


Abstract
We have recently reported a disease-causing substitution (+5G > C) at the donor site of NF-1 exon 3 that produces its skipping. We have now studied in detail the splicing mechanism involved in analyzing RNA–protein complexes at several 5′ splice sites. Characteristic protein patterns were observed by pulldown and band-shift/super-shift analysis. Here, we show that hnRNP H binds specifically to the wild-type GGGgu donor sequence of the NF-1 exon 3. Depletion analyses shows that this protein restricts the accessibility of U1 small nuclear ribonucleoprotein (U1snRNA) to the donor site. In this context, the +5G > C mutation abolishes both U1snRNP base pairing and the 5′ splice site (5′ss) function. However, exon recognition in the mutant can be rescued by disrupting the binding of hnRNP H, demonstrating that this protein enhances the effects of the +5G > C substitution. Significantly, a similar situation was found for a second disease-causing +5G > A substitution in the 5′ss of TSHβ exon 2, which harbors a GGgu donor sequence. Thus, the reason why similar nucleotide substitutions can be either neutral or very disruptive of splicing function can be explained by the presence of specific binding signatures depending on local contexts.
INTRODUCTION
One of the earlier events in the recognition of eukaryotic exons by the splicing machinery is the binding of U1 small nuclear ribonucleoprotein (U1snRNP) (1–3) to the 5′ splice site (5′ss) consensus sequence (4). This recognition, initially shown to be heavily influenced by base pair complementarity between the 5′ss and the 5′ end of U1snRNA (5–9) has been shown to be rather more complex. In fact, much still remains to be uncovered regarding the role of U1snRNP in the preliminary steps of U1snRNA–pre-mRNA complex formation, (10,11), in 5′ss definition (12–16), in intronic splicing control (17), and its interaction with the other snRNP complexes or proteins (18–21). The emerging fact in all these studies is the presence of recognition mechanisms that go beyond the simple U1snRNA–RNA complementarity and is preceded by a great number of ‘exploratory’ RNA–protein interactions to identify and select the correct 5′ss targets on the pre-mRNA. Besides the intrinsic value of characterizing these novel aspects of the splicing process, the potential importance of these studies is highlighted by the ever-increasing number of diseases that can be attributed to splicing defects, a number that has recently been estimated to be as high as 15% (22–24). As expected, many of these defects involve alterations in 5′ and 3′ splice site recognition and several therapeutic strategies have already been developed to inhibit the generation of undesired splicing events such as those arising from the activation of cryptic splice sites (25). More recently, research has also begun to make headway in repairing splicing defects that follow the loss of binding by positive effectors such as SR proteins (26–28). However, identification of these splicing mutations is not always straightforward. In fact, unless the splicing-affecting mutation occurs in the highly conserved 2 bp at the donor and acceptor sites of each exon–intron junction, the simple inspection of the genomic DNA sequence in the affected individual may not be sufficient to establish conclusively the effect(s) of a given nucleotide change.
We have recently identified as a disease-causing mutation a sequence variation (+5G > C) in the vicinity of the 5′ss of IVS3 of the NF1 gene (29) (Figure 1A), which was responsible for complete exon 3 skipping. This observation was confirmed by minigene expression studies and by the addition of a modified U1snRNA complementary to the observed mutation which was able to rescue the splicing defect (29). However, according to simple sequence analysis the U1 complementarity of the mutated +5G > C exon 3 donor site should be quite sufficient to allow 5′ss usage as is the case, e.g. in NF-1 exon 37 and exon 7 donor sites (see Table 1). In order to reconcile why +5 deviations from consensus in the NF-1 gene are effective in some contexts and not in others we have investigated the interaction between U1snRNP and NF-1 exon 3 donor site at the molecular level. Using pulldown analysis coupled with electronspray mass spectrometry we show that exon 3 skipping in the presence of the +5G > C change can be principally ascribed to a hnRNP H binding motif localized in correspondence to the wild-type −3 to +2 sequence (GGGgu) of NF-1 exon 3 donor site sequence. Most importantly, we describe here that hnRNP H binding can also be found in the donor site of exon 2 in the TSHβ gene. Significantly, also in this case, a substitution in the +5 position (+5G > A) has been associated with exon skipping and occurrence of disease (30).
Figure 1.
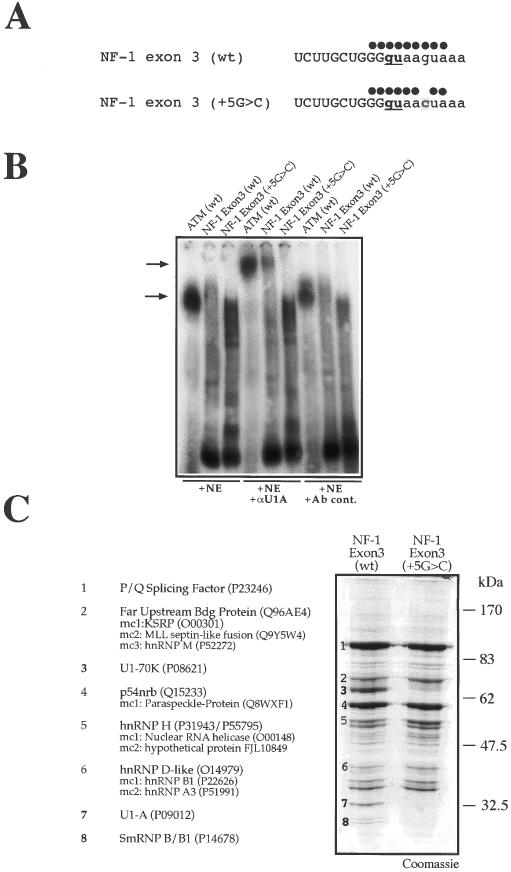
U1snRNP binds to wild type but not mutated NF-1 exon 3. (A) Schematic representation of the U1snRNA complementarity with NF-1 exon 3 wild-type (wt) and containing the NF-1 exon 3 +5G > C substitution. Full circles above each 5′ splice site sequence represent base pair matches with U1snRNA. (B) Super-shift analysis of nuclear complexes using a monoclonal antibody against U1-A protein (αU1A). The different RNAs are incubated with nuclear extract (NE) either alone or in the presence of an anti-U1A mAb (αU1A) or a control antibody (Ab cont.). The shifted complexes are indicated by the lower arrow while super-shifted αU1A-U1snRNP–RNA complexes by the upper arrow. As positive control a 20mer synthetic ribonucleotide (ATM WT) that has been previously shown to be a high affinity U1snRNP binder is used. (C) Coomassie Blue staining of a pulldown assay using adipic acid dehydrazide beads coated with NF-1 exon 3 (wt) and NF-1 exon 3 (+5G > C) RNAs following incubation with HeLa nuclear extract. In the lane from the exon 3 (wt) the numbers in boldface indicate the 70, 32.5 and 25 kDa protein bands that are absent in the NF-1 exon 3 (+5G > C) lane and which belong to the U1snRNP complex. Major protein identities are reported on the left including their Swiss-Prot Accession number. When present, minor protein components (mc) are also reported in the order of occurrence.
Table 1. Comparison of the wild-type RNA sequences (including their complementarity with U1snRNA) from the NF-1, ATM, TSHβ and ApoAII genes used in this work.

aFull circles above each 5′ splice site sequence represent base pair matches with U1snRNA.
bUpper case indicates exonic sequences and lower case indicates intronic sequences. Each exon/intron dinucleotide donor site is underlined and in boldface.
MATERIALS AND METHODS
Minigene construction
NF-1 minigene constructs were prepared as described previously (29). Briefly, wild-type and mutated genomic DNAs were inserted in a modified version of the alpha-globin–fibronectin EDB minigene in which this alternatively spliced exon had been removed to generate a unique NdeI site suitable for the insertion of exons under study. Then, 0.5 μg of each minigene were transfected in 3 × 105 HeLa cells using Qiagen Effectene transfection reagents. RNA extraction and RT–PCR analysis were performed using primers complementary to sequences in the flanking fibronectin exonic sequences. Splice site scores were calculated by the ‘Splice Site Prediction by Neural Network Site’ available at http://www.fruitfly.org/seq_tools/splice.html (31).
Preparation of RNA templates for pulldown and band-shift analysis
RNA templates were obtained by amplifying the respective exon/intron sequences using a forward primer carrying a T7 polymerase target sequence (5′-TACgTAATACgACTCACTATAg-3′) with 12 nt complementary to the specific exon and a reverse primer carrying 18 nt of the target sequence (see Table 1 for full exon/intron sequences of the amplified products). The amplified products were then purified and ∼2 μg of DNA was transcribed using T7 RNA Polymerase (Stratagene) as described previously (32). When necessary, to the reaction mix [α-32P]UTP was added to obtain a labeled RNA. The ATM WT RNA (5′-UGGCCAGGUAAGUGAUAUAU-3′) is a 20mer synthetic oligonucleotide (MWG Biotech, Firenze, Italy), which specifically interacts with U1snRNA (17). Labeling was performed by phosphorylation with [γ-32P]ATP and T4 polynucleotide kinase (PNK, Stratagene) for 1 h at 37°C.
Purification of RNA-bound protein complexes, super-shift and band-shift assays, and electronspray mass spectrometry have all been described in detail in (17,33).
Depletion of hnRNP H from the nuclear extract and band-shift analysis of U1snRNP–RNA complexes
In order to deplete hnRNP H from total nuclear extract, we used polyclonal antibodies raised in rabbit against a glutathione S-transferase (GST)–hnRNP H fusion protein. The antibodies were bound to three 30 μl aliquots of protein A/G PLUS-Agarose beads (Santa Cruz Biotechnology) for 1 h at RT and then incubated in succession for 30 min at RT with 70 μl of total nuclear extract (C4 Biotech). The depleted extract was then used in subsequent western blot and band-shift reactions. U1snRNP–RNA interactions with normal and hnRNP H-depleted extract was determined by band-shift analysis using an anti-U1A monoclonal antibody to obtain a ‘super-shift’ as described previously (17). Super-shifted complexes were quantified using the Quantity One program (Bio-Rad). Recombinant hnRNP H production has been described in detail by Romano et al. (33).
In all depletion or add-back experiments the probe was added in slight excess to make sure that all the U1snRNP–NF-1 exon 3 RNA interactions had reached their normal equilibrium. Then, in order to allow an exact quantification following the addition of the nuclear extract + antibody (or recombinant protein), we have subtracted the background density, obtained in each individual case from the equivalent region of the mock depleted lane, from the density of each super-shifted complex, and calculated the increase in super-shift density with respect to lane NE-H (mock) + αU1A. Each experiment was repeated three times to obtain standard deviation values.
RESULTS
U1snRNP binds to wild type but not mutated NF-1 exon 3
Our main objective was to determine the exact mechanism by which the disease-causing +5G > C mutation occurring in NF-1exon 3 induced the skipping of this exon. As shown in Figure 1A, the effect of this mutation was predicted to lower U1snRNA complementarity with the RNA at the exon/intron junction. The observation that a U1snRNA complementary to the mutation could rescue exon 3 skipping suggested that loss of U1snRNP binding to this 5′ss was indeed responsible for the observed exclusion (29).
In vitro confirmation that the +5G > C substitution destabilized U1snRNP interaction was obtained by super-shift analysis using a monoclonal antibody (αU1A) directed against the U1-A-specific component of U1snRNP. Figure 1B shows that only exon 3 (wt) RNA but not exon 3 (+5G > C) RNA can yield a super-shifted complex (upper arrow) in the presence of the αU1A antibody. As a positive control we used a synthetic 20mer RNA oligo (ATM WT) from intron 20 of the ATM gene that we have previously shown to be a very efficient binder of U1snRNP (17). It should be noted that for the exon 3 RNA templates we decided to include 21 nt upstream and 34 nt downstream of the GU core donor site sequences, the reason being that non-conserved intronic sequences flanking the 5′ splice site have been previously shown to affect U1snRNP binding (21).
The formation of macromolecular aggregates related to the early (E) spliceosomal complex have been described in past studies for RNAs containing just an isolated 5′ splice site, such as the very early E5′ complex (34). In order to further identify additional protein factors that might specifically interact with the NF-1 exon 3 donor site sequences, it was then decided to perform a pulldown analysis using both the wild-type (wt) and mutated (+5G > C) exon 3 RNAs. The results of this analysis are reported in Figure 1C. The wild-type exon 3 sequence was observed to contain four protein bands, of ∼70, 32.5 and a 25 kDa doublet, which were no longer present following the introduction of the +5G > C mutation. The protein bands were excised from the gel and sequenced using electronspray mass spectrometric analysis. This allowed their unambiguous identification as U1-70K and U1-A, which are U1snRNP-specific components, and snRNP B/B1 which is a common component of all UsnRNPs (3). Presumably, the other protein components of U1snRNP (U1-C, E, F, G, D1-D3) were not detected because of their low molecular weights (ranging from 17.5 to 8.5 kDa). These results were consistent with those obtained from the super-shift analysis (Figure 1B).
Sequencing of all the other major bands that appeared in the Coomassie gel also revealed several RNA binding proteins which did not vary in intensity between NF-1 exon 3 (wt) and NF-1 exon 3 (+5G > C). Their identity, however, represented an encouraging validation of our experimental approach in consideration of the fact that they have been recently isolated in splicing complex assembly. In fact, our pulldown analyses of these different 5′ splice sites included p54nrb protein, which has been recently described to associate with the 5′ splice sites within large transcription/splicing complexes (35) and KSRP (also known as FUSE binding protein 2), a splicing factor described in another system to bind an intronic splicing enhancer element (36). In particular, the KSRP/FUSE2 protein (a component of band 2, Figure 1C) has also been recently identified by site-specific protein–RNA crosslink as a specific component of complex H assembled on a synthetic pre-mRNA splicing system (37). Among these proteins, one of the most interesting result is represented by the detection of hnRNP H, a well-known splicing factor which has been shown in a variety of experimental systems to be a component of both splicing enhancers (38,39) and silencers (33,40–42). The presence of this protein was potentially very interesting because its putative binding site could be identified in correspondence to the central GU dinucleotide of the NF-1 exon 3 donor site (Table 1). In fact, binding requirements for hnRNP H involve the presence of G-rich sequences (33,42–44). Moreover, we have already demonstrated in the context of CFTR exon 9 that mutating the similar, but slightly longer GGCGGU motif to GGGGGU creates a very efficient hnRNP H binding site (the mutated base is underlined) (45).
Mutation of the putative hnRNP H binding sequence results in recovery of splicing activity
It was therefore decided to further investigate this presence by interrupting the GGGgu run. This was performed by introducing a −2G > A mutation in the 5′ splice sites with and without the +5G > C transversion (Figure 2A). This choice of mutation was also decided after a careful assessment of nucleotide frequencies in different 5′ donor sites (4) which showed that in position −2 the presence of an A was greatly favored over the presence of a G (which is rather surprising, as both A and G allow base pairing to occur with the corresponding U residue in U1snRNA). The modified exon 3 (−2G > A) and exon 3 (−2G > A, +5G > C) sequences were then inserted in the minigene system used for the original mutational analysis (29) and transfected in HeLa cells. As expected, the −2G > A mutation did not have any effect on the inclusion of NF-1 exon 3 in the wild-type sequence but succeeded in rescuing completely the aberrant splicing caused by the +5G > C mutation (Figure 2B). Moreover, a pulldown analysis performed using the NF-1 exon 3 (−2G > A) and NF-1 exon 3 (−2G > A, +5G > C) RNAs followed by western blot with an anti-hnRNP H antibody showed that the GGGgu > GAGgu change resulted in the almost complete abolishment of hnRNP H binding to both RNAs (Figure 2C), thus validating the initial considerations regarding the most likely hnRNP H binding site.
Figure 2.
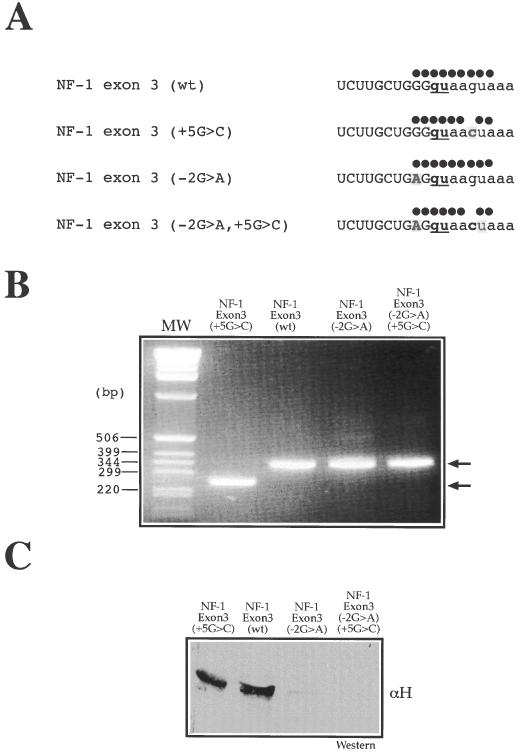
Removal of putative hnRNP H binding site results in recovery of splicing activity. (A) Nucleotide sequences around the 5′ss of NF-1 exon 3 (wt) and NF-1 exon 3 (+5G > C) following the introduction of the −2G > A substitution. Full circles above each 5′ splice site sequence represent base pair matches with U1snRNA. (B) Effect of the −2G > A substitution both in NF-1 exon 3 (wt) and in the NF-1 exon 3 (+5G > C) mutant following introduction in the EDB minigene and transfection in HeLa cells. Two products (see arrows) are seen after RNA extraction and RT–PCR analysis on agarose gel electrophoresis: the upper band (323 nt) includes exon 3 while the lower band (239 nt) lacks this exon. Note that the −2G > A mutation completely rescues NF-1 exon 3 splicing inhibition in the mutant bearing the +5G > C substitution. (C) Pulldown analysis of NF-1 exon 3 (−2G > A) and NF-1 exon 3 (−2G > A, +5G > C) RNAs followed by western blot analysis to determine the extent of hnRNP H binding to each RNA.
Decreasing and increasing the amount of hnRNP H in the nuclear extract can affect the binding of U1snRNP to the NF-1 exon 3 donor site
It was then of interest to determine whether the decrease (or increase) of hnRNP H concentration could affect directly the binding of U1snRNP to the NF-1 exon 3 (wt) RNA. The effect of decreasing hnRNP H concentration was tested in vitro by depleting this protein from the nuclear extract (Figure 3A, lower right panel). It should be noted that the polyclonal antibody against hnRNP H does not crossreact with the hnRNP H/H′-related proteins hnRNP F and hnRNP 2H9, and in western blot assays only recognizes the hnRNP H/H′ subunits (33). This characteristic ensured that no other G-rich RNA binding proteins were eventually depleted from the extract. The levels of NF-1 exon 3 (wt) RNA binding to U1snRNP were then evaluated using mAb αU1A (Figure 3A, left panel). The results demonstrate that depletion of hnRNP H from the nuclear extract results in a net increase (up to 30%) of detectable interaction between the NF-1 exon 3 (wt) donor site and U1snRNP with respect to the lane NE-H (mock)+αU1A. The specificity of hnRNP H binding for the NF-1 exon 3 (wt) sequence was also confirmed using recombinant hnRNP H protein in a band-shift experiment using labeled NF-1 exon 3 (wt) RNA (Figure 3B). This result also shows that hnRNP H binding to NF-1 exon 3 does not require the assistance of additional nuclear factors (and that it is not the result of eventual secondary interactions occurring on the donor site). Accordingly, the NF-1 exon 3 (−2G > A) RNA where the G-stretch is interrupted cannot bind this protein anymore. Finally, addition of recombinant hnRNP H could significantly reduce the levels of U1snRNP binding to the NF-1 exon 3 (wt) RNA (Figure 3C). Taken together, these results indicate that NF-1 exon 3 (wt) donor site is a finely balanced system that can normally accommodate U1snRNP and hnRNP H (as shown in Figure 1B where both proteins appear in the pulldown), although a certain degree of overlapping occurs in their binding sites.
Figure 3.
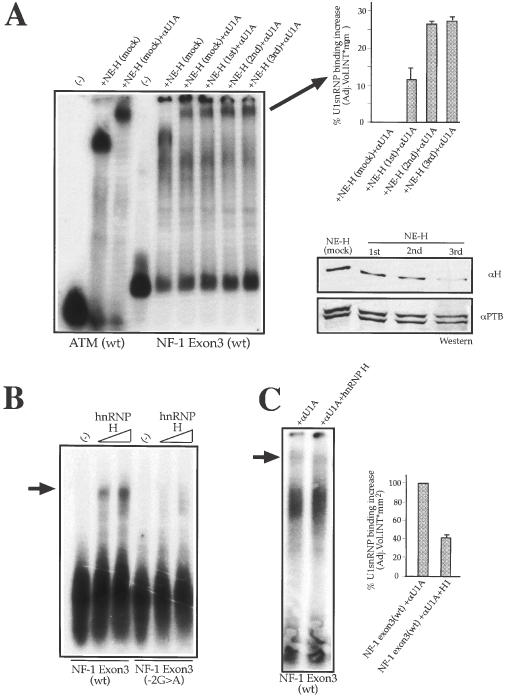
Depletion/addition of hnRNP H from the nuclear extract can affect U1snRNP binding to NF-1 exon 3 (wt) donor site. (A) Left panel shows a super-shift analysis of NF-1 exon 3 (wt) donor site RNA using a monoclonal antibody against U1snRNP U1-A protein (αU1A). The exon 3 (wt) RNA was incubated with a mock-depleted nuclear extract NE (mock) and with equal amounts of nuclear extract treated with a polyclonal antibody against hnRNP H (αH) in three successive rounds of depletion: NE-H (first), NE-H (second), and NE-H (third). As control, the ATM (wt) RNA is incubated with NE (mock) in the absence or presence of (αU1A). The super-shifted αU1A-U1snRNP–RNA complexes were quantified using the Quantity One program (Bio-Rad) (upper right panel). SD bars are shown. (A) Lower right panel shows the levels of hnRNP H from the nuclear extract with the three successive round of depletion with αH. As control, a polyclonal antibody against Polypyrimidine Tract Binding protein (αPTB) was used to test each depleted extract for aspecific depletion. (B) Shows a band-shift experiment using labeled NF-1 exon 3 (wt) and NF-1 exon 3 (−2G > A) RNAs incubated with increasing quantities of recombinant hnRNP H (500 ng and 1 μg respectively). The arrow on the left indicates the hnRNP H–RNA complex. (C) Shows the results of adding 1 μg of recombinant hnRNP H to NF-1 exon 3 (wt) RNA in the presence of nuclear extract and an antibody against U1snRNP (αU1). The arrow indicates the super-shifted αU1A–U1snRNP–RNA complex which was quantified using the Quantity One program (Bio-Rad). SD bars are shown.
The presence of hnRNP H is a distinctive feature of NF-1 exon 3 donor site as opposed to other NF-1 donor sites which do not carry a G at +5
The findings described above prompted us to perform an additional analysis regarding the eventual presence of hnRNP H in other NF-1 donor sites (Table 1). To achieve this end, we selected other NF-1 exons donor sites (such as 37 and 7) which also carry a non-G nucleotide in the +5 position but which lack the presence of G runs in the vicinity of the 5′ splice site (Figure 4A). As a control for exons carrying a G in the +5 position (besides exon 3), but which nonetheless lack a G run, we also included the donor site for NF-1 exon 10b. Pulldown experiments followed by western blots with an antibody against hnRNP H (αH) show that this protein only binds in the context of exon 3 but not exons 7, 37 and 10b (Figure 4B). Taken together, these findings indicate that hnRNP H binding in the context of exon 3 is a highly specific event.
Figure 4.
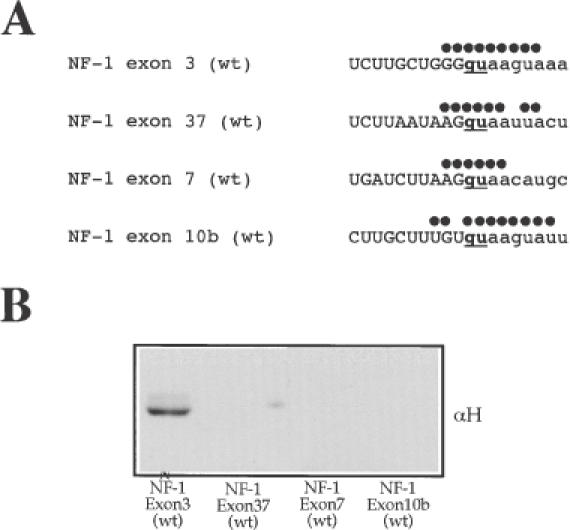
U1snRNP and hnRNP H do not bind to NF-1 exon 37, 7 and 10b donor sites carrying substitutions in the +5 or −1 position. (A) Nucleotide sequences around the 5′ss of NF-1 exon 3 (wt), NF-1 exon 7 (wt), NF-1 exon 37 (wt) and NF-1 exon 10b (wt) (see Table 1 for complete sequence) showing the region of complementarity with U1snRNA (indicated by black dots). (B) Western blot of pulldown analyses of all these RNAs to determine the eventual presence of hnRNP H.
Analysis of the 5′ splice site ribonucleoprotein complexes in other NF-1 and Apo AII exons
It was then of interest to determine whether these different donor sites could pulldown, besides hnRNP H, different complements of nuclear proteins. Figure 5 shows that pulldown analyses, extended to other 5′ splice sites, revealed both similarities but also interesting differences. In fact, although the protein signature for hnRNP H is present only in NF-1 exon 3 (wt and +5G > C), this is not the only protein band that is unique to NF-1 exon 3. For example, in Figure 5A bandnumber 6 shows that at least another protein (an hnRNP D-like factor) is unique to this sequence whilst others such as band-number 2 (corresponding to a variety of proteins, including KSRP) appear to be pulled down with higher efficiency in the exon 3 sequences as opposed to other NF-1 exon donor sites (37 and 7). Even more strikingly, the pulldown analysis of the unrelated Apo AII exon 1 (wt) (Figure 5B) shows an entire cluster of protein bands that could not be detected in the various NF-1 splice sites (such as SF2/ASF, hnRNP A2/B1, hnRNP A3). This finding highlights our hypothesis that nucleotide sequences in different 5′ splice sites can determine the binding of distinct sets of nuclear factors.
Figure 5.
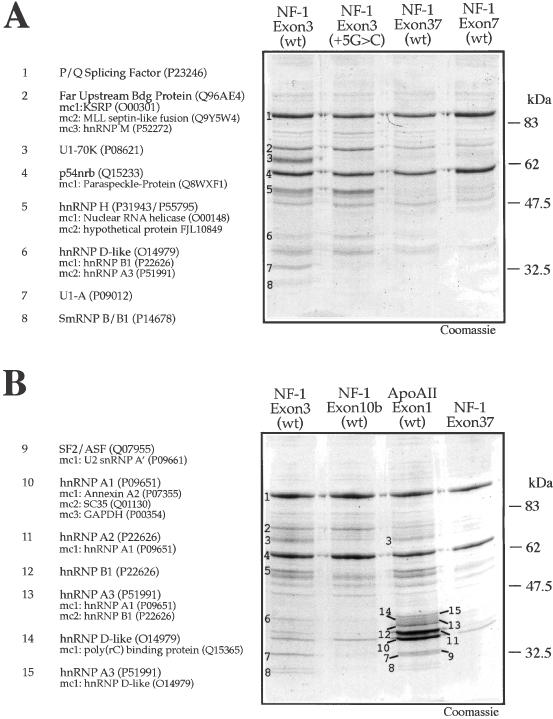
Ribonucleoprotein complexes in the 5′ss of NF-1 and Apo AII donor sites. (A) Coomassie Blue staining of a pulldown assay using beads coated with NF-1 exon 3 (wt), NF-1 exon 3 (+5G > C), NF-1 exon 7 (wt), and NF-1 exon 37 (wt) RNAs following incubation with HeLa nuclear extract. The numbers on the NF-1 exon 3 (wt) lane indicate the sequenced protein bands. The identity of each numbered band is shown to the left of the gel. (B) Coomassie Blue staining of a pulldown assay using beads derivatized with NF-1 exon 3 (wt), NF-1 exon 10b (wt) and Apo AII exon 1 (wt), and a control pulldown using exonic sequence from NF-1 exon 37 lacking a 5′ splice site. The numbers on the NF-1 exon 3 (wt) lane indicate the sequenced protein bands. On the left of the gel the identity of each band together with its Swiss-Prot Accession number is reported. Minor components (mc), when present, are also reported (in decreasing order of detection).
An interesting observation is that the U1snRNP signatures (U1-70K, U1-A, and U1-B/B1) can be detected only in two donor sites, NF-1 exon 3 (wt) and Apo AII exon 1 (wt) (Figure 5B). This is probably due to the fact that in these two donor sites the potential base pairing with U1snRNA is very high (see Table 1 for a comparison) and therefore binding to this complex can be detected efficiently using this technique. On the other hand, base complementarity for the other NF-1 donor sites analyzed (37, 7 and 10bis) is much lower (Table 1) and this may explain the failure to detect lower levels of U1snRNP interaction. In fact, although correct splicing in the absence of U1snRNP binding at least if measured by pulldown/super-shift analysis has been described in a variety of experimental systems (14,46–48), failure to detect U1snRNP in these donor sites which are efficiently recognized by the splicing machinery suggests that in these cases pulldown efficiency may simply be too low. Alternatively, these donor sites may recruit U1snRNP through the use of nearby enhancer elements which are lacking in our RNA constructs.
Presence of hnRNP H protein in unrelated pathological events that involve 5′ splice sites: the TSHβ gene
Although most disease-causing single nucleotide substitutions in donor sites involve the +1/+2 position (49), several pathological splicing alterations also involve the +5 position. This has been described in the past for the collagen genes (50–54), the ELA2 gene (55), the WT1 gene (56,57), the TSHβ-subunit gene (30) and, most recently, in the DMD gene (58). Although many other mechanisms and interactions can be potentially involved in these pathological events such as U6snRNP binding, or presence of specific TIA-1 (59), U1C interactions (11,20), it was possible that hnRNP H was also involved in some of them.
It was thus of interest to analyze the eventual presence of G runs in the donor sites of the affected exons also in these genes. Interestingly, the exon 2 donor site of the TSHβ-subunit gene involved in congenital secondary hypothyroidism (30) displayed a GGgu sequence that represented a potential candidate for an hnRNP H binding site (Table 1). Significantly, minigene analysis performed by Pohlenz et al. (30) showed clearly that the full inclusion observed for the TSHβ exon 2 donor site was abolished by the introduction of a disease-causing +5G > A substitution.
Pulldown analysis confirmed that the TSHβ exon 2 donor site was indeed capable of binding hnRNP H (Figure 6A), although with a slightly lesser efficiency than NF-1 exon 3, probably due to the fact that the GGgu sequence in TSHβ exon 2 is not as good an hnRNP H binding site as the GGGgu in NF-1 exon 3. Furthermore, pulldown analysis of the wild-type and +5G > A mutated TSHβ donor site regions (see Table 1 for the sequence) showed a protein pattern and U1snRNP signatures very similar to that observed for NF-1 exon 3. In fact, Figure 6B shows that U1snRNP binding signatures, although significantly weaker than in NF-1 exon 3, can also be observed for the TSHβ exon 2 wild-type RNA. The reason for this difference in U1snRNP binding most probably resides in the lower degree of RNA complementarity between TSHβ exon 2 donor site and U1snRNA than the one present for NF-1 exon 3 (Table 1). These characteristic U1snRNP signatures in TSHβ exon 2 (wt) disappear completely following the introduction of the +5G > A substitution (see, e.g. the U1-A protein band). As expected, a western blot analysis of the same Coomassie stained gel showed that the introduction of the +5G > A mutation had no effect on the hnRNP H binding levels (Figure 6B, lower panel).
Figure 6.
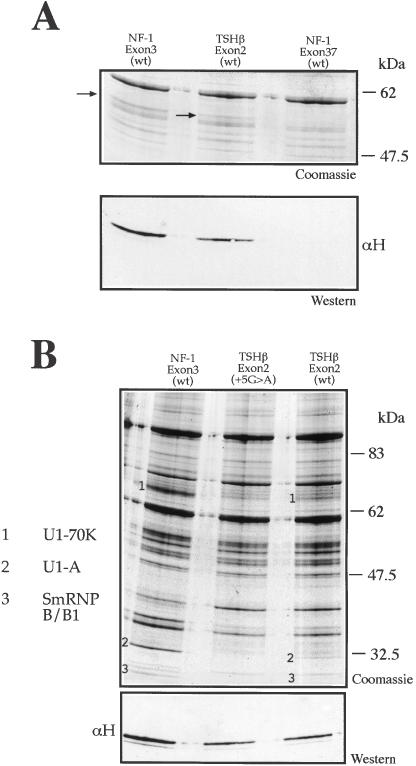
hnRNP H binds to the donor site of TSHβ exon 2 type. (A) Coomassie and western blot analysis of a pulldown assay using NF-1 exon 3 (wt), TSHβ exon 2 (wt) and NF-1 exon 37 (wt) RNAs. The arrows indicate the protein signatures corresponding to hnRNP H in the Coomassie gel. These signatures were confirmed by western blot analysis using an anti-hnRNP H antibody (αH) in the lower panel. (B) Coomassie Blue staining of a pulldown assay using adipic acid dehydrazide beads coated with NF-1 exon 3 (wt), TSHβ exon 2 (+5G > A), and TSHβ exon 2 (wt) RNAs following incubation with HeLa nuclear extract. The numbers indicate the U1snRNP components previously identified: U1-70K (1), U1-A (2) and SmRNP B/B1 (3) and the lower panel contains a western blot performed on the Coomassie gel to determine hnRNP H binding to the different RNAs.
It was thus of interest to analyze the effect of a −2G > A substitution in the binding profiles of both TSHβ exon 2 constructs (Figure 7A). Binding of hnRNP H to these constructs was tested by pulldown analysis of the transcribed RNAs followed by western blot (Figure 7B). Figure 8B shows that, consistent with the results obtained for NF-1 exon 3, both TSHβ exon 2 (−2G > A) and TSHβ exon 2 (−2G > A, +5G > A) lose the ability to bind hnRNP H. In addition, in the TSHβ exon 2 (−2G > A) construct there is a distinct increase in the U1snRNP protein signatures, consistent with the fact that hnRNP H is now prevented from binding in correspondence to the donor site. Finally, it should be noted that the TSHβ exon 2 (−2G > A, +5G > A) RNA displays levels of U1snRNP binding comparable with TSHβ exon 2 (wt) and above its total absence in the TSHβ exon 2 (+5G > A) RNA (Figure 6B).
Figure 7.
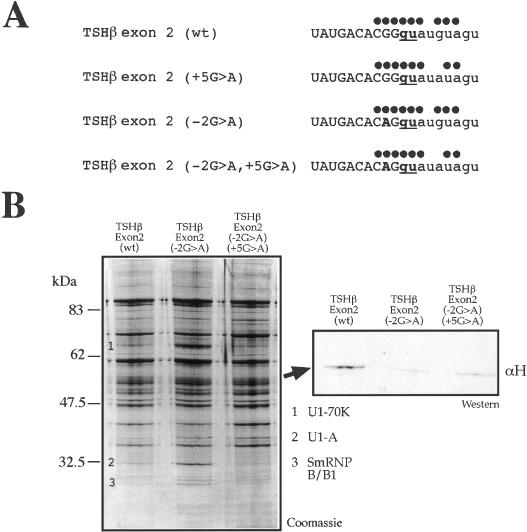
Removal of putative hnRNP H binding site in TSHβ exon 2 (wt) and TSHβ exon 2 (+5G > A). (A) Nucleotide sequences around the 5′ss of TSHβ exon 2 (wt) and TSHβ exon 2 (+5G > A) following the introduction of the −2G > A substitution. Dots above each 5′ splice site sequence represent base pair matches with U1snRNA. (B) Pulldown and western blot analysis performed on TSHβ exon 2 (wt), TSHβ exon 2 (−2G > A), and TSHβ exon 2 (−2G > A, +5G > A) RNAs to determine the extent of U1snRNP binding to each RNA. The numbers indicate the U1snRNP components previously identified: U1-70K (1), U1-A (2) and SmRNP B/B1 (3). The Coomassie gel was used in western blot analysis (panel on the right) to determine the effects of the −2G > A substitution on binding of hnRNP H to these RNAs.
Figure 8.
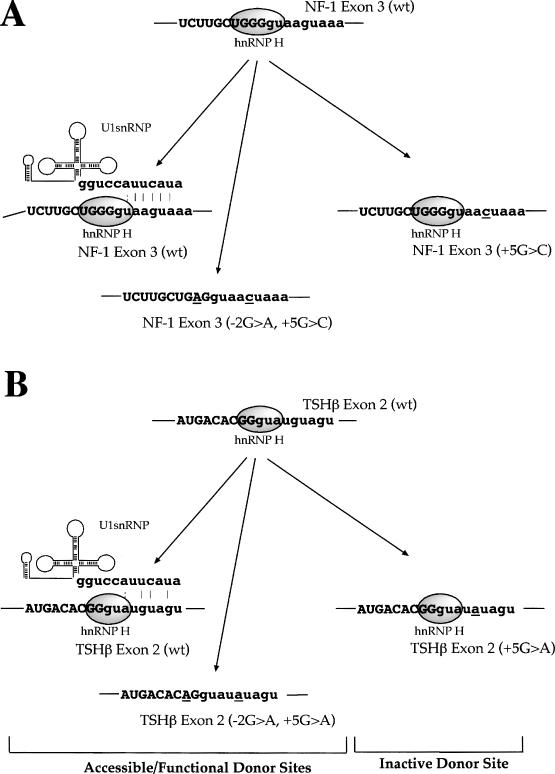
Model of donor site inhibition by hnRNP H. The diagrams represent the proposed model of splicing inhibition mediated by hnRNP H binding in correspondence to the NF-1 exon 3 (wt) donor site when the +5G > C mutation occurs and in the TSHβ exon 2 donor site following the +5G > A substitution. Both wild-type donor site sequences are capable of binding U1snRNP stably owing to the complementarity still present toward the U1snRNA sequence even in the presence of hnRNP H binding near the central GU dinucleotide. Donor site recognition can be abolished by the introduction of the +5G > C or +5G > A mutations which in both cases do not affect hnRNP H binding but lower U1snRNA complementarity with the donor site below a critical threshold. Finally, removal of hnRNP H binding site is not sufficient to recover U1snRNP binding (owing to the presence of the +5 substitutions) but is sufficient to allow recognition of the donor site by the splicing machinery.
DISCUSSION
In this work, we describe the functional characterization of a mutation in the NF-1 exon 3 donor site (+5G > C) which causes exon 3 skipping and can be associated with occurrence of disease (29).
As noted previously, most disease-causing single nucleotide substitutions in donor sites involve the +1/+2 position (49). However, pathological splicing alterations have also been observed to involve the +5 position in a variety of cases (30,50–58). The functional reason why alterations in this position may cause aberrant splicing is represented by its well-known interactions with U1snRNP and U6snRNP in the splicing process (46). However, alterations in this position do not always disrupt the splicing process and several separate lines of evidence show that the +5 position can harbor nucleotides other than a Guanosine. First, replacement of a G in the +5 position has also been observed to occur in 14 and 25% (in low and high G + C loci, respectively) of all functional splice sites (4). In the NF-1 gene, e.g. +5 substitutions can be detected in exons 1 and 7 donor sites. Second, it has been recently observed in U1snRNP binding studies that a G in the +5 position is not obligatory if the two preceding positions are purines (as in NF-1 exon 3) (13). Finally, mutations of the +5 position in the ATM system that abolish U1snRNA–RNA base pairing only reduce, but do not eliminate, ISPE usage by U1snRNP (17).
Indeed, in the NF-1 exon 3 (+5G > C) mutation, simple sequence analysis showed that the residual U1snRNA–5′ss complementarity following the introduction of the +5G > C mutation was comparable to that of other perfectly functional donor sites in the NF-1 gene which also carry a nucleotide in the +5 position which prevents complementarity, such as IVS 7 and IVS 37 (Table 1). This is also reflected in the strengths of these NF-1 donor sites according to the ‘Splice Site Prediction by Neural Network Site’ (31), which fail to reveal a reason why exon 3 (+5G > C) is skipped. In fact, by analyzing the different NF-1 5′splice sites we obtain a 5′ss score for the wild-type NF-1 exon 3 of 1.00 and for the +5G > C mutant of 0.83. This Donor Site Score (0.83) is similar to that of exon 10b (0.86) and much higher than those of exon 19b (0.13) or exon 12b (0.19) which are normally included in all NF-1 transcripts as reported previously (60).
Nonetheless, the observation that a U1snRNA complementary to the mutation could rescue NF-1 exon 3 skipping (29) suggests that it is indeed the U1snRNP-exon 3 5′ss interaction which is involved in the observed exon skipping rather than the potential disruption of U6snRNA binding at this position. This hypothesis was confirmed using pulldown and band-shift analyses which demonstrated that U1snRNP was indeed capable of binding to the NF-1 exon 3 (wt) donor site and that this binding was abolished by the +5G > C mutation. Interestingly, the pulldown analysis also showed that the GGGgu donor site sequence of NF-1 exon 3 could also bind another protein, hnRNP H, which has a well-known ability to affect the splicing process (33,38–41).
We have shown using nuclear extracts depleted of hnRNP H that binding of this protein to the GGGgu sequence can reduce, but not abolish, U1snRNP binding. In this circumstance, the +5G > C substitution weakens further the U1snRNA–5′ss complementarity hence inhibiting the 5′ splice site recognition (Figure 8 shows our proposed model). When the hnRNP H binding site of the 5′ss carrying the +5G > C is removed by the −2G > A substitution splicing function is restored and correct 5′ss recognition takes place again. Most significantly, our search for additional splicing systems in which U1snRNP/hnRNP H binding at the donor site might play a role in the splicing process uncovered a second donor site in IVS2 of the TSHβ gene, where a +5G > A substitution had also been described to be associated with occurrence of disease and exon skipping in a minigene system (30). Pulldown analyses performed on this system yielded results that are consistent with the proposed role of hnRNP H in the NF-1 donor site (see Figure 8 for a comparison of the two systems).
One question that still remains unanswered is the role of hnRNP H binding to both the NF-1 and TSHβ donor sites. Direct approaches such as RNA interference or overexpression studies will be helpful in clarifying this issue. Our attempts to knock down hnRNP H by siRNA has been unsuccessful despite using several high-score target sequences. In any case, it has to be considered that G-four and G-three runs so near to donor sites are not at all frequent in nature. In fact, a careful appraisal of 5′ss nucleotide frequencies in loci with high G + C content shows that there is a bias in nucleotide use in the −2 position that is unrelated to U1snRNA complementarity: in the −2 position G is detected 15% of the times (against 56% for A) while in position +3 it is detected 56% of the times (against 38% for A) (4). Even more striking is the fact that in the −2 position the presence of a C (15%) or T (14%) nucleotide (which disrupts base pairing with U1snRNA) is preferred to a G (that will make a wobble pairing). It is thus clear that even in high G + C loci the presence of a G in position −2 upstream of the donor site is actively selected against, probably because a G run will be created (being right next to the almost invariant Ggu sequence). On the other hand, the presence of a G in position +3 (where an eventual G run would be invariably interrupted by the U residue in Ggu) is not selected against. This could explain why in position +3 its frequency is so high and why the C and T nucleotides are seldom present at this position (9 and 7%, respectively).
An interesting observation related to our NF-1 and TSHβ experimental models lies in the relationship between the occurrence of G runs in the database of 29 853 splice site entries (http://www.ebi.ac.uk/asd/altextron/index.html) and variations in the +5 position. The results of this analysis are reported in Table 2 and it is clear that as the number of Gs increases in the −3 to −1 positions so does the frequency of a conserved G residue in the +5 position with 94% of donor site which harbor a four G run (−3 to +1) retaining a G in position +5 as opposed to 74% when there are only two Gs in positions −1 and +1. This may point toward a selective pressure owing to the presence of hnRNP H (which could bind to these Gs) to maintain a more stringent donor site definition in the +5 position.
Table 2. Correlation between the number of Gs at donor site junctions and conservation of the intronic nucleotide at +5 position.
| NNNG | gt**a | gt**c | gt**g | gt**t |
|---|---|---|---|---|
| –4–3–2–1 | ||||
| NNNG (23 461) | 9.87 (2315) | 7.03 (1649) | 74.74 (17 534) | 8.37 (1963) |
| NNGG (2405) | 3.74 (90) | 1.66 (40) | 91.73 (2206) | 2.87 (69) |
| NGGG (416) | 3.37 (14) | 0.96 (4) | 93.99 (391) | 1.68 (7) |
| GGGG (73) | 2.73 (2) | 0 (0) | 95.89 (70) | 1.37 (1) |
The human release of the AltExtron database (http://www.ebi.ac.uk/asd/altextron/index.html) containing 29 853 exons entries was used.
The percentages are referred to the available 23 461 entries for the NNNGgt**n pattern, 2405 entries for the NNGGgt**n pattern, 416 entries for the NGGGgt**n pattern and 73 entries for the GGGGgt**n pattern. NNNG indicates the exonic part (−4 to −1) of the 5′ junction whereas gt**n indicates the intronic part of the 5′ junction (+1 to +5). Asterisks signify any nucleotide. N and n are the permutated nucleotides taken into consideration for the survey.
These considerations, of course, do not rule out the possibility that the presence of these G runs in correspondence to the GU-conserved dinucleotide and a G in the +5 position may be just a chance occurrence. However, the fact that both the NF-1 and TSHβ donor sites can bind at the same time to U1snRNP and hnRNP H may well indicate that these donor sites represent a finely balanced system where both splicing factors play a role. It is thus very probable that in both cases the presence of hnRNP H (hence of a G run) in that particular position may serve some functional purpose regarding pre-mRNA processing although at the same time weakening the ability of the donor site to withstand chance substitutions in U1snRNA complementarity.
In conclusion, our data show that considerable protein heterogeneity can be observed even in the relatively small RNA region that surrounds a donor site. This may have been an expected result as the number of factors that are suspected to be involved in pre-mRNA splicing and spliceosome formation has increased considerably (61,62). The experimental observations in this work, besides their intrinsic importance for the NF-1- and TSHβ-related pathologies, suggest that this great complexity observed at the level of the entire spliceosome (63–65) may well be present, and active, even during the earliest stages of splicing complex assembly. Considering the ever-increasing need of distinguishing innocuous polymorphisms from potential disease-causing mutations this finding highlights the need for careful ‘local context’ analyses when predicting the possible effects of mutations introduced in donor sites.
Acknowledgments
ACKNOWLEDGEMENTS
We thank I. W. Mattaj and A. Segref for the anti-U1A monoclonal antibody. This work was supported by grants from the Telethon Onlus Foundation (Italy) (grant n. GGP02453), FIRB (RBNE01W9PM) and Action Research (UK).
REFERENCES
- 1.Burge C.B., Tuschl,T. and Sharp,P.A. (1999) Splicing of Precursors to mRNAs by the Spliceosome. Cold Spring Laboratory Harbor Press, Cold Spring Harbor, NY. [Google Scholar]
- 2.Mount S.M., Pettersson,I., Hinterberger,M., Karmas,A. and Steitz,J.A. (1983) The U1 small nuclear RNA–protein complex selectively binds a 5′ splice site in vitro. Cell, 33, 509–518. [DOI] [PubMed] [Google Scholar]
- 3.Stark H., Dube,P., Luhrmann,R. and Kastner,B. (2001) Arrangement of RNA and proteins in the spliceosomal U1 small nuclear ribonucleoprotein particle. Nature, 409, 539–542. [DOI] [PubMed] [Google Scholar]
- 4.Zhang M.Q. (1998) Statistical features of human exons and their flanking regions. Hum. Mol. Genet., 7, 919–932. [DOI] [PubMed] [Google Scholar]
- 5.Michaud S. and Reed,R. (1991) An ATP-independent complex commits pre-mRNA to the mammalian spliceosome assembly pathway. Genes Dev., 5, 2534–2546. [DOI] [PubMed] [Google Scholar]
- 6.Seraphin B., Kretzner,L. and Rosbash,M. (1988) A U1 snRNA:pre-mRNA base pairing interaction is required early in yeast spliceosome assembly but does not uniquely define the 5′ cleavage site. EMBO J., 7, 2533–2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Seraphin B. and Rosbash,M. (1989) Identification of functional U1 snRNA-pre-mRNA complexes committed to spliceosome assembly and splicing. Cell, 59, 349–358. [DOI] [PubMed] [Google Scholar]
- 8.Zhuang Y. and Weiner,A.M. (1986) A compensatory base change in U1 snRNA suppresses a 5′ splice site mutation. Cell, 46, 827–835. [DOI] [PubMed] [Google Scholar]
- 9.Siliciano P.G. and Guthrie,C. (1988) 5′ splice site selection in yeast: genetic alterations in base-pairing with U1 reveal additional requirements. Genes Dev., 2, 1258–1267. [DOI] [PubMed] [Google Scholar]
- 10.Du H. and Rosbash,M. (2001) Yeast U1 snRNP–pre-mRNA complex formation without U1snRNA–pre-mRNA base pairing. RNA, 7, 133–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Du H. and Rosbash,M. (2002) The U1 snRNP protein U1C recognizes the 5′ splice site in the absence of base pairing. Nature, 419, 86–90. [DOI] [PubMed] [Google Scholar]
- 12.Eperon I.C., Makarova,O.V., Mayeda,A., Munroe,S.H., Caceres,J.F., Hayward,D.G. and Krainer,A.R. (2000) Selection of alternative 5′ splice sites: role of U1 snRNP and models for the antagonistic effects of SF2/ASF and hnRNP A1. Mol. Cell. Biol., 20, 8303–8318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lund M. and Kjems,J. (2002) Defining a 5′ splice site by functional selection in the presence and absence of U1 snRNA 5′ end. RNA, 8, 166–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tarn W.Y. and Steitz,J.A. (1994) SR proteins can compensate for the loss of U1 snRNP functions in vitro. Genes Dev., 8, 2704–2717. [DOI] [PubMed] [Google Scholar]
- 15.Roca X., Sachidanandam,R. and Krainer,A.R. (2003) Intrinsic differences between authentic and cryptic 5′ splice sites. Nucleic Acids Res., 31, 6321–6333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Freund M., Asang,C., Kammler,S., Konermann,C., Krummheuer,J., Hipp,M., Meyer,I., Gierling,W., Theiss,S., Preuss,T. et al. (2003) A novel approach to describe a U1 snRNA binding site. Nucleic Acids Res., 31, 6963–6975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pagani F., Buratti,E., Stuani,C., Bendix,R., Dork,T. and Baralle,F.E. (2002) A new type of mutation causes a splicing defect in ATM. Nature Genet., 30, 426–429. [DOI] [PubMed] [Google Scholar]
- 18.Malca H., Shomron,N. and Ast,G. (2003) The U1 snRNP base pairs with the 5′ splice site within a penta-snRNP complex. Mol. Cell. Biol., 23, 3442–3455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Forch P., Merendino,L., Martinez,C. and Valcarcel,J. (2003) U2 small nuclear ribonucleoprotein particle (snRNP) auxiliary factor of 65 kDa, U2AF65, can promote U1 snRNP recruitment to 5′ splice sites. Biochem. J., 372, 235–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Forch P., Puig,O., Martinez,C., Seraphin,B. and Valcarcel,J. (2002) The splicing regulator TIA-1 interacts with U1-C to promote U1 snRNP recruitment to 5′ splice sites. EMBO J., 21, 6882–6892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Puig O., Gottschalk,A., Fabrizio,P. and Seraphin,B. (1999) Interaction of the U1 snRNP with nonconserved intronic sequences affects 5′ splice site selection. Genes Dev., 13, 569–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Caceres J.F. and Kornblihtt,A.R. (2002) Alternative splicing: multiple control mechanisms and involvement in human disease. Trends Genet., 18, 186–193. [DOI] [PubMed] [Google Scholar]
- 23.Cartegni L., Chew,S.L. and Krainer,A.R. (2002) Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nature Rev. Genet., 3, 285–298. [DOI] [PubMed] [Google Scholar]
- 24.Faustino N.A. and Cooper,T.A. (2003) Pre-mRNA splicing and human disease. Genes Dev., 17, 419–437. [DOI] [PubMed] [Google Scholar]
- 25.Friedman K.J., Kole,J., Cohn,J.A., Knowles,M.R., Silverman,L.M. and Kole,R. (1999) Correction of aberrant splicing of the cystic fibrosis transmembrane conductance regulator (CFTR) gene by antisense oligonucleotides. J. Biol. Chem., 274, 36193–36199. [DOI] [PubMed] [Google Scholar]
- 26.Buratti E., Baralle,F.E. and Pagani,F. (2003) Can a ‘patch’ in a skipped exon make the pre-mRNA splicing machine run better? Trends Mol. Med., 9, 229–232. [DOI] [PubMed] [Google Scholar]
- 27.Cartegni L. and Krainer,A.R. (2003) Correction of disease-associated exon skipping by synthetic exon-specific activators. Nature Struct. Biol. [DOI] [PubMed] [Google Scholar]
- 28.Skordis L.A., Dunckley,M.G., Yue,B., Eperon,I.C. and Muntoni,F. (2003) Bifunctional antisense oligonucleotides provide a trans-acting splicing enhancer that stimulates SMN2 gene expression in patient fibroblasts. Proc. Natl Acad. Sci. USA, 100, 4114–4119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baralle M., Baralle,D., De Conti,L., Mattocks,C., Whittaker,J., Knezevich,A., Ffrench-Constant,C. and Baralle,F.E. (2003) Identification of a mutation that perturbs NF1, a gene splicing using genomic DNA samples and a minigene assay. J. Med. Genet., 40, 220–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pohlenz J., Dumitrescu,A., Aumann,U., Koch,G., Melchior,R., Prawitt,D. and Refetoff,S. (2002) Congenital secondary hypothyroidism caused by exon skipping due to a homozygous donor splice site mutation in the TSHbeta-subunit gene. J. Clin. Endocrinol. Metab., 87, 336–339. [DOI] [PubMed] [Google Scholar]
- 31.Reese M.G., Eeckman,F.H., Kulp,D. and Haussler,D. (1997) Improved splice site detection in Genie. J. Comput. Biol., 4, 311–323. [DOI] [PubMed] [Google Scholar]
- 32.Buratti E., Dork,T., Zuccato,E., Pagani,F., Romano,M. and Baralle,F.E. (2001) Nuclear factor TDP-43 and SR proteins promote in vitro and in vivo CFTR exon 9 skipping. EMBO J., 20, 1774–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Romano M., Marcucci,R., Buratti,E., Ayala,Y.M., Sebastio,G. and Baralle,F.E. (2002) Regulation of 3′ splice site selection in the 844ins68 polymorphism of the cystathionine Beta-synthase gene. J. Biol. Chem., 277, 43821–43829. [DOI] [PubMed] [Google Scholar]
- 34.Michaud S. and Reed,R. (1993) A functional association between the 5′ and 3′ splice site is established in the earliest prespliceosome complex (E) in mammals. Genes Dev., 7, 1008–1020. [DOI] [PubMed] [Google Scholar]
- 35.Kameoka S., Duque,P. and Konarska,M.M. (2004) p54(nrb) associates with the 5′ splice site within large transcription/splicing complexes. EMBO J., 23, 1782–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Min H., Turck,C.W., Nikolic,J.M. and Black,D.L. (1997) A new regulatory protein, KSRP, mediates exon inclusion through an intronic splicing enhancer. Genes Dev., 11, 1023–1036. [DOI] [PubMed] [Google Scholar]
- 37.Rhode B.M., Hartmuth,K., Urlaub,H. and Luhrmann,R. (2003) Analysis of site-specific protein–RNA cross-links in isolated RNP complexes, combining affinity selection and mass spectrometry. RNA, 9, 1542–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Caputi M. and Zahler,A.M. (2002) SR proteins and hnRNP H regulate the splicing of the HIV-1 tev-specific exon 6D. EMBO J., 21, 845–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chou M.Y., Rooke,N., Turck,C.W. and Black,D.L. (1999) hnRNP H is a component of a splicing enhancer complex that activates a c-src alternative exon in neuronal cells. Mol. Cell. Biol., 19, 69–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen C.D., Kobayashi,R. and Helfman,D.M. (1999) Binding of hnRNP H to an exonic splicing silencer is involved in the regulation of alternative splicing of the rat beta-tropomyosin gene. Genes Dev., 13, 593–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fogel B.L. and McNally,M.T. (2000) A cellular protein, hnRNP H, binds to the negative regulator of splicing element from Rous sarcoma virus. J. Biol. Chem., 275, 32371–32378. [DOI] [PubMed] [Google Scholar]
- 42.Jacquenet S., Mereau,A., Bilodeau,P.S., Damier,L., Stoltzfus,C.M. and Branlant,C. (2001) A second exon splicing silencer within human immunodeficiency virus type 1 tat exon 2 represses splicing of Tat mRNA and binds protein hnRNP H. J. Biol. Chem., 276, 40464–40475. [DOI] [PubMed] [Google Scholar]
- 43.Caputi M. and Zahler,A.M. (2001) Determination of the RNA binding specificity of the heterogeneous nuclear ribonucleoprotein (hnRNP) H/H′/F/2H9 family. J. Biol. Chem., 276, 43850–43859. [DOI] [PubMed] [Google Scholar]
- 44.Arhin G.K., Boots,M., Bagga,P.S., Milcarek,C. and Wilusz,J. (2002) Downstream sequence elements with different affinities for the hnRNP H/H′ protein influence the processing efficiency of mammalian polyadenylation signals. Nucleic Acids Res., 30, 1842–1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pagani F., Buratti,E., Stuani,C. and Baralle,F.E. (2003) Missense, nonsense, and neutral mutations define juxtaposed regulatory elements of splicing in cystic fibrosis transmembrane regulator exon 9. J. Biol. Chem., 278, 26580–26588. [DOI] [PubMed] [Google Scholar]
- 46.Crispino J.D. and Sharp,P.A. (1995) A U6 snRNA:pre-mRNA interaction can be rate-limiting for U1-independent splicing. Genes Dev., 9, 2314–2323. [DOI] [PubMed] [Google Scholar]
- 47.Crispino J.D., Mermoud,J.E., Lamond,A.I. and Sharp,P.A. (1996) Cis-acting elements distinct from the 5′ splice site promote U1-independent pre-mRNA splicing. RNA, 2, 664–673. [PMC free article] [PubMed] [Google Scholar]
- 48.Brackenridge S., Wilkie,A.O. and Screaton,G.R. (2003) Efficient use of a ‘dead-end’ GA 5′ splice site in the human fibroblast growth factor receptor genes. EMBO J., 22, 1620–1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Krawczak M., Reiss,J. and Cooper,D.N. (1992) The mutational spectrum of single base-pair substitutions in mRNA splice junctions of human genes: causes and consequences. Hum. Genet., 90, 41–54. [DOI] [PubMed] [Google Scholar]
- 50.Schwarze U., Goldstein,J.A. and Byers,P.H. (1997) Splicing defects in the COL3A1 gene: marked preference for 5′ (donor) spice-site mutations in patients with exon-skipping mutations and Ehlers–Danlos syndrome type IV. Am. J. Hum. Genet., 61, 1276–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Giunta C. and Steinmann,B. (2000) Characterization of 11 new mutations in COL3A1 of individuals with Ehlers–Danlos syndrome type IV: preliminary comparison of RNase cleavage, EMC and DHPLC assays. Hum. Mutat., 16, 176–177. [DOI] [PubMed] [Google Scholar]
- 52.Bateman J.F., Chan,D., Moeller,I., Hannagan,M. and Cole,W.G. (1994) A 5′ splice site mutation affecting the pre-mRNA splicing of two upstream exons in the collagen COL1A1 gene. Exon 8 skipping and altered definition of exon 7 generates truncated pro alpha 1(I) chains with a non-collagenous insertion destabilizing the triple helix. Biochem. J., 302 (Pt 3), 729–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tiller G.E., Weis,M.A., Polumbo,P.A., Gruber,H.E., Rimoin,D.L., Cohn,D.H. and Eyre,D.R. (1995) An RNA-splicing mutation (G+5IVS20) in the type II collagen gene (COL2A1) in a family with spondyloepiphyseal dysplasia congenita. Am. J. Hum. Genet., 56, 388–395. [PMC free article] [PubMed] [Google Scholar]
- 54.Feshchenko S., Brinckmann,J., Lehmann,H.W., Koch,H.G., Muller,P.K. and Kugler,S. (1998) Identification of a new heterozygous point mutation in the COL1A2 gene leading to skipping of exon 9 in a patient with joint laxity, hyperextensibility of skin and blue sclerae. Mutations in brief no. 166. Online. Hum. Mutat., 12, 138. [DOI] [PubMed] [Google Scholar]
- 55.Horwitz M., Benson,K.F., Person,R.E., Aprikyan,A.G. and Dale,D.C. (1999) Mutations in ELA2, encoding neutrophil elastase, define a 21-day biological clock in cyclic haematopoiesis. Nature Genet., 23, 433–436. [DOI] [PubMed] [Google Scholar]
- 56.Bruening W., Bardeesy,N., Silverman,B.L., Cohn,R.A., Machin,G.A., Aronson,A.J., Housman,D. and Pelletier,J. (1992) Germline intronic and exonic mutations in the Wilms' tumour gene (WT1) affecting urogenital development. Nature Genet., 1, 144–148. [DOI] [PubMed] [Google Scholar]
- 57.Barbaux S., Niaudet,P., Gubler,M.C., Grunfeld,J.P., Jaubert,F., Kuttenn,F., Fekete,C.N., Souleyreau-Therville,N., Thibaud,E., Fellous,M. et al. (1997) Donor splice-site mutations in WT1 are responsible for Frasier syndrome. Nature Genet., 17, 467–470. [DOI] [PubMed] [Google Scholar]
- 58.Tuffery-Giraud S., Saquet,C., Chambert,S. and Claustres,M. (2003) Pseudoexon activation in the DMD gene as a novel mechanism for Becker muscular dystrophy. Hum. Mutat., 21, 608–614. [DOI] [PubMed] [Google Scholar]
- 59.Forch P., Puig,O., Kedersha,N., Martinez,C., Granneman,S., Seraphin,B., Anderson,P. and Valcarcel,J. (2000) The apoptosis-promoting factor TIA-1 is a regulator of alternative pre-mRNA splicing. Mol. Cell, 6, 1089–1098. [DOI] [PubMed] [Google Scholar]
- 60.Vandenbroucke I., Callens,T., De Paepe,A. and Messiaen,L. (2002) Complex splicing pattern generates great diversity in human NF1 transcripts. BMC Genomics, 3, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jurica M.S. and Moore,M.J. (2003) Pre-mRNA splicing: awash in a sea of proteins. Mol. Cell, 12, 5–14. [DOI] [PubMed] [Google Scholar]
- 62.Nilsen T.W. (2003) The spliceosome: the most complex macromolecular machine in the cell? Bioessays, 25, 1147–1149. [DOI] [PubMed] [Google Scholar]
- 63.Zhou Z., Licklider,L.J., Gygi,S.P. and Reed,R. (2002) Comprehensive proteomic analysis of the human spliceosome. Nature, 419, 182–185. [DOI] [PubMed] [Google Scholar]
- 64.Rappsilber J., Ryder,U., Lamond,A.I. and Mann,M. (2002) Large-scale proteomic analysis of the human spliceosome. Genome Res., 12, 1231–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Neubauer G., King,A., Rappsilber,J., Calvio,C., Watson,M., Ajuh,P., Sleeman,J., Lamond,A. and Mann,M. (1998) Mass spectrometry and EST-database searching allows characterization of the multi-protein spliceosome complex. Nature Genet., 20, 46–50. [DOI] [PubMed] [Google Scholar]