFO or M2 control oligonucleotide and/or plasmid DNA as described previously (13). After 4 h, the transfection medium was replaced with complete medium. Luciferase reporter assays were performed using the dual luciferase system (Promega) as described previously (13).
Western blot analysis
Cells were plated in 6-well plates and transfected with TFO or M2 control oligonucleotide as described above. Cells were washed with phosphate-buffered saline (PBS) and then lysed in PBS buffer containing 1% NP40, 0.5% sodium deoxycolate, 0.1% SDS, 1 mM sodium orthovanadate and protease inhibitor cocktail (Sigma) for 30 min on ice. Protein concentration was determined using the BCA assay (Pierce, Rockford, Ill). Aliquots of 40 μg protein were separated on 10% polyacrylamide–SDS gels, transferred to PVDF membranes and probed with antibodies against Ets2, Ets1, Bcl-xL, Bcl-2, c-myc, β-actin (Santa Cruz Biotechnology, Santa Cruz, CA) and α-tubulin (Oncogene Research Products). Blots were developed using the enhanced chemiluminescence system (ECL, Amersham).
RT–PCR
RT–PCR was performed using the SuperScript One Step RT–PCR system (Invitrogen) and gene-specific primers as described previously (13).
Cell proliferation assays
For cell proliferation assays cells were seeded in 96-well plates at a density of 1 × 103 cells/well. After 24 h, cells were transfected with TFO and M2 control oligonucleotide using DOTAP for 4 h and then allowed to grow for up to 96 h. The number of viable cells was measured at the indicated time points by adding either MTT tetrazolium salt (Sigma) or MTS (Promega) and measuring absorbance in a microplate reader as described previously (10). To assess colony forming ability, DU145 cells transfected with ETS2-TFO or M2 control oligonucleotide were plated at a density of 1 × 103 per dish in 35 mm tissue culture dishes. Colonies were stained with crystal violet and counted after 8–10 days. To measure anchorage-independent growth, DU145 cells transfected with ETS2-TFO or M2 oligonucleotide were plated in 0.3% agar on the top of a pre-coated layer of 0.7% agar (SeaPlaque Agarose). Colonies were counted under a microscope after 21–24 days. Data were expressed as percentage of surviving cells or colony number compared to untreated control cells. Each experiment was repeated at least three times. Student's t-tests were perfomed to determine statistical significance of the observed differences among treatment groups.
Cell cycle distribution and apoptosis
Cells were plated in 6-well plates, transfected with TFO or control oligonucleotide and harvested after 48 h. Both adherent and floating cells were collected by centrifugation at 800 g for 5 min and fixed with 70% ice-cold ethanol. Cells were resuspended in 100 μl PBS containing 100 μg/ml RNase A and incubated for 15 min at room temperature. Nuclei were stained by adding 400 μl of propidium iodide solution (50 μg/ml in 0.1% sodium citrate) and analyzed on a flow cytometer (FACSCalibur, Becton Dickinson). Cell cycle distribution and percentage of apoptotic cells were determined using ModFit software (Verity). TUNEL assays were done using the in situ Cell Death Detection system (Boehringer Mannheim-Roche). Cells were transfected with TFO and control oligonucleotide as described above. After 72 h, cells were harvested and fixed in 4% paraformaldeyde for 45 min at room temperature. After rinsing with PBS, cells were permeabilyzed in a solution of 0.1% Triton X-100 and 0.1% sodium citrate for 2 min on ice, washed with PBS and incubated in the TUNEL reaction buffer for 1 h at 37°C in the dark. Samples were analyzed on a flow cytometer. To examine DNA fragmentation by gel electrophoresis, cells transfected with TFO or control oligonucleotide were lysed in 10 mM Tris–HCl (pH 8.0), 10 mM EDTA and 0.5% Triton X-100 on ice for 15 min. Cell lysates were centrifuged at 12 000 g for 15 min to separate fragmented (soluble) DNA from intact genomic DNA. Soluble DNA was treated with RNase A (50 μg/ml) at 37°C for 1 h and then with proteinase K (100 μg/ml in 0.5% SDS) for 2 h at 50°C. After phenol–chloroform extraction and ethanol precipitation, DNA was size-fractionated in 1.2% agarose gels in 1× TBE buffer and visualized by ethidium bromide staining. Nuclear morphology of TFO and control-treated cells were assessed by staining with DAPI (1 μg/ml in methanol) for 15 min at 37°C and examining the cells under a fluorescence microscope.
RESULTS
Inhibition of prostate cancer cell growth by the Ets2-TFO
Recent evidence indicates that Ets2 has a role in development and progression of several types of cancer, including prostate cancer (17,19–21). Ets2 may stimulate proliferation and/or protect cancer cells from apoptotic cell death. We have previously shown that an Ets2-targeting TFO inhibited selectively transcription of the Ets2 gene and induced a dose-, time- and sequence-dependent downregulation of Ets2 RNA (13). The present study was conducted to assess the consequences of downregulation of Ets2 in prostate cancer cells and understand the role that this transcription factor might have in the pathogenesis of prostate cancer. We began by determining whether the TFO-mediated inhibition of Ets2 expression would have any effect on the growth of prostate cancer cells. DU145 cells were transfected with the Ets2-TFO or control oligonucleotide for 4 h and then incubated for an additional 96 h without further addition of the oligonucleotides. As shown in Figure 2A, the Ets2-TFO inhibited growth of DU145 cells with an IC50 < 250 nM. The number of viable cells was reduced by >90% when cells were incubated with 500 nM of Ets2-TFO. Similar results were obtained in prostate cancer cells PC3 with an IC50 < 250 nM. The M2 control oligonucleotide had only a minimal effect on cell growth at these concentrations. Also, incubation of cells with the transfection reagent DOTAP at concentrations identical to those used in combination with the oligonucleotides did not affect cell growth. In a separate experiment, DU145 cells were treated with Ets2-TFO, control oligonucleotide or DOTAP alone and the number of viable cells was measured at 24 h intervals after transfection (Figure 2B). The number of viable cells decreased considerably at 24 h post-transfection (∼50% inhibition) and was further reduced at the later time points in samples incubated with Ets2-TFO compared to control samples.
Figure 2.
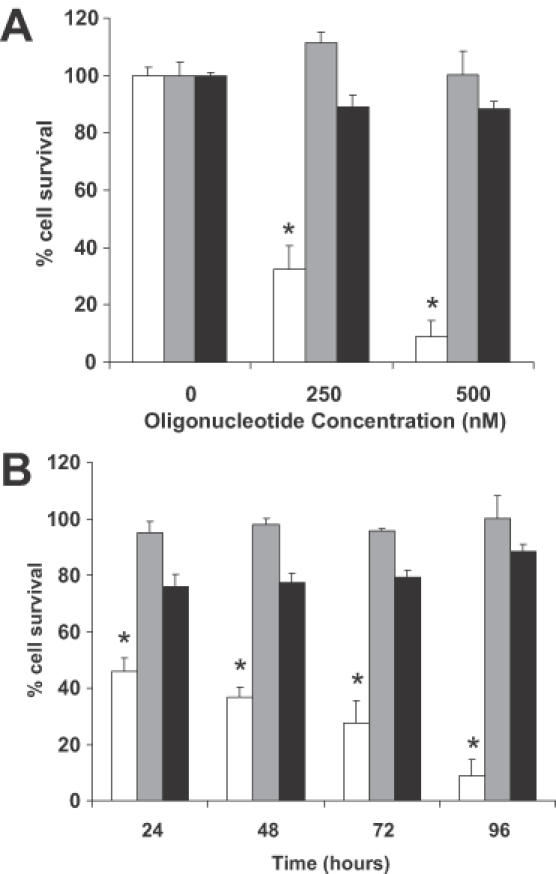
TFO-mediated inhibition of prostate cancer cell growth. DU145 cells were transfected for 4 h with Ets2-TFO or M2 control oligonucleotide in the presence of DOTAP or DOTAP alone. Viable cell number was determined using the MTT assay. Data are presented as percentage of viable cell number compared to untreated control samples. Data are mean ± SD of triplicate samples from a representative experiment. Similar results were obtained in three separate experiments. (A) Fraction of surviving cells was determined at 96 h post-transfection with the indicated concentrations of oligonucleotides. Final concentrations of DOTAP in culture medium were 0, 10 and 20 μg/ml, respectively. (B) Fraction of surviving cells was measured at 24 h intervals after transfection with 500 nM TFO and M2 control oligonucleotide or incubation with 20 μg/ml of DOTAP alone. Empty bars, Ets2-TFO; gray bars, M2 control oligonucleotide; black bars, DOTAP alone. Asterisk indicates p < 0.001 compared to DOTAP- and M2-treated cells.
Colony forming ability of prostate cancer cells is impaired by treatment with the Ets2-TFO
Studies were carried out to determine whether inhibition of Ets2 expression affected colony forming ability of prostate cancer cell both in anchorage-dependent and anchorage-independent conditions. In the first set of experiments, DU145 cells were transfected with Ets2-TFO or M2 control oligonucleotide for 4 h and then plated in tissue culture dishes at a low cell density to assess colony forming ability in anchorage-dependent conditions. As shown in Figure 3A, DU145 cells treated with the TFO gave rise to a reduced number of colonies (∼80% inhibition) compared to cells treated with M2 control oligonucleotide or DOTAP alone in the anchorage-dependent growth assay. In addition, the few colonies visible in TFO-treated dishes were considerably smaller than those observed in control dishes. We assessed also, the effects of the TFO under anchorage-independent growth conditions. DU145 cells were transfected with Ets2-TFO or M2 control oligonucleotide and then plated in soft agar. DU145 cells treated with Ets2-TFO formed a considerably lower number of colonies in soft agar (>90% inhibition) compared to cells treated with M2 control oligonucleotide or DOTAP alone (Figure 3B). Interestingly, the effect on anchorage-independent growth was constantly more pronounced than the effect on anchorage-dependent growth. Taken together, these results indicated that the Ets2-TFO had a long term inhibitory effect on proliferation and colony formation of prostate cancer cells, an effect that generally correlates well with reduced in vivo tumorigenicity of cancer cells.
Figure 3.
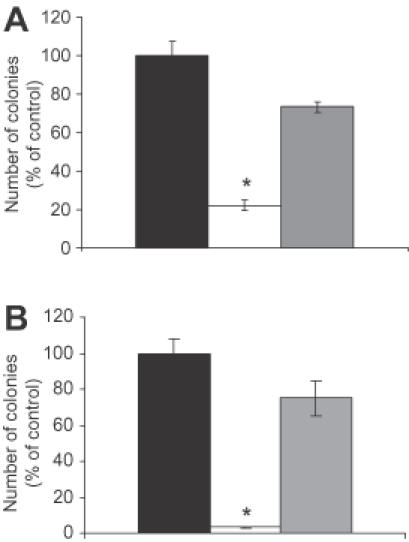
Inhibition of colony forming ability of prostate cancer cells by the Ets2-targeting TFO. DU145 cells were transfected for 4 h with 500 nM Ets2-TFO or M2 control oligonucleotide in the presence of DOTAP or DOTAP alone. Cells were counted and plated to determine colony foming ability in anchorage-depedent and anchorage-independent growth conditions. (A) Cells (1 × 103 per well) were plated in 35 mm tissue culture dishes and colonies were counted after 8–10 days. (B) Cells (2 × 103 per well) were plated in 0.3% soft agar as described in Materials and Methods. Colonies were counted after 21–24 days. Data are mean ± SD of triplicate samples from a representative experiment. Black bars, DOTAP alone, empty bars, Ets2-TFO; gray bars, M2 control oligonucleotide. Asterisk indicates p < 0.001 compared to DOTAP- and M2-treated cells.
Ets2-TFO alters cell cycle progression and induces apoptosis of prostate cancer cells
Downregulation of Ets2 expression had a dramatic effect on proliferation and clonogenic potential of prostate cancer cells. To investigate the mechanisms underlying the Ets2-TFO mediated growth inhibition, we examined DNA content and cell cycle distribution of control and TFO-treated cells by flow cytometry. Figure 4 shows the results of the analysis of cells transfected with 1 μM of Ets2-TFO or M2 control oligonucleotide and then grown for 48 h. Incubation with Ets2-TFO resulted in multiple changes in the flow cytometric profile. First, a population of cells with sub-G1 DNA content was present in samples treated with Ets2-TFO (14%). The sub-G1 population was virtually absent (0.4%) in M2-treated and M2-untreated samples. In addition, the fraction of S phase cells was increased in Ets2-treated samples (21– 36%), with a corresponding decrease of the percentage of cells in G1 (68–54%). Similar changes were observed in cells treated with 0.5 μM of TFO (G1, 59%; G2-M, 11%; S, 31%; apoptotic cells, 11%). No changes in cell cycle distribution were observed in cells treated with 0.5 and 1 μM of M2 control oligonucleotide. Thus, treatment of prostate cancer cells with the Ets2-TFO delayed the progression of cells throughout the S phase with a concomitant loss of cells from the G1 compartment. The appearance of a sub-G1 population indicated that incubation of cells with Ets2-TFO increased the rate of apoptotic cell death and probably accounted for the loss of cells from the G1 compartment. Induction of apoptotic cell death in Ets2-TFO-treated cells was confirmed by examining genomic DNA fragmentation using the TUNEL assay and gel electrophoresis. In the TUNEL assay, FITC-positive apoptotic cells were considerably increased in samples of TFO-treated cells (∼32% of apoptotic cells at 72 h) compared to control-treated and -untreated cells (Figure 5A). At the same time, flow cytometry showed 17% of cells with a sub-G1 DNA content (Figure 5B). DNA laddering typical of apoptotic cells was also observed by gel electrophoresis in Ets2-TFO-treated cells (Figure 5C). Furthermore, nuclei with morphological features characteristic of apoptosis, such as condensed chromatin and fragmented nuclei were observed upon DAPI staining of TFO-treated cells (data not shown). Neither DNA fragmentation nor apoptotic nuclei were present in control-treated and -untreated cells.
Figure 4.
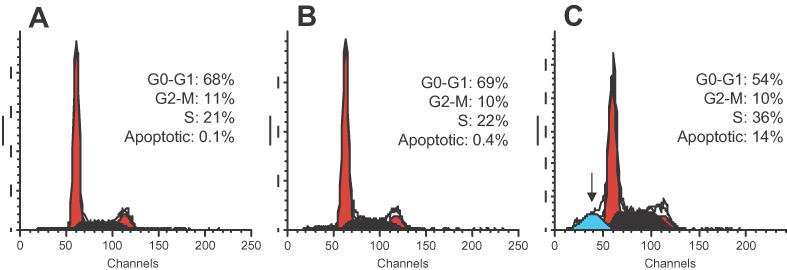
Cell cycle effects of Ets2 downregulation in prostate cancer cells. DU145 cells were left untreated or transfected with 1 μM Ets2-TFO or M2 control oligonucleotide. At 48 h post-transfection, cells were collected, fixed, stained with propidium iodide and analyzed for DNA content by flow cytometry. Percentages of cells in different phase of cell cycle or showing sub-G1 DNA content (arrow) are indicated for each panel. (A) untreated control; (B) Ets2-TFO; (C) M2 control oligonucleotide.
Figure 5.
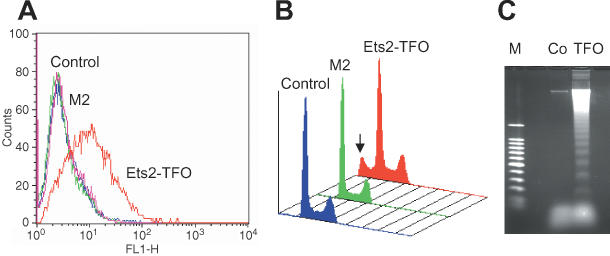
Apoptosis and DNA fragmentation in Ets2-TFO-treated prostate cancer cells. DU145 cells were incubated with 500 nM Ets2-TFO or M2 control oligonucleotide or left untreated (control). After 72 h, cells were harvested and analyzed. (A) Apoptotic cells were detected by terminal deoxynucleotidyl-transferase-mediated nick end labeling (TUNEL assay) and flow cytometry. (B) Percentage of cells with sub-G1 DNA content (arrow) was determined by flow cytometry analysis of propidium iodide stained cells. (C) Soluble DNA was isolated and analyzed on a 1.2% agarose gel to determine the presence of internucleosomal DNA fragmentation. Co, untreated control; M, 100 bp DNA ladder.
Downregulation of Ets2 expression is responsible for growth inhibition induced by the Ets2-TFO
To ascertain that the anti-proliferative activity of the Ets2-TFO was a consequence of the selective downregulation of Ets2, we determined whether expression of the Ets2 gene under the control of a heterologous promoter, which could not be affected by the Ets2-TFO, could prevent these effects in prostate cancer cells. Transient transfection of DU145 cells with the Ets2 expressing plasmid resulted in increased level of Ets2 protein compared to cells transfected with an empty vector (Figure 6A). A similar band was observed upon transfection of the pSGK-Ets2 in MCF-7 breast cancer cells, which had a very low level of endogenous Ets2. Furthermore, transfection efficiency, as monitored by co-transfection of an enhanced green fluorescent protein (EGFP) expressing plasmid, was consistently high in these experiments with 70–90% EGFP-positive cells (data not shown). Next, DU145 prostate cancer cells were transfected with Ets2-TFO and M2 control oligonucleotide along with the Ets2 expressing vector or an empty vector. Cell growth was assessed by a colorimetric assay after 72 h or by colony forming assay after 8–10 days. As shown in Figure 6B, growth inhibition induced by the Ets2-TFO was almost completely abolished by co-transfection with the Ets2 expressing vector (80% surviving cells compared to 20% in control-transfected cells). A similar protective effect of the Ets2 expressing plasmid against the Ets2-TFO was also observed in the colony forming assays (Figure 6C). Western blot analysis confirmed a higher Ets2 level in TFO-treated cells transfected with the pSGK-Ets2 compared to cells transfected with the empty vector at 72 h post-transfection (Figure 6D). Thus, forced expression of Ets2 conferred significant protection against the effects of the Est2- TFO on cell proliferation, providing evidence that these effects were, in fact, due to downregulation of Ets2 expression. It is interesting that transfection of the pSGK-Ets2 plasmid in M2-treated cells had a mild growth inhibitory effect, not observed in cells treated with the empty vector pSGK. Apparently, forced expression of Ets2 might be toxic to cells that already express high levels of this transcription factor as previously suggested (23).
Figure 6.
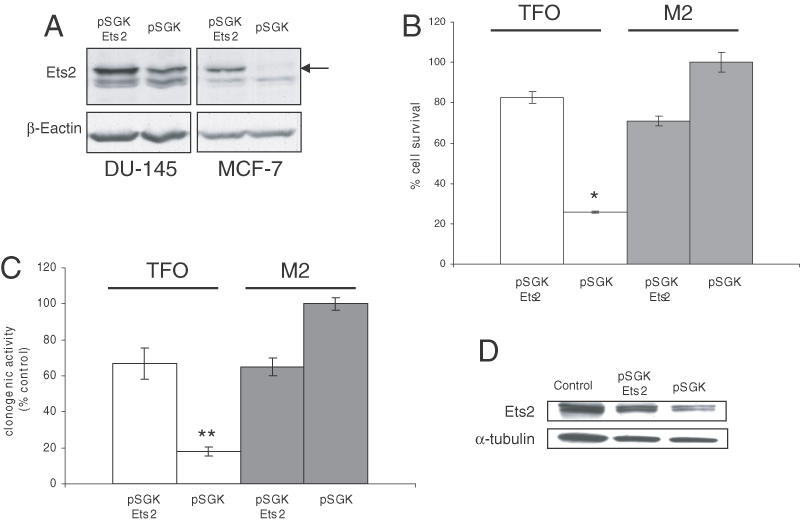
Forced expression of Ets2 rescues cells from growth inhibition induced by the Ets2-TFO. (A) Expression of Ets2 in transiently transfected DU145 and MCF-7 cells was determined by western blot at 48 h post-transfection. (B) DU145 were co-transfected with the Ets2 expression vector (pSGK-Ets2) or an empty vector (pSGK) along with 250 nM of Ets2-TFO or M2 control oligonucleotide for 4 h. Percentage of surviving cells was determined with MTS assay after 72 h. *P < 0.001 compared to cells transfected with pSGK-Ets2 and Ets2-TFO. (C) Cells were transfected with Ets2-TFO or M2 oligonucleotide with or without pSGK-Ets2 and empty vector (pSGK) as described above and plated in 35 mm tissue culture dishes. Colonies were counted after 8–10 days. Data are mean ± SD of triplicate samples from a representative experiment. **P < 0.01 compared to cells transfected with pSGK-Ets2 and Ets2-TFO. (D) Western blot analysis of DU145 cells left untreated (control) or transfected with Ets2-TFO along with pSGK-Ets2 or pSGK for 4 h as described above and then harvested after 72 h.
Normal human cells that express low levels of Ets2 are not affected by the Ets2-TFO
We hypothesized that growth of normal cells, which express Ets2 at low levels and may not depend on Ets2-regulated pathways for cell proliferation and survival, would not be affected by inhibition of Ets2 transcription. To address this issue, we studied the effect of the Ets2-TFO on normal human fibroblast. We confirmed by RT–PCR that the Ets2 RNA level was considerably lower (∼10-fold lower) in normal human fibroblasts than DU145 cells (Figure 7A). Next, normal human fibroblasts were transfected with Ets2-TFO or M2 control oligonucleotide and growth measured by a colorimetric assay (Figure 7B). The Ets2-TFO failed to inhibit the growth of normal human fibroblasts at concentrations that greatly reduced the growth of prostate cancer cells. Furthermore, flow cytometry showed no evidence of cell cycle alterations or induction of apoptotic cell death in normal human fibroblasts treated with Ets2-TFO (data not shown). This lack of effect of the Ets2-TFO was not due to inefficient uptake of the oligonucleotide by fibroblasts. In fact, the Ets2-TFO was able to reduce Ets2 promoter activity in luciferase reporter assays, indicating that cell permeability was not a limiting factor in these experiments (Figure 7C). These data suggest that the anti-proliferative and pro-apoptotic effects of the Ets2-TFO were selective toward cancer cells expressing the target gene. This observation may be important for development of Ets2 inhibitors as therapeutic agents, since it suggests that Ets2 is not essential for proliferation and survival of normal cells, such as normal human fibroblasts.
Figure 7.
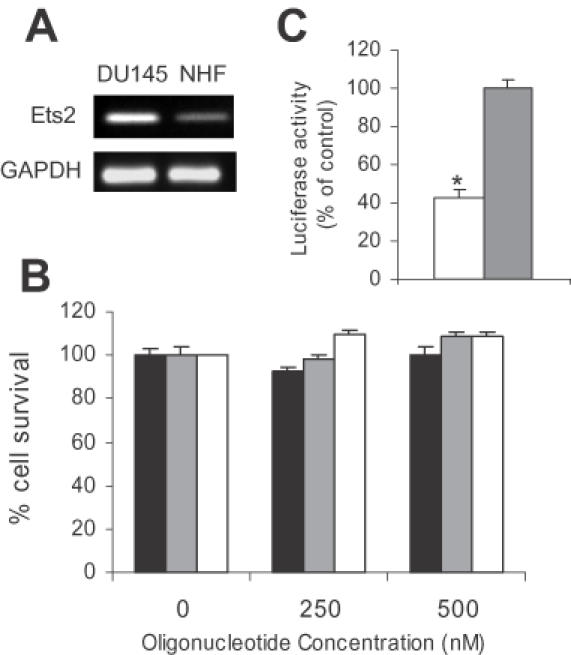
Insensitivity of normal human fibroblasts to the growth inhibitory effects of the Ets2-TFO. (A) Expression of Ets2 in normal human fibroblasts (NHF) and DU145 cells was determined by RT–PCR. GAPDH was used as control. (B) Human fibroblasts were transfected for 4 h with the indicated concentrations of Ets2-TFO or M2 control oligonucleotide in the presence of DOTAP or DOTAP alone. Viable cell number was determined after 96 h as indicated in the legend to Figure 2. (C) Human fibroblasts were transfected with pGL3-Ets2 luciferase reporter construct along with 500 nM Ets2-TFO or M2 oligonucleotide. Luciferase assays were performed after 24 h post-transfection. Data are mean ± SD of triplicate samples. P < 0.01 compared to M2-treated cells.
Downregulation of expression of Ets2-target genes in Ets2-TFO-treated prostate cancer cells
The anti-proliferative and pro-apoptotic effects induced by the Ets2-TFO were due to the downregulation of Ets2 expression and were probably associated with concomitant downregulation of the expression of multiple genes controlled by this transcription factor and involved in critical functions in prostate cancer cells. To determine whether the Ets2-TFO acted by inhibiting expression of critical downstream target genes, we examined the levels of proteins known to be involved in cell cycle, proliferation and apoptosis and to be regulated by Ets factors in other cell types. We performed western blot analysis of DU145 cells transfected with Ets2-TFO or M2 control oligonucleotide. As shown in Figure 8, a reduced level of Ets2 was associated with reduced levels of Bcl-xL, c-myc and cyclin D1 in Ets2-TFO-treated cells compared to M2-treated cells. In contrast, Ets1 and Bcl-2 protein levels were unchanged in TFO- and M2-treated cells. Thus, Ets2 controls specifically the expression of Bcl-xL, c-myc and cyclin D1 in prostate cancer cells and its downregulation reduced the levels of these important cell death and proliferation regulatory proteins.
Figure 8.

Expression of Ets2 downstream target genes in Ets2-TFO-treated prostate cancer cells. DU145 cells were transfected with 500 nM Ets2-TFO or M2 control oligonucleotide for 4 h. After 48 h, western blots were performed on cell lysates from Ets2-TFO- and M2-treated cells using antibodies against the indicated proteins. β-actin and α-tubulin were used as loading controls.
DISCUSSION
Cancer of the prostate is the most frequently diagnosed cancer and the second leading cause of cancer death among men in western countries (24,25). Current therapies for advanced androgen independent prostate cancer are only marginally effective (26). Thus, it is important to understand the molecular mechanisms involved in the pathogenesis and progression of this disease in order to identify novel therapeutic targets and develop more effective treatment strategies. Deregulated expression and activity of Ets-domain transcription factors may be involved in prostate cancer development. Ets factors are known to play a role in various aspects of cell proliferation and differentiation (15,16,27). A large number of genes, including those coding for cell cycle regulators, transcription factors, matrix metalloproteinases, extracellular matrix receptors, growth factors and growth factor receptors, are known to contain Ets binding sites (22). Ets factors are downstream effectors of multiple signaling pathways, including the Ras/Raf/MAPK and PI3K/Akt pathways, which mediate the response to growth factor receptor stimulation and are often activated during oncogenic transformation (28–34). The oncogenic potential of Ets factors has been demonstrated in several experimental systems. Overexpression of a number of Ets factors (e.g. Ets1 and Ets2) increases proliferation, anchorage-independent growth and tumorigenicity in nude mice (35,36). Interfering with Ets factors, in contrast, has been shown to reduce growth and tumorigenicity (19–21,33,37,38). At the transcriptional level, while most Ets factors act as activators, some Ets factors act preferentially as repressors and others have been shown to be both activators and repressors (15,16,27). Indeed, the functions of Ets factors may vary in different cell types depending on the specific cellular context, regulatory signaling pathways and crosstalk between Ets proteins and other protein partners (15,16,27). Similarities in protein structure and DNA binding specificity complicate the identification of the specific roles of individual Ets factors in mediating malignant transformation. Selective inactivation of individual Ets family members may provide a way to analyze the role of each factor in the cell type of interest. Ets2 function was previously shown to be required for maintenance of the malignant phenotype in several tumor cell types. Ets2 antisense or dominant negative constructs reduced anchorage-independent growth of prostate, breast and thyroid cancer cells (19–21). However, antisense and dominant negative constructs might not be specific for an individual Ets factor and it is quite possible that they block the activity of multiple Ets factors. Thus, the precise role of Ets2 in these cancers could not be clearly defined. Here, we used a PS-modified TFO directed to a unique sequence in the Ets2 promoter to selectively inhibit transcription of this gene. Our study shows that Ets2 regulates the expression of genes critical for cell proliferation and apoptosis, such as bcl-xL, c-myc and cyclin D1, in prostate cancer cells and downregulation of Ets2 leads to cell growth inhibition and cell death. These data demonstrate that Ets2, which is often overexpressed in advanced prostate cancers, may have an important role in the pathogenesis of this disease. In addition to the short-term effects on cell growth and survival, the Ets2-TFO also reduced the ability of DU145 cells to form colonies in anchorage-dependent and anchorage-independent growth assays. To the best of our knowledge, this is the first report showing target-related and long-term activity of a TFO in cell cultures. These long-term effects generally correlate well with ability of cancer therapeutics to reduce tumor growth in vivo. The activity and selectivity of the Ets2-TFO suggest that triplex-mediated gene targeting might be an effective strategy for the treatment of prostate cancer. The concomitant downregulation of multiple genes, which are involved in different cellular functions and are all controlled by Ets2, may explain the marked effect of the Ets2-TFO on prostate cancer cell proliferation and survival. Indeed, a transcription factor, such as Ets2, appears as a very attractive therapeutic target because, by blocking it several downstream genes can be affected. In addition, signaling by several growth factor receptors, such as EGFR and IGF-1R, and multiple intracellular signal transduction pathways would be inhibited. Therefore, the Ets2-TFO described here may represent a very effective tool to antagonize multiple oncogenic pathways in cancer cells.
In our study, the Ets2-targeting TFO had a dramatic effect on growth and survival of DU145 prostate cancer cells. Triplex-mediated downregulation of Ets2 expression was associated with changes in the expression of the anti-apoptotic protein Bcl-xL and the growth-promoting genes cyclin D1 and c-myc. Although these were known Ets target genes in other cell types, control of their expression by Ets2 in prostate cancer cells had not been reported yet (22). Ets2 was shown to regulate expression of Bcl-xL in murine macrophages and forced expression of Ets2 protected these cells from death induced by growth factor deprivation (39). The promoters of cyclin D1 and c-myc also contain Ets binding sites and Ets factors have been shown to activate transcription of both these genes (40,41). Cyclin D1 and c-myc are positive regulators of cell growth and critical targets of mitogenic signals (42,43). Thus, downregulation of Ets2 in prostate cancer cells can disrupt important mitogenic signals mediated by cyclin D1 and c-myc, consistent with the effects on cell cycle progression and proliferation seen upon treatment with the Ets2-TFO. Both cyclin D1 and c-myc are required for progression of cells from G1 to S phase of the cell cycle (42). However, we did not observe a G1 arrest but rather an accumulation of cells in S phase of the cell cycle. This is probably due to the concomitant induction of apoptotic cell death and downregulation of other cell cycle regulatory proteins, since it is likely that other cell cycle and cell death regulatory factors are affected by the reduced levels of Ets2. Collectively, these observations appear particularly relevant to understanding the role of Ets2 in advanced prostate cancer. Bcl-xL is expressed in a large number of human prostate cancers and higher levels of Bcl-xL are generally associated with higher grade tumors and presence of metastases (44). Overexpression and amplification of c-myc is frequent in advanced and androgen independent prostate cancer and forced expression of c-myc confers androgen-independent growth properties to androgen dependent prostate cancer cells (45). Cyclin D1 overexpression also increases proliferation in vitro and induce growth of androgen independent prostate cancers in vivo (46). However, the role of cyclin D1 in androgen-independent prostate cancer is unclear. Cyclin D1 has been shown to act as a corepressor of the androgen receptor (47), while expression of cyclin D1, which is infrequent in primary prostate tumors, is found increasingly in bone mestatases and lesions with high proliferative index (48). Thus, higher levels of cyclin D1 might have a role in suppressing androgen receptor signaling and perhaps favoring selection of highly metastatic, androgen independent cell clones.
One of the problems associated with agents, such as AOs, siRNA and TFOs, directed to a specific molecular target, is the difficulty to determine whether their biological effects are truly due to the inhibition of the intended target rather than to non-specific interactions with other cell components. In our study, the PS modification, which considerably increases the half-life and enhances cellular uptake of oligonucleotides, may increase the risk of non-specific cellular effects (49). Also, G-rich oligonucleotides have a tendency to form self-aggregates, such as homoduplex and quadruplex structures, and formation of these secondary structures may be an additional source of both sequence- and non-sequence-specific activity of AOs and TFOs (50–53). To address these concerns, we showed that expression of Ets2 under the control of a heterologous promoter protected prostate cancer cells from the antiproliferative effects of the Ets2-TFO. This protective effect was observed both in short- and long-term cell growth assays, indicating that sustained levels of Ets2 were sufficient to prevent the effects of the TFO. The ability of the Ets2 expression plasmid to rescue cells from the effects of the TFO indicated both that these effects were dependent on Ets2 downregulation and that the Ets2-TFO acted specifically on its target. In a previous study, we showed that the Ets2-TFO inhibited transcription of the Ets2 gene by a mechanism involving triplex-DNA formation at the Ets2 promoter site (13). Control oligonucleotides with mismatched sequences were used to rule out non-sequence-dependent effects that might be due to chemical or nucleotide composition of the TFO. Furthermore, oligonucleotide duplexes and luciferase reporter constructs with mutations that prevented binding of the TFO to the sequence in the Ets2 promoter excluded non-triplex mediated and sequence-dependent effects of the TFO both in vitro and in cells (13). Importantly, the effects of the Ets2-TFO on Ets2 transcription could not be attributed to a decoy or aptamer effect of the oligonucleotide on transcription factors, such as Sp1 and Sp3, binding to the Ets2 promoter. In fact, we could not detect any effect of the Ets2-TFO on the binding of nuclear proteins to the Ets2 target site unless the correct triplex target sequence was present and, in cells, the Ets2-TFO did not affect promoter–reporter activity and transcription of other genes known to have very similar purine-rich sequences and Sp1 sites in their regulatory regions (13). We have further tested the biological specificity of the TFO toward its intended target by examining its effects on normal cells, such as normal human fibroblasts, which expressed a very low level of Ets2. Therefore, it was important to find that growth of human fibroblasts was not affected by the Ets2-TFO. This result shows the absence of non-target specific toxicity of the PS-modified Ets2-TFO and suggests that normal cells that express low levels of Ets2 and do not depend on Ets2-regulated pathways for growth and survival might not be affected by this approach. The present observations are relevant for the future development of this approach for cancer therapy. However, the long-term effects and potential mutagenicity of TFOs in normal tissues will need to be carefully examined in the future. The high degree of activity and specificity of the Ets2-targeting TFO that we have observed in these cell culture studies have important implications in view of the possible use of this approach to investigate the mechanisms of cancer pathogenesis and for prostate cancer therapy.
Acknowledgments
ACKNOWLEDGEMENTS
This work was supported by grants from NIH (CA-70735, C.V.C.), DOD (N00014-96-1-1298, C.V.C. and D.K.W.) Oncosuisse (OCS 01264-08-2002, C.V.C.), Fondazione Ticinese per la Ricerca sul Cancro (G.M.C.) and Functional Genetics Program, Hollings Cancer Center (G.M.C. and D.K.W.).
REFERENCES
- 1.Gewirtz A.M., Sokol,D.L. and Ratajczak,M.Z. (1998) Nucleic acid therapeutics: state of the art and future prospects. Blood, 92, 712–736. [PubMed] [Google Scholar]
- 2.Dorsett Y. and Tuschl,T. (2004) siRNAs: applications in functional genomics and potential as therapeutics. Nature Rev. Drug Discov., 3, 318–329. [DOI] [PubMed] [Google Scholar]
- 3.Chan P.P. and Glazer,P.M. (1997) Triplex DNA: fundamentals, advances, and potential applications for gene therapy. J. Mol. Med., 75, 267–282. [DOI] [PubMed] [Google Scholar]
- 4.Praseuth D., Guieysse,A.L. and Helene,C. (1999) Triple helix formation and the antigene strategy for sequence-specific control of gene expression. Biochim. Biophys. Acta, 1489, 181–206. [DOI] [PubMed] [Google Scholar]
- 5.Seidman M.M. and Glazer,P.M. (2003) The potential for gene repair via triple helix formation. J. Clin. Invest., 112, 487–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Frank-Kamenetskii M.D. and Mirkin,S.M. (1995) Triplex DNA structures. Annu. Rev. Biochem., 64, 65–95. [DOI] [PubMed] [Google Scholar]
- 7.Goni J.R., de la Cruz,X. and Orozco,M. (2004) Triplex-forming oligonucleotide target sequences in the human genome. Nucleic Acids Res., 32, 354–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Winters T.A. (2000) Gene targeted agents: new opportunities for rational drug development. Curr. Opin. Mol. Ther., 2, 670–681. [PubMed] [Google Scholar]
- 9.Uil T.G., Haisma,H.J. and Rots,M.G. (2003) Therapeutic modulation of endogenous gene function by agents with designed DNA-sequence specificities. Nucleic Acids Res., 31, 6064–6078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Catapano C.V., McGuffie,E.M., Pacheco,D. and Carbone,G.M. (2000) Inhibition of gene expression and cell proliferation by triple helix-forming oligonucleotides directed to the c-myc gene. Biochemistry, 39, 5126–5138. [DOI] [PubMed] [Google Scholar]
- 11.McGuffie E.M., Pacheco,D., Carbone,G.M. and Catapano,C.V. (2000) Antigene and antiproliferative effects of a c-myc-targeting phosphorothioate triple helix-forming oligonucleotide in human leukemia cells. Cancer Res., 60, 3790–3799. [PubMed] [Google Scholar]
- 12.Carbone G.M., McGuffie,E., Napoli,S., Flanagan,C.E., Dembech,C., Negri,U., Arcamone,F., Capobianco,M.L. and Catapano,C.V. (2004) DNA binding and antigene activity of a daunomycin-conjugated triplex-forming oligonucleotide targeting the P2 promoter of the human c-myc gene. Nucleic Acids Res., 32, 2396–2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carbone G.M., McGuffie,E.M., Collier,A. and Catapano,C.V. (2003) Selective inhibition of transcription of the Ets2 gene in prostate cancer cells by a triplex-forming oligonucleotide. Nucleic Acids Res., 31, 833–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sharrocks A.D. (2001) The ETS-domain transcription factor family. Nature Rev. Mol. Cell. Biol., 2, 827–837. [DOI] [PubMed] [Google Scholar]
- 15.Hsu T., Trojanowska,M. and Watson,D.K. (2004) Ets proteins in biological control and cancer. J. Cell. Biochem., 91, 896–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oikawa T. and Yamada,T. (2003) Molecular biology of the Ets family of transcription factors. Gene, 303, 11–34. [DOI] [PubMed] [Google Scholar]
- 17.Liu A.Y., Corey,E., Vessella,R.L., Lange,P.H., True,L.D., Huang,G.M., Nelson,P.S. and Hood,L. (1997) Identification of differentially expressed prostate genes: increased expression of transcription factor ETS-2 in prostate cancer. Prostate, 30, 145–153. [DOI] [PubMed] [Google Scholar]
- 18.Liu A.Y., True,L.D., LaTray,L., Nelson,P.S., Ellis,W.J., Vessella,R.L., Lange,P.H., Hood,L. and van den Engh,G. (1997) Cell–cell interaction in prostate gene regulation and cytodifferentiation. Proc. Natl Acad. Sci. USA, 94, 10705–10710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sementchenko V.I., Schweinfest,C.W., Papas,T.S. and Watson,D.K. (1998) ETS2 function is required to maintain the transformed state of human prostate cancer cells. Oncogene, 17, 2883–2888. [DOI] [PubMed] [Google Scholar]
- 20.Sapi E., Flick,M.B., Rodov,S. and Kacinski,B.M. (1998) Ets-2 transdominant mutant abolishes anchorage-independent growth and macrophage colony-stimulating factor-stimulated invasion by BT20 breast carcinoma cells. Cancer Res., 58, 1027–1033. [PubMed] [Google Scholar]
- 21.de Nigris F., Mega,T., Berger,N., Barone,M.V., Santoro,M., Viglietto,G., Verde,P. and Fusco,A. (2001) Induction of ETS-1 and ETS-2 transcription factors is required for thyroid cell transformation. Cancer Res., 61, 2267–2275. [PubMed] [Google Scholar]
- 22.Sementchenko V.I. and Watson,D.K. (2000) Ets target genes: past, present and future. Oncogene, 19, 6533–6548. [DOI] [PubMed] [Google Scholar]
- 23.Foos G. and Hauser,C.A. (2000) Altered Ets transcription factor activity in prostate tumor cells inhibits anchorage-independent growth, survival, and invasiveness. Oncogene, 19, 5507–5516. [DOI] [PubMed] [Google Scholar]
- 24.Gronberg H. (2003) Prostate cancer epidemiology. Lancet, 361, 859–864. [DOI] [PubMed] [Google Scholar]
- 25.Nelson W.G., De Marzo,A.M. and Isaacs,W.B. (2003) Prostate cancer. N. Engl. J. Med., 349, 366–381. [DOI] [PubMed] [Google Scholar]
- 26.Shaffer D.R. and Scher,H.I. (2003) Prostate cancer: a dynamic illness with shifting targets. Lancet Oncol., 4, 407–414. [DOI] [PubMed] [Google Scholar]
- 27.Sharrocks A.D. (2001) The ETS-domain transcription factor family. Nature Rev. Mol. Cell. Biol., 2, 827–837. [DOI] [PubMed] [Google Scholar]
- 28.Yang B.S., Hauser,C.A., Henkel,G., Colman,M.S., Van Beveren,C., Stacey,K.J., Hume,D.A., Maki,R.A. and Ostrowski,M.C. (1996) Ras-mediated phosphorylation of a conserved threonine residue enhances the transactivation activities of c-Ets1 and c-Ets2. Mol. Cell. Biol., 16, 538–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McCarthy S.A., Chen,D., Yang,B.S., Garcia Ramirez,J.J., Cherwinski,H., Chen,X.R., Klagsbrun,M., Hauser,C.A., Ostrowski,M.C. and McMahon,M. (1997) Rapid phosphorylation of Ets-2 accompanies mitogen-activated protein kinase activation and the induction of heparin-binding epidermal growth factor gene expression by oncogenic Raf-1. Mol. Cell. Biol., 17, 2401–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wasylyk C., Maira,S.M., Sobieszczuk,P. and Wasylyk,B. (1994) Reversion of Ras transformed cells by Ets transdominant mutants. Oncogene, 9, 3665–3673. [PubMed] [Google Scholar]
- 31.Wasylyk B., Hagman,J. and Gutierrez-Hartmann,A. (1998) Ets transcription factors: nuclear effectors of the Ras-MAP-kinase signaling pathway. Trends Biochem. Sci., 23, 213–216. [DOI] [PubMed] [Google Scholar]
- 32.Galang C.K., Der,C.J. and Hauser,C.A. (1994) Oncogenic Ras can induce transcriptional activation through a variety of promoter elements, including tandem c-Ets-2 binding sites. Oncogene, 9, 2913–2921. [PubMed] [Google Scholar]
- 33.Galang C.K., Garcia-Ramirez,J., Solski,P.A., Westwick,J.K., Der,C.J., Neznanov,N.N., Oshima,R.G. and Hauser,C.A. (1996) Oncogenic Neu/ErbB-2 increases ets, AP-1, and NF-kappaB-dependent gene expression, and inhibiting ets activation blocks Neu-mediated cellular transformation. J. Biol. Chem., 271, 7992–7998. [DOI] [PubMed] [Google Scholar]
- 34.Yordy J.S. and Muise-Helmericks,R.C. (2000) Signal transduction and the Ets family of transcription factors. Oncogene, 19, 6503–6513. [DOI] [PubMed] [Google Scholar]
- 35.Seth A., Watson,D.K., Blair,D.G. and Papas,T.S. (1989) c-ets-2 protooncogene has mitogenic and oncogenic activity. Proc. Natl Acad. Sci. USA, 86, 7833–7837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Seth A. and Papas,T.S. (1990) The c-ets-1 proto-oncogene has oncogenic activity and is positively autoregulated. Oncogene, 5, 1761–1767. [PubMed] [Google Scholar]
- 37.Man A.K., Young,L.J., Tynan,J.A., Lesperance,J., Egeblad,M., Werb,Z., Hauser,C.A., Muller,W.J., Cardiff,R.D. and Oshima,R.G. (2003) Ets2-dependent stromal regulation of mouse mammary tumors. Mol. Cell. Biol., 23, 8614–8625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Neznanov N., Man,A.K., Yamamoto,H., Hauser,C.A., Cardiff,R.D. and Oshima,R.G. (1999) A single targeted Ets2 allele restricts development of mammary tumors in transgenic mice. Cancer Res., 59, 4242–4246. [PubMed] [Google Scholar]
- 39.Sevilla L., Aperlo,C., Dulic,V., Chambard,J.C., Boutonnet,C., Pasquier,O., Pognonec,P. and Boulukos,K.E. (1999) The Ets2 transcription factor inhibits apoptosis induced by colony-stimulating factor 1 deprivation of macrophages through a Bcl-xL-dependent mechanism. Mol. Cell. Biol., 19, 2624–2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Albanese C., Johnson,J., Watanabe,G., Eklund,N., Vu,D., Arnold,A. and Pestell,R.G. (1995) Transforming p21ras mutants and c-Ets-2 activate the cyclin D1 promoter through distinguishable regions. J. Biol. Chem., 270, 23589–23597. [DOI] [PubMed] [Google Scholar]
- 41.Roussel M.F., Davis,J.N., Cleveland,J.L., Ghysdael,J. and Hiebert,S.W. (1994) Dual control of myc expression through a single DNA binding site targeted by ets family proteins and E2F-1. Oncogene, 9, 405–415. [PubMed] [Google Scholar]
- 42.Sherr C.J. (1996) Cancer cell cycles. Science, 274, 1672–1677. [DOI] [PubMed] [Google Scholar]
- 43.Facchini L.M. and Penn,L.Z. (1998) The molecular role of Myc in growth and transformation: recent discoveries lead to new insights. FASEB J., 12, 633–651. [PubMed] [Google Scholar]
- 44.Krajewska M., Krajewski,S., Epstein,J.I., Shabaik,A., Sauvageot,J., Song,K., Kitada,S. and Reed,J.C. (1996) Immunohistochemical analysis of bcl-2, bax, bcl-X, and mcl-1 expression in prostate cancers. Am. J. Pathol., 148, 1567–1576. [PMC free article] [PubMed] [Google Scholar]
- 45.Bernard D., Pourtier-Manzanedo,A., Gil,J. and Beach,D.H. (2003) Myc confers androgen-independent prostate cancer cell growth. J. Clin. Invest., 112, 1724–1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen Y., Martinez,L.A., LaCava,M., Coghlan,L. and Conti,C.J. (1998) Increased cell growth and tumorigenicity in human prostate LNCaP cells by overexpression to cyclin D1. Oncogene, 16, 1913–1920. [DOI] [PubMed] [Google Scholar]
- 47.Petre-Draviam C.E., Cook,S.L., Burd,C.J., Marshall,T.W., Wetherill,Y.B. and Knudsen,K.E. (2003) Specificity of cyclin D1 for androgen receptor regulation. Cancer Res., 63, 4903–4913. [PubMed] [Google Scholar]
- 48.Drobnjak M., Osman,I., Scher,H.I., Fazzari,M. and Cordon-Cardo,C. (2000) Overexpression of cyclin D1 is associated with metastatic prostate cancer to bone. Clin. Cancer Res., 6, 1891–1895. [PubMed] [Google Scholar]
- 49.Crooke S.T. and Bennett,C.F. (1996) Progress in antisense oligonucleotide therapeutics. Annu. Rev. Pharmacol. Toxicol., 36, 107–129. [DOI] [PubMed] [Google Scholar]
- 50.Benimetskaya L., Berton,M., Kolbanovsky,A., Benimetsky,S. and Stein,C.A. (1997) Formation of a G-tetrad and higher order structures correlates with biological activity of the RelA (NF-kappaB p65) ‘antisense’ oligodeoxynucleotide. Nucleic Acids Res., 25, 2648–2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bates P.J., Kahlon,J.B., Thomas,S.D., Trent,J.O. and Miller,D.M. (1999) Antiproliferative activity of G-rich oligonucleotides correlates with protein binding. J. Biol. Chem., 274, 26369–26377. [DOI] [PubMed] [Google Scholar]
- 52.Basu S. and Wickstrom,E. (1997) Temperature and salt dependence of higher order structure formation by antisense c-myc and c-myb phosphorothioate oligodeoxyribonucleotides containing tetraguanylate tracts. Nucleic Acids Res., 25, 1327–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cogoi S., Quadrifoglio,F. and Xodo,L.E. (2004) G-rich oligonucleotide inhibits the binding of a nuclear protein to the Ki-ras promoter and strongly reduces cell growth in human carcinoma pancreatic cells. Biochemistry, 43, 2512–2523. [DOI] [PubMed] [Google Scholar]