

Abstract
AIM
To characterize the role of apolipoprotein B100 (apoB100) in hepatitis C viral (HCV) infection.
METHODS
In this study, we utilize a gene editing tool, transcription activator-like effector nucleases (TALENs), to generate human hepatoma cells with a stable genetic deletion of APOB to assess of apoB in HCV. Using infectious cell culture-competent HCV, viral pseudoparticles, replicon models, and lipidomic analysis we determined the contribution of apoB to each step of the viral lifecycle. We further studied the effect of mipomersen, an FDA-approved antisense inhibitor of apoB100, on HCV using in vitro cell-culture competent HCV and determined its impact on viral infectivity with the TCID50 method.
RESULTS
We found that apoB100 is indispensable for HCV infection. Using the JFH-1 fully infectious cell-culture competent virus in Huh 7 hepatoma cells with TALEN-mediated gene deletion of apoB (APOB KO), we found a significant reduction in HCV RNA and protein levels following infection. Pseudoparticle and replicon models demonstrated that apoB did not play a role in HCV entry or replication. However, the virus produced by APOB KO cells had significantly diminished infectivity as measured by the TCID-50 method compared to wild-type virus. Lipidomic analysis demonstrated that these virions have a fundamentally altered lipidome, with complete depletion of cholesterol esters. We further demonstrate that inhibition of apoB using mipomersen, an FDA-approved anti-sense oligonucleotide, results in a potent anti-HCV effect and significantly reduces the infectivity of the virus.
CONCLUSION
ApoB is required for the generation of fully infectious HCV virions, and inhibition of apoB with mipomersen blocks HCV. Targeting lipid metabolic pathways to impair viral infectivity represents a novel host targeted strategy to inhibit HCV.
Keywords: Apolipoprotein, Lipid, Hepatitis C virus, Gene silencing, Viral replication
Core tip: Hepatitis C virus (HCV) circulates as a very-low-density lipoprotein (VLDL)-like lipoviral particle. Apolipoprotein B100 (apoB100) is the core protein of VLDL, buts its role in HCV has remained incompletely characterized. Use of gene-editing with transcription activator-like effector nucleases permits the characterization of the role of apoB100 in HCV. We demonstrate that apoB100 is required for HCV infection. Loss of apoB100 results in the secretion of HCV virions with an altered lipid composition and limited ability to infect naive cells. Mipomersen, an FDA-approved antisense inhibitor of apoB100, has an anti-HCV effect and limits the viral infectivity.
INTRODUCTION
Hepatitis C virus (HCV) infection is a major global health problem, affecting 185 million individuals worldwide and 3 million in the United States[1,2]. While highly effective direct-acting antivirals (DAAs) for HCV have expanded therapeutic armamentarium against HCV, these drugs may be limited by genotype specificity, limited success in some subpopulations, and the potential for development of multiclass resistance in treatment failures. One successful strategy to address treatment failures is the targeting host factors required for HCV infection, which possess a high barrier to the development of virologic resistance[3].
The development of direct-acting and host-targeted antiviral medications emerged on the heels of a vastly enhanced understanding of the HCV viral lifecycle, enabled by the development of robust in vitro models of HCV. Even prior to characterization beyond non-A non-B hepatitis, the virus was observed to physically associate with the low density fraction of human sera suggesting an association with human lipoproteins. Indeed, viral RNA could be precipitated with antibodies against apolipoprotein B100 and apolipoprotein E[4-6]. More recent data has demonstrated that the virus circulates as a highly lipidated lipoviral particle (LVP), which contains both apoE and apoB, and the lipid composition of this LVP very closely resembles human very-low-density lipoprotein (VLDL)[7,8].
Despite these observations, the exact role of apoB in HCV infection remains incompletely characterized. Data have been conflicting, with some pharmacologic studies suggesting an important role for apoB, but RNAi experiments suggesting that apoB does not play a function at all in HCV infection[9-11]. An important limitation of these in vitro studies has been their use of hepatoma cells lines which are highly permissive to HCV, but which do not fully recapitulate the production of human VLDL.
Novel and specific gene editing tools have been developed to better understand gene function in cellular and animal models. One such tool is the use of transcription activator-like effector nucleases (TALENs), derived from plant nucleases, which can be specifically designed to bind target genomic sequences and result in loss of gene expression. This strategy generates stable cellular genetic deletions without requiring antibiotics or transfection, and has minimal off-target effects. We used this technique to generate a hepatoma cell line lacking APOB expression and found HCV infection to be inhibited in the absence of apoB[12]. Following these findings, an additional study utilized zinc-finger nucleases and clarified that apoB and apolipoprotein E (apoE) both likely play a role in infectious HCV particle formation and that there is HCV core accumulation on lipid droplets without apoB and E expression. Further, additional data has additionally suggested that apoB is important for cell-free transmission of the virus[13,14].
In this study, we characterize the specific contribution of human apolipoprotein B 100 to the HCV lifecycle and determine the effect of an FDA-approved inhibitor of apoB on the virus. The cells used for this study were Huh7 human hepatoma cells which over-express the HCV entry co-receptor CD81. Huh7 cells do model human VLDL secretion[15], and overexpression of CD81 renders them more permissive to HCV. Using these novel APOB knockout cells, we confirm that the loss of APOB inhibits HCV infection[12] and that apoB expression is indispensable for HCV. Specifically, its absence results in virus that has a fundamentally altered lipidome and is significantly impaired in its ability to infect other cells. Further, and importantly, we demonstrate a novel use and potent and dose-dependent anti-HCV effect of an FDA-approved compound which inhibits apoB expression, mipomersen.
MATERIALS AND METHODS
Cell culture
TALEN-induced APOB KO Huh 7/CD81 cells were generated and maintained as previously described[12]. All experiments were conducted in triplicate.
HCV infection
JFH1, a genotype 2a HCV isolate, and the Jc1e2FLAG JFH1 chimera were used for HCV infection. Naive cells were incubated with the virus for 4-6 h, after which the viral supernatant was removed. Virus of identical multiplicity of infection (MOI) was used for all experiments. Cell lysates were isolated for protein and/or RNA at the time points indicated. Lysate RNA was collected using RNEasy isolation kits (QIAGEN, Valencia, CA). Viral RNA was isolated from the supernatant using a viral nucleic acid isolation kit (Roche, Indianapolis, IN). LDL rescue was performed using commercially available LDL extracted from human plasma (Sigma, St. Louis, MO). In rescue experiments, LDL was delivered at a concentration of 2.5 pg/mL following infection with JFH1.
Immunofluorescence
Cells were cultured on coverslips and fixed with 4% PFA at indicated time points. Primary antibodies (LDL-R, Ab-Cam, Cambridge, MA, and HCV core hybridoma) were diluted in 10% Donkey Serum, and were stained with IgG Alexa Fluor 488 secondary antibodies (Life Technologies, Carlsbad, CA).
Pseudoparticle experiments
HCV E1 E2 and control VZV pesudoparticles were a kind gift of Dr. Francois-Loic Cosset (Lyon, France). Naive cells were plated in 96-well plate and exposed to HCVpp or VZV for 72 h. The infectivity was determined using immunofluorescence microscopy.
Replicon experiments
Genotype 2a (JFH-1) full genomic and subgenomic replicons were provided courtesy of Professor Takaji Wakita (Tokyo, Japan). The constructs were electroporated into naive cells using the BioRad Gene PulseXcell electroporation system and then selected using G418 supplemented media for approximately three weeks. The G418-resistant colonies were then pooled and cultured on 10cm dishes. HCV levels were determined using RNA and protein isolation from cell lysates.
Infectivity
Virus generated in WT or KO cells was used to infect highly permissive Huh 7.5.1 cells. Dilutions were performed to normalize for HCV RNA titer in the supernatant. The tissue culture infectious dose (TCID50) per mL was performed using methods previously described[16].
Lipid extraction
Jc1E2FLAG HCV particles were isolated and the lipid fraction of the purified Jc1E2FLAG HCV particles was extracted and purified using the methods previously described[7]. The organic phase was collected for use in LC-MS analysis. Analyses of polar and non-polar lipids were conducted using an LC-MS system comprised of an Open Accela 1250 U-HPLC and a Q Exactive hybrid quadruple orbitrap mass spectrometer (Thermo Fisher Scientific; Waltham, MA).
Mipomersen experiments
Huh 7/CD81 cells were treated with escalating doses of mipomersen as indicated, or mock, at the time of plating as well as 24 and 48 h following infection. Cell lysate and viral supernatant were harvested for analysis at 72 h following infection. Cell viability was monitored using Cell Titer Glo (Promega, Madison, WI).
Statistical analysis
Statistical analysis was performed using GraphPad Prism 5 software (GraphPad software, Inc, La Jolla, CA) and by a researcher trained in biostatistics. RT-QPCR data is represented as the mean with 95% confidence intervals. Two tailed student’s t-test was used. A P value of < 0.05 was considered statistically significant.
RESULTS
APOB KO cells inefficiently support HCV infection
APOB KO cells were generated as previously described using TALEN-mediated gene knockout. The parent cells were Huh7 hepatoma cells over-expressing the HCV entry co-factor CD81 (Huh7/CD81). The APOB-/- knockout (APOB KO) cells have a < 3% expression of APOB at the mRNA level and have no detectable apoB100 protein[12].
As we have previously reported, APOB deletion impaired the ability of the hepatoma cells to support HCV infection. Three days following infection with the fully infectious tissue culture-competent hepatitis C virus (HCVcc) strain JFH-1, we observed a 72% (95% CI: 67%-77%, P = 0.01) reduction in HCV RNA and no detectable core protein (Figure 1A). We then confirmed this finding in three additional knockout clones to ensure this finding was not a clone-dependent phenotype and found that HCV was inhibited in all studied clones (Figure 1B).
Figure 1.
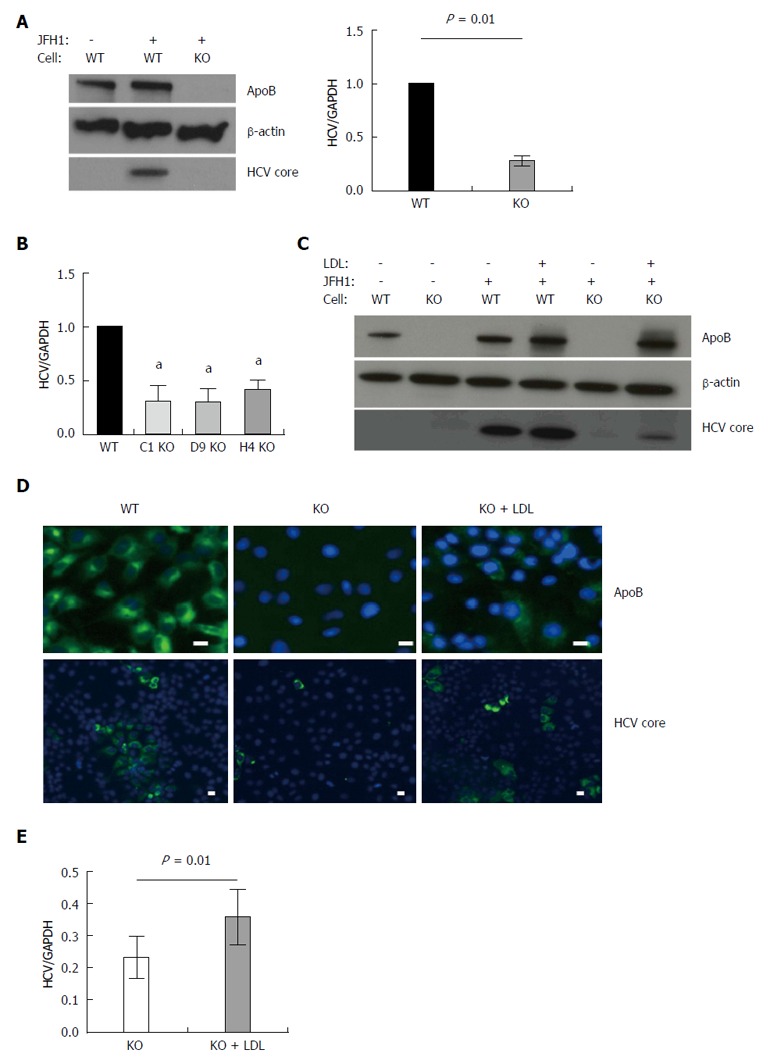
APOB genetic knockout Huh 7 hepatoma cells inefficiently support hepatitis C virus. A: Using TALEN genetic deletion of APOB, knockout huh7/CD81 (high) cells were generated (APOB-/-, KO). We demonstrated that these cells inefficiently support hepatitis C virus (HCV) infection with the fully infectious tissue culture HCV virus, HCVcc (JFH1). There was markedly decreased JFH1 72 h following infection both at the level of HCV core protein and RNA. Data is shown as mean with 95% confidence interval, normalized to WT; B: To confirm this was not a clone-specific effect, three additional clones were infected with the JFH1 virus and intracellular HCV RNA assessed 72 h following infection. We again found a decrease in HCV RNA in the APOB KO clones (aP < 0.05); C, D: Rescue experiments were performed by treating cells with human LDL at the same time as infection with the JFH1 virus. We found partial restoration of intracellular apoB expression at 72 h following exposure (D), and there was a partial restoration of HCV infection, with increased expression of HCV core protein (C), HCV RNA (E), and IF demonstrating increased HCV core expression in the LDL-treated cells (D). Scale bar: 20 μm.
We next sought to determine whether restoring intracellular apoB100 to the APOB KO cells would, in turn, allow them to again support HCV infection. The large size of the full apoB100 protein (550 kda) made plasmid-based transfection challenging; we therefore restored intracellular apoB expression via administration of LDL into the cell media, allowing for LDL-R mediated uptake of apoB. While it is held that apoB is degraded following LDL-R mediated endocytosis[17], the fate of apoB in deficient cells is not known. We confirmed that reintroducing apoB protein into the APOB KO cells resulted in sustained intracellular levels of apoB via western blot and immunofluorescence (Figure 1C and D). Further, reintroduction of apoB led to a significant increase in the intracellular levels of HCV RNA and HCV core protein 72 h following the addition of LDL. These findings were confirmed with immunofluorescence staining, which demonstrated cytoplasmic expression of apoB100 protein following LDL exposure, and enhanced HCV core protein expression in the LDL-treated knockout cells (Figure 1C-E).
Taken together, these findings suggested that loss of APOB expression rendered the Huh7 hepatoma cells non-permissive to the full HCV lifecycle, a state that was reversed upon reintroduction of apoB into the cytoplasm.
Further, we confirmed that these changes were not due to impaired entry using HCV pseudoparticles, a model of HCV entry. We found that entry of HCV into the APOB KO cells was not impaired (Figure 2A), and that the cells were not deficient in known HCV entry factors (Figure 2B). We next sought to understand whether replication was affected in APOB KO cells.
Figure 2.
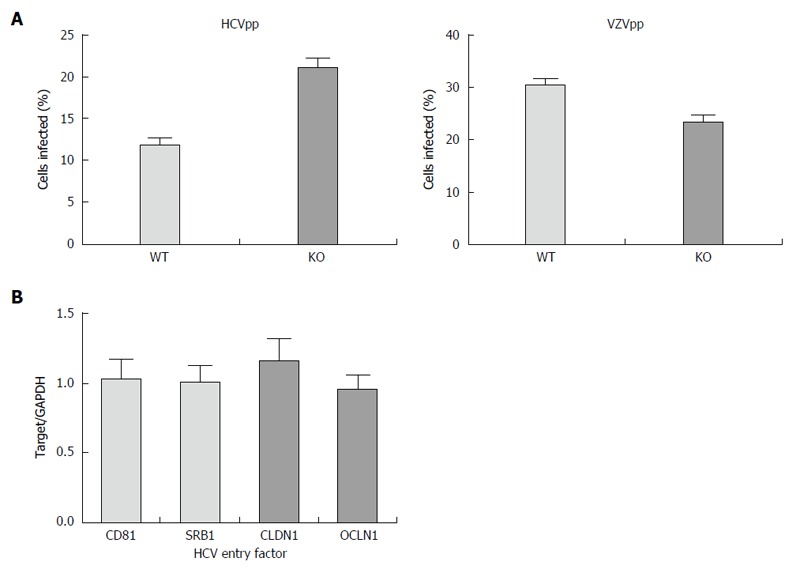
Hepatitis C virus entry is not impaired in APOB KO cells. A: Using an hepatitis C virus (HCV) genotype 1b pseudoparticle (HCVpp), cells were exposed to the pseudoparticle and incubated for 72 h at which time the degree of HCV entry was assessed using GFP expression; a control VZV pseudoparticle (VZVpp) was also assessed. We that entry of neither HCV nor VZV was impaired in the APOB KO cells; B: We confirmed that the expression levels of known HCV entry factors CD81, SRB1, CLDN1, OCLN1 were preserved in the KO cells (normalized to WT cell expression level, 1).
HCV Replication is not impaired in APOB deficient hepatoma cells
HCV viral replication takes place on a highly specialized membranous web formed through the combined effects of both host and viral proteins, and which is closely apposed to cytoplasmic lipid droplets. While apolipoproteins have not been previously suggested to be directly involved in viral RNA replication, the lipid droplet is known to play an important role as a bridge between replication and early assembly. Approximately 20% of the total cellular HCV RNA is localized on the lipid droplet, where it has been demonstrated to co-localize with the HCV core protein and the non-structural protein NS5A[18]. ApoB100 has been described, in the absence of HCV infection, to localize to lipid droplets in a so-called “apoB crescent” while awaiting either lipidation or degradation[19]. We therefore hypothesized that apoB100 might play a role in replication, potentially bridging replication and assembly, possibly through interactions with core, NS5A and the lipid droplet.
To determine whether apoB is required for HCV replication, we utilized both subgenomic and full genomic replicons derived from the genotype 2a JFH1 virus. The replicons are introduced into the cell by electroporation and model the intracellular steps of HCV propagation, but no live virus is secreted[20]. The subgenomic replicon lacks the structural HCV proteins (E1, E2 and core), whereas the full genomic replicon produces all the HCV proteins. Both replicons were introduced into the APOB WT and APOB KO hepatoma cells as previously described[21], and the intracellular HCV levels were determined after antibiotic-mediated selection for replicon-containing cells. We found no difference in HCV RNA between the APOB WT and KO cells using either cellular model, and detected no difference in the expression of core protein in the full genomic replicon model (Figure 3A and B).
Figure 3.
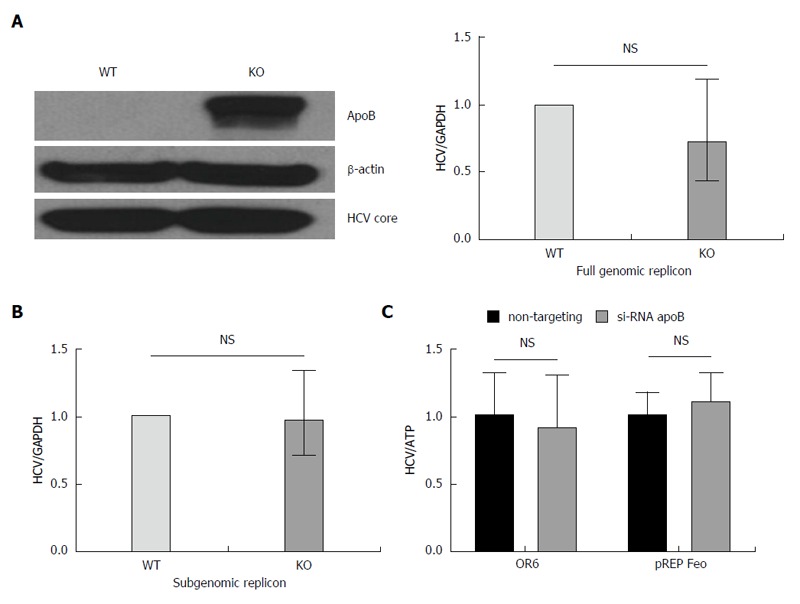
Loss of apoB does not impair replication in APOB KO cells. A: A full genomic replicon derived from the JFH1 hepatitis C virus (HCV)cc strain was electroporated into WT and KO cells which were then selected in culture based on antibiotic resistance. We demonstrated that there was no decrease in intracellular HCV core or RNA in the KO compared to WT cells; B: A subgenomic replicon, which lacks expression of the E1, E2 and core proteins, was also electroporated into the cells and again there was no difference in HCV RNA. Data is shown as mean with 95%CI, normalized to WT; C: To confirm these findings, we used replicon cells with a full genome of a different viral genotype (OR6, 1b and pREP-Feo, 1b) and subsequently knocked down apoB expression using RNAi. There was no difference in HCV replication in the setting of apoB knockdown. Data is shown as mean ± SD.
To further verify these findings, we repeated these experiments in two additional replicon-based cell lines: the OR6 cell, which stably harbors a full length, genotype 1b HCV genome[22], and pRep-Feo cells which contains a subgenomic, genotype 1b HCV replicon[23]. Using RNAi-mediated gene silencing, we knocked down apoB100 expression, and detected no difference in HCV RNA in either cellular model (Figure 3C). These data provide firm evidence that apoB100 does not play an important role in the replication step of the HCV lifecycle.
There is decreased production of HCV virion in APOB KO human hepatoma cells
There are several lines of evidence demonstrating an association between the HCV virion and VLDL production. First, there is prior evidence for a protein-protein interaction between the HCV core protein and the microsomal triglyceride transfer protein (MTTP), a protein critical for the lipidation of apoB100, in the early stages of HCV assembly[24]. Further, HCV follows the pathway of VLDL secretion[25]. However, whether apoB100 is important for the production of fully infectious HCV virions has remained controversial[26-28]. Some data have demonstrated that pharmacologic inhibition of MTTP blocks HCV assembly[10], but inhibition of apoB100 itself has produced disparate effects on HCV LVP production[26,29]. The parent cell line for the KO cells is competent for VLDL production, and the knockouts do not display decreased intracellular levels of either MTTP or apolipoprotein E (data not shown). We determined that the APOB KO cells have diminished HCV secretion.
We first examined the quantity of viral RNA released into the supernatant 72 h following the infection of the APOB WT and KO with JFH-1. There was a substantial and significant reduction in the quantity of HCV RNA released from the KO cells (Figure 4A).
Figure 4.
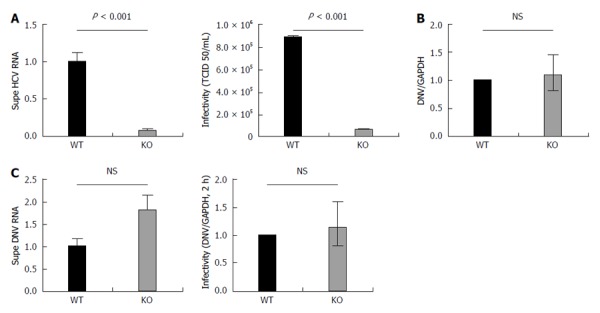
APOB KO cells have diminished hepatitis C virus production and KO virus has impaired infectivity, but APOB KO has no effect on dengue viral infection. A: 72 h following infection with JFH1 hepatitis C virus (HCV)cc, supernatant was harvested and viral RNA was isolated and quantified using qPCR. The supernatant harvested from infected KO cells had significantly lower levels of HCV RNA compared to WT supernatant. Data are normalized to WT RNA titer and standard error of the mean shown. After normalizing for the differences in HCV RNA by diluting the WT virus, the infectivity of the two viruses in highly HCV-permissive Huh 7.5.1 cells was then determined using the TCID50 method. The KO generated virus had significantly reduced infectivity in the permissive Huh 7.5.1 cells; B: Dengue replication: WT and KO cells were infected with Dengue virus, and the level of intracellular RNA was assessed at 72 h. No difference was observed between WT and KO cells. Data is shown as mean with 95% confidence interval, normalized to WT; C: Supernatant of dengue-infected cells was harvested at 72 h and dengue viral RNA quantified. RNA levels were slightly higher in the supernatant of the KO cells, but the difference was not significant. A similar trend was observed with the infectivity of the KO generated dengue virus, assessed at DNV RNA at 2 h following infection.
We then sought to determine whether the virus that was successfully produced by the APOB KO cells differed from wild type virus with respect to its ability to infect naive cell (infectivity). To do this, we exposed uninfected, highly permissive, Huh 7.5.1 cells to virus generated by either APOB WT (WT virus) or APOB KO (KO virus) cells. We normalized the quantity of virus used for inoculation by the viral RNA titer (to account for the decreased levels of HCV RNA detected from the KO cells), and assessed infectivity using the previously described TCID50 method[16]. We demonstrated that the KO virus was significantly less infectious in the highly permissive Huh7.5.1 cells than the WT Virus (Figure 4A).
Lipids play an important role in flavivirus production and entry, and have been implicated in replication of the dengue virus[30-32]. Dengue has a lipid-rich envelope, and interruption of cholesterol biosynthesis impairs its replication and infectivity[33]. To determine whether the diminished infectivity observed in the KO HCV virus was related to a defect in lipid metabolism that is extendable to other flaviviridae, we examined viral production and infectivity of the Dengue virus using the apoB cells.
After infecting WT and KO cells with dengue virus and assessing the levels of viral RNA 72 h following inoculation, we found no difference in the ability of the APOB KO cells to support dengue infection (Figure 4B). We analyzed the viral supernatant and additionally demonstrated that there was no decrease in DNV RNA produced by the KO cells. Further, after normalizing for RNA titers, there was no decrease in dengue viral infectivity (Figure 4C). We thus concluded that the perturbations observed following deletion of APOB are not generalizable to dengue virus, and may be specific to HCV.
HCV virions produced by APOB KO human hepatoma cells have a fundamentally altered lipidome
The density of viral particles has been described to be an important predictor of viral infectivity, and the lipid composition is the major determinant of buoyant density. The lipid composition of the HCVcc produced by Huh 7.5.1 cells has recently been characterized[7]; however, the buoyant density of Huh 7.5 cells and tissue culture-generated virus is lower than that observed in human serum[34,35], and Huh 7.5 and Huh 7.5.1 cells have not been demonstrated to recapitulate the human VLDL machinery, unlike Huh7 cells[15]. Virus generated by VLDL-competent Huh7/CD81 cells is more likely to reflect the human LVP, and we hypothesized that the observed impaired infectivity reflected an altered lipid composition of the KO virus.
Prior to characterizing the lipidome, we sought to confirm the presence of apoE in the virus, since apoE has also been well-described to be important for generation of viral particles and viral infectivity[36] . Using an ELISA-based assay, we determined that the KO virus, as expected, had barely detectable apoB100 secreted into the media. We also determined, however, that there was a modest, but significant, reduction in the amount of apoE (Figure 5A). This reduction however was far less than the observed decrease in infectivity, suggesting that apoE reduction is not the primary cause of the loss of infectivity.
Figure 5.
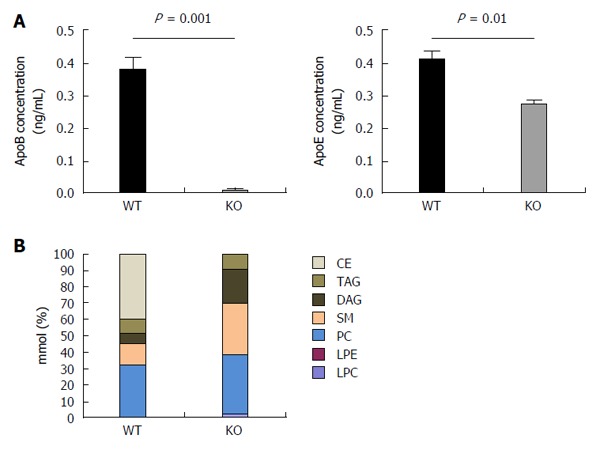
Hepatitis C virus produced by APOB KO cells has a fundamentally altered lipidome. A: The concentrations of apoB and apoE were determined of the viral supernatant using ELISA. ApoB levels were barely detected in the viral supernatant of the KO cells. ApoE concentrations were also found to be lower in KO compared to the WT cells; B: Hepatitis C virus (HCV)cc generated in APOB KO cells has an altered lipidome. Using a Jc1E2FLAG HCV tissue culture fully infectious virus, viral particles were purified using affinity purification particles were extracted and the lipidome was purified using liquid chromatography - mass spectrometry (LC-MS). LC-MS demonstrated that the viral particles generated in the APOB knockout cells are completely depleted in cholesterol esters. CE: Cholesterol and cholesterol esters; TAG: Triacylglycerols; DAG: Diacylglycerols; SM: Sphingomyelins; PC: Phosphatidylcholines; LPE: Lysophosphatidylethanolamines; LPC: Lysophosphatidylcholines.
To characterize the lipidome of HCVcc produced in WT and KO cells, we utilized a JFH-1 derived, JC-1 virus with a FLAG-tag on the N-terminus of the E2 protein (JC1E2FLAG). After harvesting JC-1 virus generated in the APOB WT and APOB KO cells, we performed affinity purification of HCV virion. The purified virus was then analyzed using liquid chromatography/mass spectrometry (LCMS) to determine the lipid composition of the virions.
KO virus had a profoundly altered lipidome compared to WT virus. Specifically, triacylglycerols and diacylglycerols comprised approximately 15% of the lipid composition in the WT virus, compared to close to 30% in the KO virus. Most strikingly, the primary lipids (accounting for approximately 40% of all lipids) of WT virus were cholesterol esters, which was consistent with prior data; however, the KO virus was completely lacking in cholesterol esters (Figure 5B). This perturbation alone may account for a substantial amount of the impaired infectivity of the KO virus: indeed, two of the known HCV entry receptors, SRB1 and the Niemann-Pick-C1-Like-Receptor1 (NPC1L1) are described to recognize cholesterol esters[37,38].
We therefore concluded that apoB plays a critical role in HCV infection, primarily due to its deleterious effect on generation of infectious virus. Without apoB, production and secretion of new virion is impaired, and those virions which are secreted have a markedly diminished infectivity, which is likely related to the depletion of cholesterol esters from the lipidome.
An FDA-approved anti-sense oligonucleotide against apoB inhibits HCV virion infectivity
Based on these findings, we hypothesized that a drug targeting apoB expression or synthesis would have potent anti-HCV effect. Mipomersen is an antisense oligonucleotide against apoB, and inhibits apoB synthesis at the level of transcription from mRNA. It has been demonstrated to significantly reduce production of apoB-containing lipoproteins, most notably circulating LDL, in phase III clinical trials[39], and is currently approved by the FDA for the treatment of homozygous familial hypercholesterolemia. For its treatment of hypercholesterolemia, it is delivered as a once-weekly subcutaneous injection and has been demonstrated to have no significant drug-drug interactions.
We treated Huh7/CD81 WT cells with mipomersen at escalating doses and infected the cells with HCVcc (JFH1). At 72 h following infection, there was an 80%-85% reduction in intracellular HCV RNA with the higher mipomersen doses (800 and 1000 μg/mL), and we observed a dose-response effect. We confirmed these findings at the level of HCV core protein expression (Figure 6A). To confirm that the inhibition of HCV did not occur at the level of HCV RNA replication, assessed the effect of mipomersen on the OR6 replicon model, and confirmed that mipomersen had no effect on viral replication (Figure 6B). We observed no significant effect on cell viability with escalating doses of mipomersen (Figure 6C).
Figure 6.
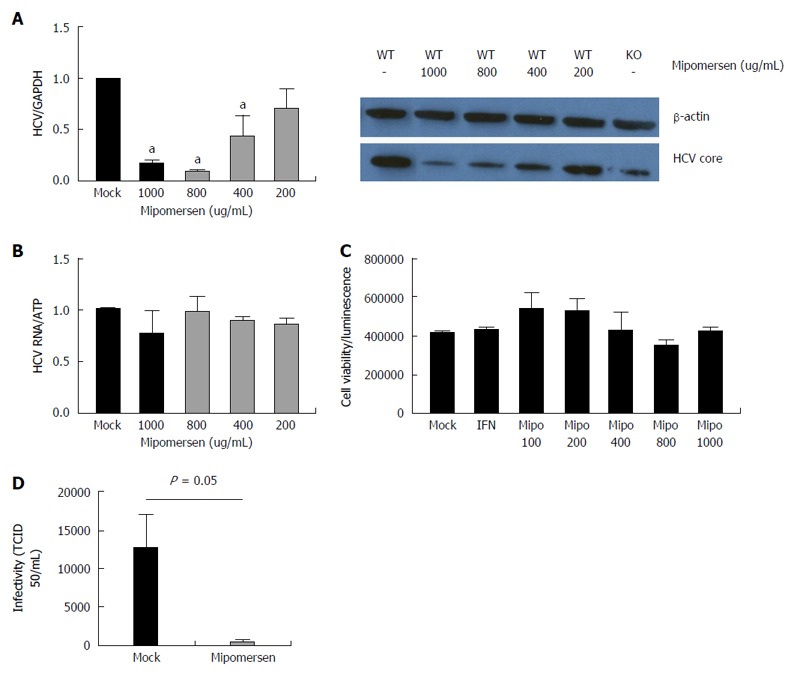
Mipomersen exerts a dose-dependent and potent anti-hepatitis C virus effect. A: Huh7/CD81 high cells were treated with escalating doses of mipomersen from 200 ug/mL to 1000 ug/mL and then infected with JFH1. At 72 h following infection, there was a dose-dependent inhibition of hepatitis C virus (HCV), demonstrated at both the RNA and protein (HCV core) level. Data are shown as means with 95%CI (aP < 0.05); B: OR6 genotype 1b replicon cells were treated with mipomersen for 72 h and no effect was observed on HCV replication; C: There was no negative impact of escalating doses of mipomersen on cell viability; D: Infectivity of the virus was assessed using the TCID50 method. Virus generated in cells treated with mipomersen had a significant reduction in infectivity compared with virus generated in mock-treated cells. Data shown as mean TCID50/mL ± SEM.
The mechanism by which mipomersen exerts its antiviral effect was confirmed using infectivity assays. HCVcc was generated in either mock-treated or mipomersen-treated (800 μg/mL) Huh 7 cells and TCID50 of the virus assayed. We determined that the mipomersen-treated virus was significantly less infectious, with a greater than 2-log reduction in TCID50/mL (12536 vs 442 TCID50/mL, P < 0.05). Mipomersen exerts its potent anti-HCV effect by impairing the infectivity of the virus (Figure 6D).
DISCUSSION
This is the first study to comprehensively characterize the role of apolipoprotein B100, the core protein of human VLDL and LDL, in each step of the HCV lifecycle and to demonstrate that inhibition of apoB100 by an FDA-approved therapeutic has potent antiviral effect. Similarities between circulating HCV and VLDL have long been observed, yet whether human apolipoprotein B100 is central to the viral lifecycle has remained uncertain, in part due to the limitations imposed by the techniques and cellular models used to date. This study leverages recent advances in genome editing (TALEN), allowing for specific and complete gene knockout, combined with a cell line (Huh-7/CD81) which recapitulates human VLDL synthesis, but also supports HCV to fully assess the role of apoB in HCV.
We have established that apoB is not required for entry or replication, but rather that loss of apoB renders the Huh7 hepatoma cells deficient in their ability to support HCV by generating a virus that has a fundamentally altered lipidome and is significantly impaired in its ability to infect naive cells. We determined by mass spectrometry that, in addition to being deficient in apoB, the generated lipoviral particles are significantly depleted in cholesterol esters. It has previously been described that cholesterols are required to maintain the association between HCV and host lipoproteins[40], and are required for viral infectivity.
Depletion of the virion of cholesterol esters may additionally lead to impaired viral entry. SR-BI, a receptor which recognizes lipoproteins, is a well characterized co-entry receptor for HCV[41-43], and it has been previously characterized that both lipoproteins and cholesterol esters are important for the uptake of HDL by SR-BI[44]. Similar findings have been demonstrated in SR-BI-mediated HCV entry[45]. Thus, the KO virus lacking in cholesterol esters, may in part have diminished infectivity due to loss of SR-BI-mediated uptake.
Finally, we demonstrated the in vitro efficacy of mipomersen, an FDA-approved drug for the treatment of familial hypercholesterolemia, which inhibits apoB synthesis using antisense technology. ApoB is therefore a practical and viable pharmacologic target in anti-HCV therapy, with an inhibitor already in clinical use. We showed that treatment with mipomersen significantly inhibited infection with HCV in a dose-dependent fashion. Further, similar to what we observed in the KO cells, we demonstrated that mipomersen exerts its anti-HCV by generating virus which is significantly less infectious than untreated virus. This highlights the possibility of a class of host-targeted antivirals which would serve as infectivity inhibitors, generating virus that is crippled in its ability to infect naive cells. The major limitation of this study is that it is limited to in vitro experimentation in cell lines. This study did not include in vivo experiments, and additional studies to determine the effect of mipomersen in vivo are needed.
While the drugs to treat HCV have become highly effective as direct acting antivirals are emerging into clinical practice, the most difficult-to-treat patients, particularly those who have been treated with multiple DAA classes, may still require alternate regimens and strategies. In this regard, the “real world” experience with these novel regimens is not yet proven, and development of resistance may be a concern when the drug regimens are adopted for broader use.
Targeting host factors required for the virus is regarded as a promising avenue for the development of adjunctive therapies, since this strategy has been shown to impose a higher barrier to the development of viral resistance. ApoB targeting with an FDA-approved inhibitor would present an attractive target. Here we demonstrate the in vitro efficacy of mipomersen against HCV, and suggests an additional, readily targeted host factor required for HCV. Further, we demonstrate that blocking apoB alters viral infectivity by perturbing the lipidome required to generate fully infectious virus. The use of a drug to block this pathway highlights a novel approach towards the treatment of HCV. While we did not see effect against dengue virus, drugs which inhibit the infectivity of a virus by altering the cholesterol composition may pose a novel new strategy to combat refractory HCV and other emerging viral diseases.
ACKNOWLEDGMENTS
The investigational compound for this research was supplied by Genzyme, a Sanofi Company. We also would like to thank Ramnik Xavier, Kiran Musunuru, Manish Gala, Jakob Begun, Joanna Peloquin, Carol Lin, and Mary McGowan for their assistance and input throughout the project.
COMMENTS
Background
The hepatitis C virus (HCV) circulates in humans as a lipoviral particle (LVP) which has a very similar composition to very-low-density lipoprotein (VLDL). Despite this known relationship, the role of apolipoprotein B 100 (the core protein of VLDL) in the HCV lifecycle has remained uncertain.
Research frontiers
This study utilized genome editing technology (TALEN) to characterize the role of apoB100 in the HCV lifecycle.
Innovations and breakthroughs
The use of genome editing allowed for careful examination of several critical steps in HCV infection without the use of lipid-based or viral transfection for gene knockdown. This permitted lipidomic analysis of HCV lipoviral particles, demonstrating that apoB is critical to maintain the “VLDL”-like composition and infectivity of the lipoviral particle. Further, this study demonstrated that an FDA-approved antisense inhibitor of apoB blocks HCV infection in vitro.
Applications
The findings highlight the critical role of lipids and lipoproteins in HCV infection, and specifically in the infectivity of the lipoviral particle. Targeting host lipid metabolic pathways may be useful in resistant or difficult-to-treat HCV infection in humans.
Terminology
TALEN refers to transcription activator-like effector nucleases, which can be designed to target host genomic sequences and generate stable genetic deletions without transfection, antibiotics. These have been shown to generate stable knock-out models with minimal off-target effect.
Peer-review
The paper presents work linking hepatic apoB100 secretion with hepatitis C virion assembly and secretion, studied in vitro with Huh7/CD81 cells in which apoB expression was deleted. The work makes the point that apoB co-expression is required for infectious HCV particle formation. Further work will be required to be sure that this applies to processing of HCV in vivo.
Footnotes
Manuscript source: Invited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: United States
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): B
Grade C (Good): 0
Grade D (Fair): 0
Grade E (Poor): 0
Institutional review board statement: No patients or patient-derived samples were involved in this study.
Conflict-of-interest statement: The authors have no conflicts of interest to declare.
Data sharing statement: Technical appendix and data set available from the corresponding author.
Peer-review started: July 25, 2016
First decision: September 20, 2016
Article in press: October 27, 2016
P- Reviewer: Rudel LL S- Editor: Yu J L- Editor: A E- Editor: Wang CH
References
- 1.Zaltron S, Spinetti A, Biasi L, Baiguera C, Castelli F. Chronic HCV infection: epidemiological and clinical relevance. BMC Infect Dis. 2012;12 Suppl 2:S2. doi: 10.1186/1471-2334-12-S2-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Klevens RM, Hu DJ, Jiles R, Holmberg SD. Evolving epidemiology of hepatitis C virus in the United States. Clin Infect Dis. 2012;55 Suppl 1:S3–S9. doi: 10.1093/cid/cis393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mamedov R, Novruzov N, Baskiran A, Yetisir F, Unal B, Aydın C, Bayramov N, Kayaalp C, Yilmaz S. Living donor liver transplantation with replacement of vena cava for Echinococcus alveolaris: A case report. Int J Surg Case Rep. 2014;5:169–171. doi: 10.1016/j.ijscr.2014.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nielsen SU, Bassendine MF, Burt AD, Martin C, Pumeechockchai W, Toms GL. Association between hepatitis C virus and very-low-density lipoprotein (VLDL)/LDL analyzed in iodixanol density gradients. J Virol. 2006;80:2418–2428. doi: 10.1128/JVI.80.5.2418-2428.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thomssen R, Bonk S, Propfe C, Heermann KH, Köchel HG, Uy A. Association of hepatitis C virus in human sera with beta-lipoprotein. Med Microbiol Immunol. 1992;181:293–300. doi: 10.1007/BF00198849. [DOI] [PubMed] [Google Scholar]
- 6.André P, Komurian-Pradel F, Deforges S, Perret M, Berland JL, Sodoyer M, Pol S, Bréchot C, Paranhos-Baccalà G, Lotteau V. Characterization of low- and very-low-density hepatitis C virus RNA-containing particles. J Virol. 2002;76:6919–6928. doi: 10.1128/JVI.76.14.6919-6928.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Merz A, Long G, Hiet MS, Brügger B, Chlanda P, Andre P, Wieland F, Krijnse-Locker J, Bartenschlager R. Biochemical and morphological properties of hepatitis C virus particles and determination of their lipidome. J Biol Chem. 2011;286:3018–3032. doi: 10.1074/jbc.M110.175018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gastaminza P, Dryden KA, Boyd B, Wood MR, Law M, Yeager M, Chisari FV. Ultrastructural and biophysical characterization of hepatitis C virus particles produced in cell culture. J Virol. 2010;84:10999–11009. doi: 10.1128/JVI.00526-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang H, Sun F, Owen DM, Li W, Chen Y, Gale M, Ye J. Hepatitis C virus production by human hepatocytes dependent on assembly and secretion of very low-density lipoproteins. Proc Natl Acad Sci USA. 2007;104:5848–5853. doi: 10.1073/pnas.0700760104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goldwasser J, Cohen PY, Lin W, Kitsberg D, Balaguer P, Polyak SJ, Chung RT, Yarmush ML, Nahmias Y. Naringenin inhibits the assembly and long-term production of infectious hepatitis C virus particles through a PPAR-mediated mechanism. J Hepatol. 2011;55:963–971. doi: 10.1016/j.jhep.2011.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jiang J, Cun W, Wu X, Shi Q, Tang H, Luo G. Hepatitis C virus attachment mediated by apolipoprotein E binding to cell surface heparan sulfate. J Virol. 2012;86:7256–7267. doi: 10.1128/JVI.07222-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ding Q, Lee YK, Schaefer EA, Peters DT, Veres A, Kim K, Kuperwasser N, Motola DL, Meissner TB, Hendriks WT, et al. A TALEN genome-editing system for generating human stem cell-based disease models. Cell Stem Cell. 2013;12:238–251. doi: 10.1016/j.stem.2012.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fukuhara T, Wada M, Nakamura S, Ono C, Shiokawa M, Yamamoto S, Motomura T, Okamoto T, Okuzaki D, Yamamoto M, et al. Amphipathic α-helices in apolipoproteins are crucial to the formation of infectious hepatitis C virus particles. PLoS Pathog. 2014;10:e1004534. doi: 10.1371/journal.ppat.1004534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gondar V, Molina-Jiménez F, Hishiki T, García-Buey L, Koutsoudakis G, Shimotohno K, Benedicto I, Majano PL. Apolipoprotein E, but Not Apolipoprotein B, Is Essential for Efficient Cell-to-Cell Transmission of Hepatitis C Virus. J Virol. 2015;89:9962–9973. doi: 10.1128/JVI.00577-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meex SJ, Andreo U, Sparks JD, Fisher EA. Huh-7 or HepG2 cells: which is the better model for studying human apolipoprotein-B100 assembly and secretion? J Lipid Res. 2011;52:152–158. doi: 10.1194/jlr.D008888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lindenbach BD. Measuring HCV infectivity produced in cell culture and in vivo. Methods Mol Biol. 2009;510:329–336. doi: 10.1007/978-1-59745-394-3_24. [DOI] [PubMed] [Google Scholar]
- 17.Lodish H, Berk A, Zipursky SL. Molecular Cell Biology. New York: W.H. Freeman; 2000. [Google Scholar]
- 18.Miyanari Y, Atsuzawa K, Usuda N, Watashi K, Hishiki T, Zayas M, Bartenschlager R, Wakita T, Hijikata M, Shimotohno K. The lipid droplet is an important organelle for hepatitis C virus production. Nat Cell Biol. 2007;9:1089–1097. doi: 10.1038/ncb1631. [DOI] [PubMed] [Google Scholar]
- 19.Ohsaki Y, Cheng J, Suzuki M, Fujita A, Fujimoto T. Lipid droplets are arrested in the ER membrane by tight binding of lipidated apolipoprotein B-100. J Cell Sci. 2008;121:2415–2422. doi: 10.1242/jcs.025452. [DOI] [PubMed] [Google Scholar]
- 20.Miyamoto M, Kato T, Date T, Mizokami M, Wakita T. Comparison between subgenomic replicons of hepatitis C virus genotypes 2a (JFH-1) and 1b (Con1 NK5.1) Intervirology. 2006;49:37–43. doi: 10.1159/000087261. [DOI] [PubMed] [Google Scholar]
- 21.Rodgers MA, Villareal VA, Schaefer EA, Peng LF, Corey KE, Chung RT, Yang PL. Lipid metabolite profiling identifies desmosterol metabolism as a new antiviral target for hepatitis C virus. J Am Chem Soc. 2012;134:6896–6899. doi: 10.1021/ja207391q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ikeda M, Abe K, Dansako H, Nakamura T, Naka K, Kato N. Efficient replication of a full-length hepatitis C virus genome, strain O, in cell culture, and development of a luciferase reporter system. Biochem Biophys Res Commun. 2005;329:1350–1359. doi: 10.1016/j.bbrc.2005.02.138. [DOI] [PubMed] [Google Scholar]
- 23.Tanabe Y, Sakamoto N, Enomoto N, Kurosaki M, Ueda E, Maekawa S, Yamashiro T, Nakagawa M, Chen CH, Kanazawa N, et al. Synergistic inhibition of intracellular hepatitis C virus replication by combination of ribavirin and interferon- alpha. J Infect Dis. 2004;189:1129–1139. doi: 10.1086/382595. [DOI] [PubMed] [Google Scholar]
- 24.Perlemuter G, Sabile A, Letteron P, Vona G, Topilco A, Chrétien Y, Koike K, Pessayre D, Chapman J, Barba G, et al. Hepatitis C virus core protein inhibits microsomal triglyceride transfer protein activity and very low density lipoprotein secretion: a model of viral-related steatosis. FASEB J. 2002;16:185–194. doi: 10.1096/fj.01-0396com. [DOI] [PubMed] [Google Scholar]
- 25.Felmlee DJ, Hafirassou ML, Lefevre M, Baumert TF, Schuster C. Hepatitis C virus, cholesterol and lipoproteins--impact for the viral life cycle and pathogenesis of liver disease. Viruses. 2013;5:1292–1324. doi: 10.3390/v5051292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiang J, Luo G. Apolipoprotein E but not B is required for the formation of infectious hepatitis C virus particles. J Virol. 2009;83:12680–12691. doi: 10.1128/JVI.01476-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shiokawa M, Fukuhara T, Ono C, Yamamoto S, Okamoto T, Watanabe N, Wakita T, Matsuura Y. Novel permissive cell lines for complete propagation of hepatitis C virus. J Virol. 2014;88:5578–5594. doi: 10.1128/JVI.03839-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jammart B, Michelet M, Pécheur EI, Parent R, Bartosch B, Zoulim F, Durantel D. Very-low-density lipoprotein (VLDL)-producing and hepatitis C virus-replicating HepG2 cells secrete no more lipoviroparticles than VLDL-deficient Huh7.5 cells. J Virol. 2013;87:5065–5080. doi: 10.1128/JVI.01405-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boyer A, Dumans A, Beaumont E, Etienne L, Roingeard P, Meunier JC. The association of hepatitis C virus glycoproteins with apolipoproteins E and B early in assembly is conserved in lipoviral particles. J Biol Chem. 2014;289:18904–18913. doi: 10.1074/jbc.M113.538256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heinz FX, Stiasny K, Allison SL. The entry machinery of flaviviruses. Arch Virol Suppl. 2004;(18):133–137. doi: 10.1007/978-3-7091-0572-6_11. [DOI] [PubMed] [Google Scholar]
- 31.Rothwell C, Lebreton A, Young Ng C, Lim JY, Liu W, Vasudevan S, Labow M, Gu F, Gaither LA. Cholesterol biosynthesis modulation regulates dengue viral replication. Virology. 2009;389:8–19. doi: 10.1016/j.virol.2009.03.025. [DOI] [PubMed] [Google Scholar]
- 32.Martínez-Gutierrez M, Castellanos JE, Gallego-Gómez JC. Statins reduce dengue virus production via decreased virion assembly. Intervirology. 2011;54:202–216. doi: 10.1159/000321892. [DOI] [PubMed] [Google Scholar]
- 33.Carro AC, Damonte EB. Requirement of cholesterol in the viral envelope for dengue virus infection. Virus Res. 2013;174:78–87. doi: 10.1016/j.virusres.2013.03.005. [DOI] [PubMed] [Google Scholar]
- 34.Lindenbach BD, Meuleman P, Ploss A, Vanwolleghem T, Syder AJ, McKeating JA, Lanford RE, Feinstone SM, Major ME, Leroux-Roels G, et al. Cell culture-grown hepatitis C virus is infectious in vivo and can be recultured in vitro. Proc Natl Acad Sci USA. 2006;103:3805–3809. doi: 10.1073/pnas.0511218103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bartenschlager R, Penin F, Lohmann V, André P. Assembly of infectious hepatitis C virus particles. Trends Microbiol. 2011;19:95–103. doi: 10.1016/j.tim.2010.11.005. [DOI] [PubMed] [Google Scholar]
- 36.Lee JY, Acosta EG, Stoeck IK, Long G, Hiet MS, Mueller B, Fackler OT, Kallis S, Bartenschlager R. Apolipoprotein E likely contributes to a maturation step of infectious hepatitis C virus particles and interacts with viral envelope glycoproteins. J Virol. 2014;88:12422–12437. doi: 10.1128/JVI.01660-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sainz B, Barretto N, Martin DN, Hiraga N, Imamura M, Hussain S, Marsh KA, Yu X, Chayama K, Alrefai WA, et al. Identification of the Niemann-Pick C1-like 1 cholesterol absorption receptor as a new hepatitis C virus entry factor. Nat Med. 2012;18:281–285. doi: 10.1038/nm.2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Acton S, Rigotti A, Landschulz KT, Xu S, Hobbs HH, Krieger M. Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science. 1996;271:518–520. doi: 10.1126/science.271.5248.518. [DOI] [PubMed] [Google Scholar]
- 39.Thomas GS, Cromwell WC, Ali S, Chin W, Flaim JD, Davidson M. Mipomersen, an apolipoprotein B synthesis inhibitor, reduces atherogenic lipoproteins in patients with severe hypercholesterolemia at high cardiovascular risk: a randomized, double-blind, placebo-controlled trial. J Am Coll Cardiol. 2013;62:2178–2184. doi: 10.1016/j.jacc.2013.07.081. [DOI] [PubMed] [Google Scholar]
- 40.Yamamoto M, Aizaki H, Fukasawa M, Teraoka T, Miyamura T, Wakita T, Suzuki T. Structural requirements of virion-associated cholesterol for infectivity, buoyant density and apolipoprotein association of hepatitis C virus. J Gen Virol. 2011;92:2082–2087. doi: 10.1099/vir.0.032391-0. [DOI] [PubMed] [Google Scholar]
- 41.Zeisel MB, Koutsoudakis G, Schnober EK, Haberstroh A, Blum HE, Cosset FL, Wakita T, Jaeck D, Doffoel M, Royer C, et al. Scavenger receptor class B type I is a key host factor for hepatitis C virus infection required for an entry step closely linked to CD81. Hepatology. 2007;46:1722–1731. doi: 10.1002/hep.21994. [DOI] [PubMed] [Google Scholar]
- 42.Catanese MT, Ansuini H, Graziani R, Huby T, Moreau M, Ball JK, Paonessa G, Rice CM, Cortese R, Vitelli A, et al. Role of scavenger receptor class B type I in hepatitis C virus entry: kinetics and molecular determinants. J Virol. 2010;84:34–43. doi: 10.1128/JVI.02199-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dorner M, Horwitz JA, Donovan BM, Labitt RN, Budell WC, Friling T, Vogt A, Catanese MT, Satoh T, Kawai T, et al. Completion of the entire hepatitis C virus life cycle in genetically humanized mice. Nature. 2013;501:237–241. doi: 10.1038/nature12427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nieland TJ, Xu S, Penman M, Krieger M. Negatively cooperative binding of high-density lipoprotein to the HDL receptor SR-BI. Biochemistry. 2011;50:1818–1830. doi: 10.1021/bi101657j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dao Thi VL, Granier C, Zeisel MB, Guérin M, Mancip J, Granio O, Penin F, Lavillette D, Bartenschlager R, Baumert TF, et al. Characterization of hepatitis C virus particle subpopulations reveals multiple usage of the scavenger receptor BI for entry steps. J Biol Chem. 2012;287:31242–31257. doi: 10.1074/jbc.M112.365924. [DOI] [PMC free article] [PubMed] [Google Scholar]