


Abstract
Identification of genes required for segregation of chromosomes in meiosis (scm) is difficult because in most organisms high-fidelity chromosome segregation is essential to produce viable meiotic products. The biology of fission yeast Schizosaccharomyces pombe facilitates identification of such genes. Insertional mutagenesis was achieved by electroporation of linear ura4+ DNA into cells harboring a ura4 deletion. Approximately 1000 stable transformants were screened individually for the production of elevated frequencies of aneuploid spore colonies. Twenty-two candidates were subjected to a secondary screen for cytological defects. Five mutants exhibited significant levels of aberrant meiotic chromosome segregation, but were proficient for mating and completion of meiosis. Each mutant's phenotype cosegregated with its respective ura4+ transgene. The mutations were recessive and defined five complementation groups, revealing five distinct genes (scm1, scm2, scm3, scm4 and scm5). Southern blotting revealed single-site integration in each transformant, indicating that insertional mutagenesis is useful for generating single-locus scm mutations linked to a selectable marker. The transgene insertion points were refractory to analysis by inverse-PCR. Molecular and real-time PCR analyses revealed the presence of multiple, truncated copies of ura4+ at each integration site. Thus, electroporation-mediated insertional mutagenesis in S.pombe is preceded by exonucleolytic processing and concatomerization of the transforming DNA.
INTRODUCTION
Meiosis produces four haploid cells from a diploid premeiotic cell. This is achieved by coupling one round of premeiotic DNA replication with two rounds of chromosome segregation. Proper segregation of homologous chromosomes in the first (reductional) meiotic division requires that the paired homologs be held together and aligned on the metaphase plate of meiosis I (1). In most organisms, a combination of crossover recombination structures (chiasmata) and sister chromatid cohesion distal to chiasmata provide this glue. Dissolution of distal cohesion allows homologs to segregate from one–another in anaphase I (2,3). Centromere-proximal cohesion holds sister chromatids together on the metaphase plate of meiosis II. Dissolution of proximal cohesion permits sisters to segregate from one another in anaphase II (3,4).
In fission yeast, rec12 (spo11) mutants entirely lack meiotic recombination and suffer nondisjunction of homologous chromosomes in meiosis I (5,6). rec8 mutants lack normal meiotic sister chromatid cohesion and suffer precocious separation of sister chromatids in meiosis I and random segregation of sisters in meiosis II (7–10). Nevertheless, ∼20–50% of the spores produced by these mutants are viable, whereas in most other organisms (e.g. budding yeast) rec8 and rec12 (spo11) mutants exhibit a meiotic-lethal phenotype (11–13).
For mutations affecting meiotic chromosome segregation, the probability of obtaining viable meiotic products is inversely proportional to the power of chromosome number. Because fission yeast has only three pairs of chromosomes, aberrant or random chromosome assortment can produce, by chance, a significant fraction of spores that receive at least one copy of each chromosome and that are, consequently, viable. Viable aneuploids, such as diploid spores or spores trisomic for chromosome III, are also produced (6,8,9,14). Fission yeast therefore provides an excellent system to screen for mutations affecting meiotic chromosome dynamics.
In the presence of homology within the genome, linear DNA transformed into Schizosaccharomyces pombe can integrate either by homologous recombination with the target locus or by nonhomologous recombination elsewhere in the genome (15,16). In the absence of a homologous target, stable transformants arise exclusively by nonhomologous integration. This provides a method for insertional mutagenesis and, by screening physically or genetically for the transformed DNA, facilitates the subsequent identification of the sites of integration within the genome (17,18).
The goal of this study was to test the feasibility of a novel genetic screen to identify genes required for faithful meiotic chromosome dynamics. Mutagenesis employed nonhomologous integration of a dominant, selectable marker gene (ura4+). We directly screened ∼1000 stable transformant colonies for an initial phenotype of producing a high frequency of aneuploid meiotic products. Candidates passing the primary screen were subjected to a secondary, cytological screen for aberrant chromosome segregation. This identified five loci (scm1–scm5) that are required for proper segregation of chromosomes in meiosis (scm).
MATERIALS AND METHODS
Schizosaccharomyces pombe culture
The genotypes of the S.pombe strains used are listed in Table 1 and genetic methods were as described elsewhere (9,19). Rich yeast-extract liquid (YEL) and yeast-extract agar (YEA), sporulation liquid (SPL) and sporulation liquid agar (SPA), and minimal nitrogen base liquid (NBL) and nitrogen base agar (NBA) were as described elsewhere (19,20). YEA-B was YEA containing 2.5 μg/ml of Phloxin-B (Sigma, St Louis, MO) and 100 μg/ml of adenine. On Phloxin-B-containing plates, haploid cells produce light pink colonies whereas diploid cells produce dark pink colonies. Media were supplemented with required amino acids and uracil at 100 μg/ml and5′-FOA (American Bioorganics, Niagara Falls, NY) at 1 mg/ml, as necessary. Meiotic efficiencies and recombinant frequencies were determined as described previously (21).
Table 1. Genotypes of S.pombe strains.
| Strain | Genotype |
|---|---|
| WSP 0002 | h− wild-type |
| WSP 0607 | h− ura4-D18 arg3-D4 ade6-M26 |
| WSP 0620 | h+ ura4-D18 arg3-D4 ade6-M210 |
| WSP 0628 | h90 ura4-D18 his3-D1 arg3-D4 |
| WSP 1240 | h90 ade6-52 rec12-117 |
| WSP 1694 (T1) | h90 ura4-D18 his3-D1 arg3-D4 anon::ura4,23-6 |
| WSP 1695 (T2) | h90 ura4-D18 his3-D1 arg3-D4 anon::ura4,27-3 |
| WSP 1696 (T3) | h90 ura4-D18 his3-D1 arg3-D4 anon::ura4,28-48 |
| WSP 1697 (T4) | h90 ura4-D18 his3-D1 arg3-D4 scm1::ura4 |
| WSP 1698 (T5) | h90 ura4-D18 his3-D1 arg3-D4 anon::ura4,37-40 |
| WSP 1699 (T6) | h90 ura4-D18 his3-D1 arg3-D4 anon::ura4,38-18 |
| WSP 1700 (T7) | h90 ura4-D18 his3-D1 arg3-D4 anon::ura4,38-46 |
| WSP 1701 (T8) | h90 ura4-D18 his3-D1 arg3-D4 scm2::ura4 |
| WSP 1702 (T9) | h90 ura4-D18 his3-D1 arg3-D4 anon::ura4,40-32 |
| WSP 1703 (T10) | h90 ura4-D18 his3-D1 arg3-D4 anon::ura4,42-8 |
| WSP 1704 (T11) | h90 ura4-D18 his3-D1 arg3-D4 scm3::ura4 |
| WSP 1705 (T12) | h90 ura4-D18 his3-D1 arg3-D4 anon::ura4,42-36 |
| WSP 1706 (T13) | h90 ura4-D18 his3-D1 arg3-D4 scm4::ura4 |
| WSP 1707 (T14) | h90 ura4-D18 his3-D1 arg3-D4 anon::ura4,43-16 |
| WSP 1708 (T15) | h90 ura4-D18 his3-D1 arg3-D4 anon::ura4,47-1 |
| WSP 1709 (T16) | h90 ura4-D18 his3-D1 arg3-D4 scm5::ura4 |
| WSP 1710 (T17) | h90 ura4-D18 his3-D1 arg3-D4 anon::ura4,48-27 |
| WSP 1711 (T18) | h90 ura4-D18 his3-D1 arg3-D4 anon::ura4,52-12 |
| WSP 1712 (T19) | h90 ura4-D18 his3-D1 arg3-D4 anon::ura4,53-23 |
| WSP 1713 (T20) | h90 ura4-D18 his3-D1 arg3-D4 anon::ura4,59-42 |
| WSP 1714 (T21) | h90 ura4-D18 his3-D1 arg3-D4 anon::ura4,60-35 |
| WSP 1715 (T22) | h90 ura4-D18 his3-D1 arg3-D4 anon::ura4,68-40 |
| WSP 1813 | h− ura4-D18 ade6-M26 rec12-D13::ura4 |
| WSP 1827 | h+ ura4-D18 ade6-M210 rec12-D13::ura4 |
| WSP 1890 | h+ ura4-D18 arg3-D4 ade6-M210 scm1::ura4 |
| WSP 1892 | h− ura4-D18 arg3-D4 ade6-M26 scm1::ura4 |
| WSP 1902 | h+ ura4-D18 arg3-D4 ade6-M210 scm2::ura4 |
| WSP 1904 | h− ura4-D18 arg3-D4 ade6-M26 scm2::ura4 |
| WSP 1914 | h+ ura4-D18 arg3-D4 ade6-M210 scm3::ura4 |
| WSP 1916 | h− ura4-D18 arg3-D4 ade6-M26 scm3::ura4 |
| WSP 1925 | h+ ura4-D18 arg3-D4 ade6-M210 scm4::ura4 |
| WSP 1927 | h− ura4-D18 arg3-D4 ade6-M26 scm4::ura4 |
| WSP 1936 | h+ ura4-D18 arg3-D4 ade6-M210 scm5::ura4 |
| WSP 1938 | h− ura4-D18 arg3-D4 ade6-M26 scm5::ura4 |
Transformation
Transformations contained 1 × 108 cells, 1 μg of linear ura4+ DNA (see below) and, where indicated, 50 μg of sheared, denatured salmon sperm DNA. Lithium acetate transformation was as described elsewhere (22) using lithium acetate solutions at pH 4.9 or 7.5. Electroporation was as described elsewhere (23,24) using microcuvettes and (Bio-Rad, Hercules, CA) electroporator settings of 1.5 kV, 200 Ω, 25 μF. Transformation mixtures were plated on NBA supplemented with histidine and arginine and incubated for 3–4 days at 32°C.
Assay for stable transformation
Individual Ura+ transformants were streak-purified serially one time with selection (NBA lacking Uracil) and 3–5 times on nonselective media. They were then either streaked directly onto YEA containing 5′-FOA or patched onto YEA and replica plated onto YEA–FOA. Positive control (ura4+) and stable transformants yielded few (≤10−4) FOAr colonies; negative control (ura4-D18) and unstable transformants produced many FOAr colonies.
Preparation of genomic DNA
Cells were grown in 10 ml of YEL to a density of 1 × 107 cells/ml and were harvested by centrifugation. Cell pellets were resuspended in 0.5 ml of disruption buffer (10 mM Tris–HCl, pH 8.0, 100 mM NaCl, 1 mM EDTA, 1% SDS, 2% Triton X-100) and were placed in screw-cap tubes containing 0.65 ml of acid-washed glass beads (0.5 mm in diameter) and 0.5 ml of phenol. Cells were disrupted by beating for 1 min at medium speed in a Beadbeater 8 (Biospec Products, Bartlesville, OK). The aqueous phases were subjected to sequential extraction two times each with equal volumes of phenol, phenol–chloroform and choloroform, and nucleic acids were recovered by ethanol precipitation. Pellets were dissolved in 50 μl of TE (10 mM Tris–HCl, 1 mM EDTA, pH 7.5) containing 100 μg/ml of RNase A. Typical yields were ≥1 μg of DNA per ml of culture with DNA fragment sizes in the range of 7–15 kbp.
PCR, inverse-PCR and real-time quantitative PCR
The ura4+ primer sequences for PCR are listed in Table 2. Standard reactions (25) using primers ura4-PR3 and ura4-PR4 were used to amplify a 1663 bp fragment from pURA4 template (15). Products were purified by serial extraction with phenol, phenol–chloroform and chloroform; they were recovered by ethanol precipitation and were dissolved in TE at a final concentration of 1 μg/μl.
Table 2. ura4+ primer sequences.
| Name | Sequence |
|---|---|
| ura4-PR3 | 5′-ACGTACAAATCCCACTG-3′ |
| ura4-PR4 | 5′-ATTTTACATTCATCTACATACA-3′ |
| ura4-PR6 | 5′-TTCTTCATTAAGTAACAAA-3′ |
| ura4-PR7 | 5′-TATGTACAAAGCCAATGAAAGATG-3′ |
| ura4-PR8 | 5′-TGCCAAAAATTACACAAGATAGAA-3′ |
| ura4-PR9 | 5′-GACATTGAATAAGAAAAGAGTGAA-3′ |
| ura4-PR11 | 5′-ATAGATAAACACCTTGGGAATA-3′ |
Ligation-mediated, inverse-PCR reactions were essentially as described elsewhere (26). Genomic DNA samples (1 μg) were digested to completion with one of three restriction enzymes (HaeII, HpaII or EcoR1), were recovered by extraction and ethanol precipitation, and were resuspended in TE. Samples were treated with T4 DNA ligase according to the instructions of the manufacturer (Promega, Madison, WI) at a DNA concentration of 1 μg/ml to favor intramolecular ligation. Samples were heated to 65°C for 15 min to inactivate the DNA ligase. Inverse-PCR reactions were in 25 μl and contained 5 ng DNA template, 5 pM primers (ura4-PR7/8, −11/8, or −9/8), and 0.2 mM dNTPs. DNA was denatured at 94°C for 5 min and subjected to 25 cycles of PCR amplification (94°C for 30 sec, 55°C for 1 min, 72°C for 1 min). Products were analyzed on ethidium bromide-stained 1.5% agarose gels. For subsequent analyses, bands of interest were cut out of the gels and DNA was recovered using a QIAquick gel extraction kit (Qiagen, Valencia, CA).
To determine ura4+ copy number, genomic DNA was subjected to real-time, quantitative PCR using TaqMan technology and an Applied Biosystems Inc. PRISM 7700 fluorometric thermal cycler using established methods (27). Primers and probes were designed using the Primer Express software (version 1.5; Applied Biosystems, Foster City, CA). The gene names, forward and reverse primers, and TaqMan probe sequences are provided in Table 3. For each reaction, ura4+ signal strengths were normalized to those of a single-copy, internal control gene, cam1+. To facilitate comparison, the copy numbers of ura4+ transgenes are presented relative to that of a strain harboring a single copy of ura4+ at its endogenous locus.
Table 3. Primer and probe sequences used for real-time, quantitative-PCR (TaqMan).
| Gene | Forward primer | Reverse primer | TaqMan probe |
|---|---|---|---|
| cam1+ | 5′-CGATGGTAATGGCACAATTGAT-3′ | 5′-TTCGTTGTCGGTATCCTTCATTT-3′ | 5′-6FAM-TACCGAATTTTTGACTATGATGGCCCGA-TAMRA-3′ |
| ura4+ | 5′-TTGTAAACTCGGTAGCGATATCA-3′ | 5′-GGCTTCGACAACAGGATTACG-3′ | 5′-6FAM-TTGTTGGTCGTGGAGTCTATGGAGCTGG-TAMRA-3′ |
Screen for scm mutants
The primary screen was adapted to a 96-well microtiter plate format. Individual transformant colonies (∼3 mm in diameter) were transferred with sterile toothpicks into wells containing 200 μl of supplemented SPL. Cultures were incubated for 5–7 days at 25°C to allow self-mating and sporulation. Ten microliters of a 10% solution of glusulase (NEN, Boston, MA) were added to each well and the plates were incubated at 32°C to digest any asci that were still intact. Ninety microliters of ethanol were added to each well and the plates were incubated at 22°C for 30 min to kill the remaining vegetative cells (19). Spore suspensions were diluted by transferring 20 μl of each sample into 180 μl of H2O in a fresh microtiter plate. Thirty miroliters of each suspension (∼250 spores) were then spotted onto YEA-B. After 3 days' incubation at 32°C, the plates were examined. Candidates that produced either an elevated frequency of diploid (dark pink) spore colonies or a low viable titer were analyzed further. They were streak-purified (with selection) and tested for stability of the Ura+ phenotype as described above. Stable candidates were subjected to a secondary, cytological screen for chromosome segregation defects. Asci from meiotic cultures were fixed with 70% ethanol at −20°C for 15 min, were washed with H2O, and were stained with 4,6-diamidino-2-phenylindole (DAPI) at a final concentration of 1 μg/ml. Cells were examined by differential interference contrast (DIC) and fluorescence (DAPI) microscopy with a Zeiss Axiophot microscope (Carl Zeiss, Thornwood, NY). Images were analyzed using the MetaMorph software package (Universal Imaging, West Chester, PA).
RESULTS
Production of stable transformants by nonhomologous integration
The strategy for insertional mutagenesis is depicted in Figure 1. We transformed a 1.7 kbp long, linear DNA fragment containing the S.pombe ura4+ gene into homothallic (self-mating) cells harboring a 1.8 kbp deletion of the ura4 locus (ura4-D18) (15). Since there was no extensive homology between the ura4+ targeting fragment and sequences within the genome, and since the ura4+ fragment lacked an origin of replication and telomeres, we expected stable prototrophic Ura+ transformants to arise by nonhomologous integration of the transforming DNA (Figure 1).
Figure 1.
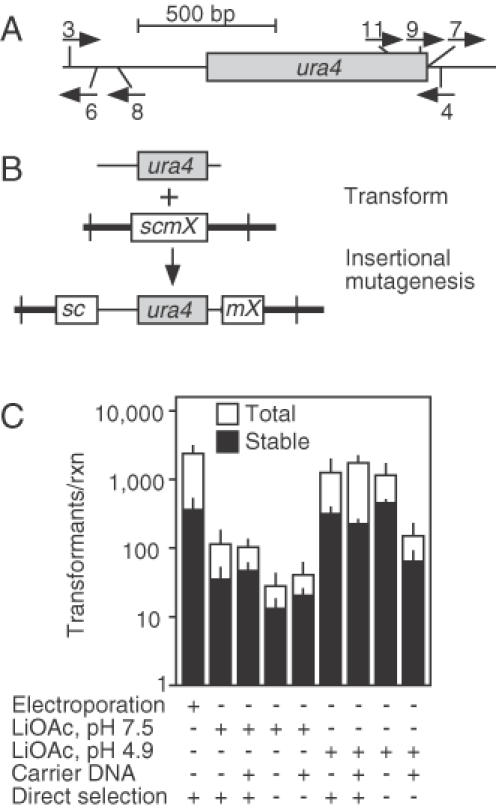
Insertional mutagenesis. (A) Schematic diagram of the ura4+ gene showing the coding region (box) and locations of the oligonucleotide primers (arrows). Oligonucleotide primers 3 and 4 were used to generate a 1.7 kbp long, linear ura4+ DNA fragment that was used for transformation. The other primers were used for subsequent analysis. (B) Transformation of a ura4-D18 (deletion) strain with linear ura4+ DNA leads to nonhomologous integration and insertional mutagenesis; stable Ura+ transformants can be screened for those which confer the desired mutant phenotype. (C) Effect of the transformation procedure on nonhomologous integration frequency. Shown are the total frequencies of Uracil prototrophic colonies (open bars) and the frequencies of stable transformants (filled bars). Data are mean ± standard deviation from three experiments.
Practical application of insertional mutagenesis requires efficient integration of the transforming DNA into the genome, so we first examined parameters affecting both the overall frequency of transformation and the frequency of stable transformation (Figure 1C). These included the protocol for delivery of DNA into cells (electroporation versus two chemical methods) and the presence or absence of carrier DNA (denatured salmon sperm DNA). Because ura4+ DNA might enter some cells and transiently confer a Ura+ phenotype without actually integrating into the chromosomes, we also tested whether a period of growth on nonselective medium prior to selection would increase the relative frequency of stable transformants.
Transformation by each of the protocols generated colonies which fell into two classes. Between 80 and 95% of the Ura+ colonies were small and, invariably, cells from those colonies lost the Ura+ phenotype upon subsequent culture in nonselective conditions. We infer that those Ura+ colonies arose from cells that had taken up ura4+ DNA but failed to integrate the ura4+ fragment into the genome. Between 5 and 20% of the colonies were larger, ranging in size from 2 to 4 mm in diameter after 4 days of selection. Among those larger colonies, roughly half were unstable for the Ura+ phenotype after subsequent culture in nonselective conditions. The remaining colonies maintained their Ura+ phenotype after a period of nonselective culture, suggesting that their respective founder cells were transformed stably with ura4+ DNA.
The pH 4.9 lithium acetate and electroporation methods produced the highest frequencies of transformants and the efficiency of transformation was unaffected by the presence of carrier DNA (Figure 1C). The relative efficiency of stable transformation was not significantly increased by an initial growth period in the absence of selection (Figure 1C), suggesting that the number of cell divisions before selection was inadequate to dilute out extrachromosomal ura4+ fragments. Based upon these findings, we chose to use electroporation, without carrier DNA, and direct selection for insertional mutagenesis.
Identification of mutants with aberrant chromosome segregation during meiosis
The rationale for the genetic screen (Figure 2) is based on the fact that mutants of fission yeast with aberrant meiotic chromosome segregation produce spores among which the frequency of viability is reduced a few-fold and the frequency of diploidy is increased markedly (8,9). Approximately 2000 individual, Ura+ transformant colonies were inoculated into microtiter wells containing SPL. The SPL cultures were incubated at room temperature for 5–7 days. Under these conditions, the cells self-mated, passed through meiosis, and produced spores. The self-mating of each transformant rendered any ura4+ insertion loci homozygous, thereby facilitating identification of recessive mutations.
Figure 2.
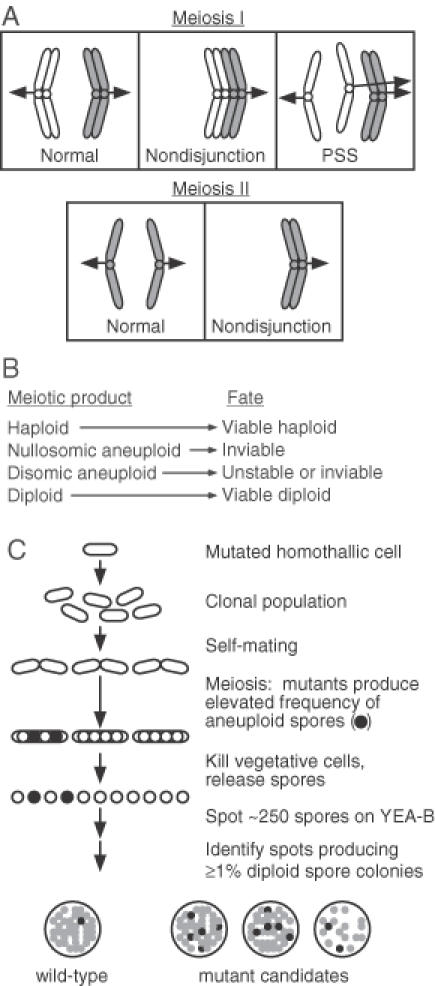
Rationale for scm mutant screen. (A) Segregation of chromosomes in meiosis. For the sake of clarity, the behavior of a single pair of homologous chromosomes is displayed. Nondisjunction during meiosis I, precocious separation of sister chromatids (PSS) in meiosis I, and meiosis II nondisjunction can produce at least one meiotic product that receives more than one copy of the chromosome (e.g. disomic). (B) Fates of the meiotic products in S.pombe. Aneuploid spores that receive at least one copy of each chromosome, including diploids, can be viable. (C) Rationale for mutant screening. Individual transformants were screened for those that produced a reduced frequency of spore viability or an elevated frequency of diploid spore colonies after self-mating, which renders the insertional mutations homozygous and permits the identification of recessive mutations.
Individual spore suspensions were diluted and ∼250 spores from each self-mating were spotted onto rich medium containing phloxin-B (YEA-B). After incubation at 32°C for 3–4 days, the spots were examined for growth and for the production of darker-staining papillae indicative of diploid spore colonies. From this primary screen, 45 candidates were identified as having either a reduced viable spore titer or as producing an elevated frequency of aneuploid (phloxin-B-staining) spore colonies relative to those of wild-type cells.
The 45 candidates that passed the primary screen were tested for stability of the Ura+ phenotype. Twenty-three of the candidates did not exhibit a stable Ura+ phenotype and were set aside. The remaining 22 stable candidates were subjected to a secondary, cytological screen for meiotic chromosome segregation defects. A rec12 (spo11) meiotic recombination mutant, which is achiasmatic and exhibits meiotic chromosome segregation defects (5,6), was included as a control.
Asci were stained with the DNA-specific stain DAPI and were examined by differential interference contrast and fluorescence microscopy. The parental, wild-type control cells produced predominantly four-spored asci with equal DNA content in each of the four spores (Figure 3). In contrast, the majority of the asci produced by the rec12 mutant controls had an aberrant distribution of DNA and some asci contained fewer than four spores (Figure 3). Of the 22 mutant candidates from the primary screen, 17 exhibited ascus phenotypes indistinguishable from that of wild-type cells. The remaining five candidates, however, produced phenotypes very similar to those of the rec12 mutant controls—the DNA distribution was clearly aberrant and some asci contained fewer than four spores (Figure 3). The frequencies of aberrant asci produced by those five mutants (21–68%) were similar to the frequency for rec12 mutant controls (60%) (Figure 4A). We conclude that each of those five transformants (T4, T8, T11, T13 and T16) harbors one or more insertions of ura4+ DNA that affect meiotic chromosome segregation.
Figure 3.
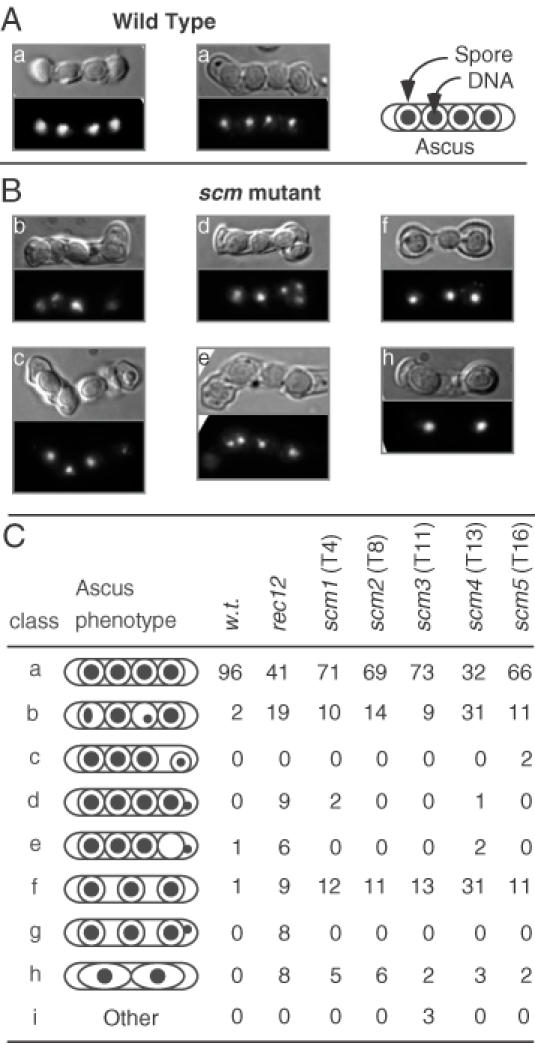
Cytological analysis of meiotic chromosome segregation and ascus formation. Representative asci from wild-type (A) and scm mutant (B) strains. Differential interference contrast microscopy was used to analyze spore and ascus morphology (upper panel) and DAPI fluorescence microscopy revealed the distribution of DNA within each ascus (lower panel). Mutant phenotypes include unequal partitioning of chromosomes (b, c, e), failure of DNA to enclose within spores (d, e) and aberrant (c, d) or missing (f, h) spores. (C) Schematic representation of the ascus phenotypes. The outer oval depicts the ascus; the inner circles and ovals depict the spore walls; the filled circles depict the location and relative amount of DNA.
Figure 4.
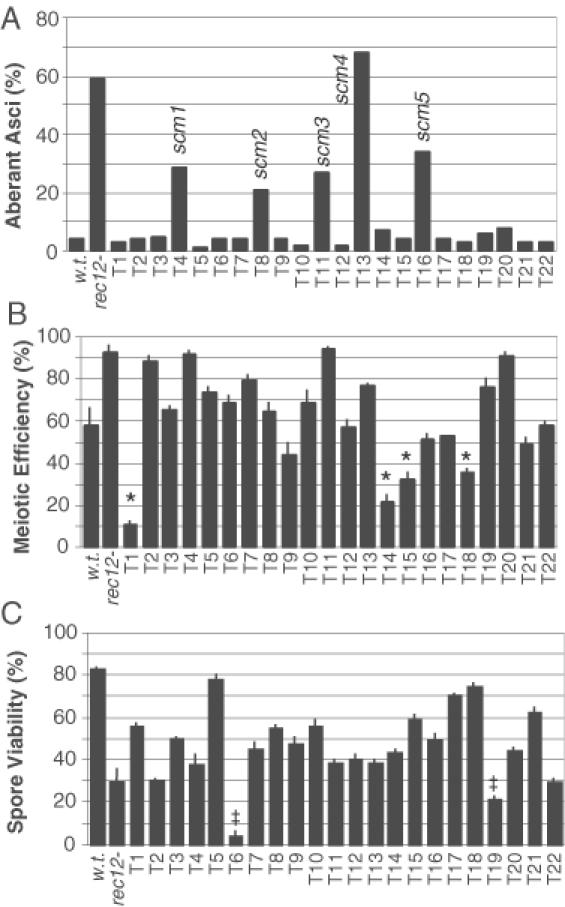
Meiotic phenotypes of stable transformants that passed the primary screen. Wild-type (WSP628), rec12 mutant (WSP1240) and candidate mutant transformants (T1–T22) were each scored for production of aberrant asci (A), proficiency of mating and meiosis (B) and frequency of viable spores (C). One hundred asci were scored for each histogram bar in panel ‘A’, data in panels ‘B’ and ‘C’ are the mean ± standard deviation of data from three separate experiments. Mutant classes include those with defects in meiotic chromosome segregation (scm), those with defects in mating and/or meiotic progression (*) and those with defects in spore viability (‡).
The mutant phenotypes cosegregate with single-locus ura4+ transgenes
The homothallic mutants were auxotrophic for histidine, but prototrophic for uracil due to the ura4+ transgene(s) (h90 ura4-D18 his3-D1 arg3-D4 anon::ura4+). The mutants were each crossed to heterothallic strains that were His+ but Ura− (h+ ura4-D18 arg3-D4 his3+ ade6-M210). Spores were plated on medium lacking histidine to select against products of homothallic self-mating. Subsequently, the segregation patterns of various markers were determined.
Crosses with the mutant T8 produced many His+ spore colonies, but <0.5% of those colonies were also Ura+. We conclude that the site of integration of the ura4+ transgene is within ∼0.5 cM (28) of the his3 locus on Chromosome II and that there are no additional, unlinked copies of ura4+ elsewhere in the T8 genome.
In crosses with the mutant T11, the ura4+ marker segregated independently of the his3 marker. However, only 24% of the Ura+ colonies were mating type h+, suggesting that the site of integration of the ura4+ transgene is ∼33 cM (28) from the mating type locus mat1 on Chromosome II. These data suggest, but do not prove, that there is a single-locus integration of the ura4+ transgene in T11. (Two copies of the ura4+ transgene, one linked to mat1 and one unlinked to mat1, would give a similar segregation pattern.)
To determine whether or not the mutant phenotype cosegregated with a single-locus ura4+ transgene in each mutant, we examined microscopically the meiotic phenotypes of between 10 and 20 homothallic Ura+ derivatives of each outcross. For each mutant, including T11, each Ura+ derivative exhibited the aberrant ascus phenotype characteristic of its respective parental candidate mutant. Similarly, each Ura− segregant analyzed had a wild-type phenotype. Thus, with resolution of ∼5 cM, the ura4+ transgenes are linked to single-locus mutations affecting meiotic chromosome behavior. We therefore classify the mutants as scm mutants.
The linkage analyses indicated that there were at least three distinct scm genes: one linked to his3, one linked to mat1 and at least one additional gene (three mutations) unlinked to his3 or mat1. To determine whether the remaining mutations were allelic or resided in separate genes, we conducted complementation studies. Heterothallic derivatives of each scm mutant were mated to those of the other scm mutants and asci were examined for the presence or absence of the scm phenotype (Table 4). For each intercross, full complementation was observed, demonstrating that the respective mutations are recessive and define five complementation groups. This indicates that the insertional mutagenesis had identified five separate scm genes, which we name scm1, scm2, scm3, scm4 and scm5.
Table 4. Complementation analysis of scm mutations.
| Frequency of aberrant asci from cross (%) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Genotype | WSP # | Wild-type 0607 | rec121813 | scm11892 | scm21904 | scm31916 | scm41927 | scm51938 |
| Wild-type | 0620 | 2 | 3 | 1 | 4 | 6 | 3 | 2 |
| rec12 | 1827 | 4 | 54 | 3 | 5 | 3 | 6 | 2 |
| scm1 | 1890 | 2 | 5 | 32 | 0 | 1 | 3 | 1 |
| scm2 | 1902 | 1 | 3 | 2 | 26 | 2 | 1 | 1 |
| scm3 | 1914 | 1 | 3 | 3 | 1 | 29 | 6 | 0 |
| scm4 | 1925 | 1 | 4 | 3 | 2 | 3 | 48 | 5 |
| scm5 | 1936 | 3 | 2 | 2 | 0 | 1 | 0 | 35 |
Haploid strains of mating type h− (top row) and mating type h+ (left column) were mixed and plated on sporulation agar to induce mating, meiosis and ascus formation. At least 100 asci from each cross were examined by DIC and DAPI microscopy as in Figure 3 to determine the frequency of asci with aberrant chromosome partitioning.
The scm mutants have aberrant chromosome segregation and compromised spore viability but are proficient for meiosis and meiotic recombination
All 22 stable candidates from the primary screen, including the 5 scm mutants, were tested for their ability to undergo sexual development and produce viable spores. The rec12 mutant, which is proficient for mating and meiotic progression but produces spores with reduced viability (6,29), was included as a control.
The five scm mutants were each as proficient for meiosis as wild-type cells (Figure 4B). We conclude that their mutations have little or no effect upon mating type switching, the mating pathway or meiotic progression. However, like the rec12 mutant, each of the scm mutants exhibited a modest but significant decrease in spore viability relative to wild-type cells (Figure 4C). The viable spore frequencies ranged from 38 to 56%, which is close to the frequency of viable spores expected (42%) following aberrant (random) segregation of three pairs of chromosomes in one of the meiotic divisions (6).
The five scm gene products could ensure faithful chromosome segregation by affecting recombination or by affecting other processes. We therefore compared the frequencies of heteroallelic meiotic recombination in the mutants to those of rec12 null mutant and wild-type cells (Table 5). Four of the scm mutants (scm1–scm4) produced recombinant frequencies that were indistinguishable statistically from that of the wild-type cells. The recombinant frequency for mutant scm5 was significantly higher (2.5-fold) than that for wild-type cells. We conclude that the chromosome segregation defects of the mutants are not caused by deficiencies in recombination.
Table 5. Effect of scm mutations on homologous recombination in meiosis.
| Relevant genotype | Strains crossed | Ade+ recombinant frequency (×106) |
|---|---|---|
| Wild-type | WSP 0607 × WSP 0620 | 44 ± 17 |
| rec12 | WSP 1813 × WSP 1827 | 0.07 ± 0.06 |
| scm1 | WSP 1890 × WSP 1892 | 38 ± 8.7 |
| scm2 | WSP 1902 × WSP 1904 | 53 ± 9.7 |
| scm3 | WSP 1914 × WSP 1916 | 50 ± 10 |
| scm4 | WSP 1925 × WSP 1927 | 68 ± 32 |
| scm5 | WSP 1936 × WSP 1938 | 106 ± 11 |
Recombination between the ade6-M26 and ade6-M210 heteroalleles was assayed by plating spores on complete minimal medium and minimal medium lacking adenine. The recombinant frequency is the titer of adenine-prototrophic spore colonies divided by the titer of viable spore colonies. Data are mean ± standard deviation from three separate experiments.
Although our principal goal was to identify scm mutants, some of the other mutants are worth noting. Mutants T1, T14, T15 and T18 exhibited a significant decrease in meiotic proficiency and produced a lower spore yield (Figure 4B). Mutants T6 and T19 were proficient for mating and meiosis (Figure 4B) but produced spores with significantly reduced viability (Figure 4C). All six of these candidates produced normal-looking asci with DNA distributions indistinguishable from those of wild-type control asci (Figures 3, 4A). The first four mutations likely affect some feature of the mating pathway or meiotic progression, and the latter two mutations likely affect some aspect of spore viability unrelated to DNA content.
The ura4+ transgene integrates in concatomeric arrays at a single locus in each transformant
To determine the organization of the ura4+ transgenes, genomic DNA was cleaved with a restriction endonuclease that does not cut within the ura4+ DNA fragment used for transformation. Southern blotting was performed using ura4+ DNA as the probe. In most transformants, a single hybridizing band was observed (Figure 5). DNA from scm3 mutant T11 produced initially two hybridization signals (Figure 5), but upon retesting a single band was produced (data not shown), indicating that the genomic DNA was not digested to completion in the first sample. Genomic DNA digested individually with each of three different restriction enzymes produced a single ura4-hybridizing band for each transformant (data not shown). We conclude that each transformant harbored a ura4+ transgene at a single locus in its genome.
Figure 5.
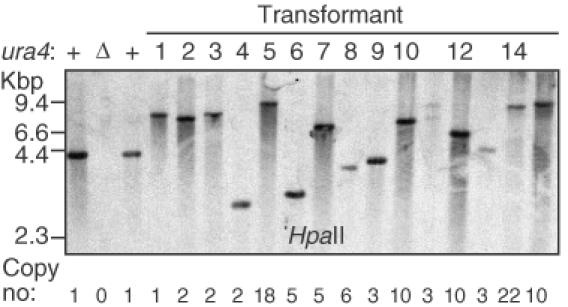
Single-locus integration of ura4+ DNA. Genomic DNA from stable transformants was digested with HpaII restriction endonuclease (4 bp recognition site) and analyzed by Southern blotting using ura4+ DNA as the probe. Controls harbored a single, wild-type copy (+) or complete deletion (Δ) of the endogenous ura4+ locus. The copy number of a DNA sequence within the ura4+ open reading frame was determined by quantitative-PCR. (See text and Table 6 for additional details.)
Because each transformant contained a single transgenic locus, one should be able to identify the sites of integration of each ura4+ transgene by using inverse-PCR and DNA sequencing (Figure 6A) (18,26). Genomic DNA samples were therefore digested with a restriction enzyme that does not cut within ura4+, were diluted, and were treated with DNA ligase under conditions that favor intramolecular ligation. The samples were then subjected to PCR with primer pairs pointed outward from the ura4+ DNA fragment (Figure 6A). Inverse-PCR products were readily obtained from 19 of the 22 samples, but rather than containing DNA molecules of one length, the products contained a mixture of DNA molecules of multiple lengths (Figure 6B). The complex patterns were observed when DNA was digested with each of three different restriction enzymes prior to ligation and PCR; they were present when each of three different ura4+ primer pairs was used and were reproducible under various experimental conditions routinely employed to optimize the fidelity of PCR (data not shown). Thus, the complex patterns of PCR products likely stem from intrinsic complexity of the ura4+ transgene loci themselves.
Figure 6.
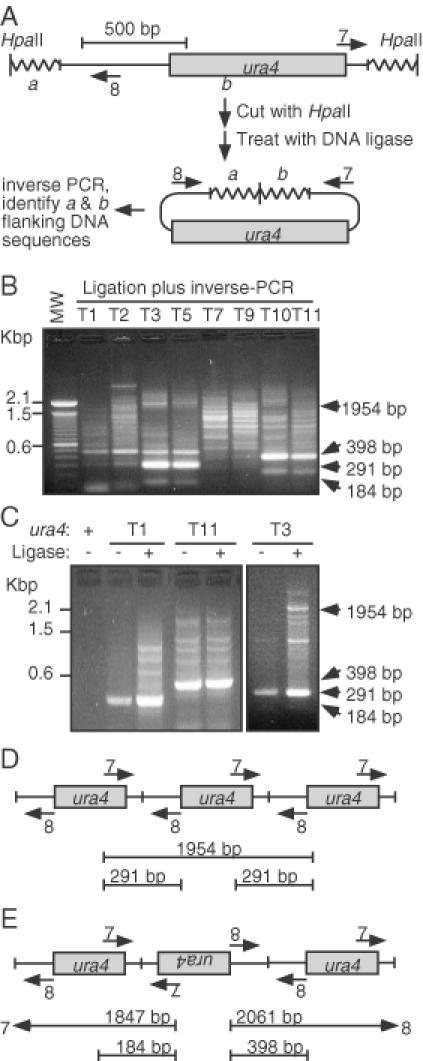
Complex structures of ura4+ transgenes. (A) Expected transgene structure and inverse-PCR method to identify its location. Genomic DNA (wavy line) from each transformant was expected to harbor a single copy of the ura4+ transgene. In that case, inverse-PCR would produce a single DNA product that could be sequenced to identify the genomic locus into which the transgene had inserted. (B) Complex structure of transgenes. Genomic DNA was digested with HpaII, treated with DNA ligase, subjected to inverse-PCR using primers in panel ‘A’, and analyzed using agarose gel electrophoresis. (C) Analysis of junction fragments. Inverse-PCR was performed using samples with or without prior treatment with DNA ligase. Ligase-independent bands of characteristic size are diagnostic for different types of concatomeric fusions. (D) Schematic diagram of unit-length head-to-tail concatomers and diagnostic inverse-PCR product sizes. (E) Schematic diagram of head-to-head and tail-to-tail concatomers and diagnostic inverse-PCR product sizes. Product sizes such as those in (D) would also be generated.
Several of the PCR product band sizes suggested that the transforming ura4+ DNA fragment became concatomeric (Figure 6B and C). The predominant bands of ∼291 sbp, ∼398 bp and ∼184 bp are consistent with the presence of head-to-tail, head-to-head and tail-to-tail fusions, respectively (Figure 6D and E). Notably, these smaller fragments were also generated in PCR reactions using genomic DNA that had not been treated with DNA ligase and they required the presence of the ura4+ transgenes (Figure 6C), confirming that concatomeric junctions were produced by transformation.
The Southern blot analysis employed restriction enzymes with 4 bp recognition sites that should statistically cleave within flanking DNA close to the ends of the inserted ura4+ transgenes. However, some of the hybridizing DNA fragments were significantly larger than expected for single-copy integrants (Figure 5), also suggesting that the transformants harbored tandem insertions of ura4+. We therefore used real-time, quantitative PCR (27) to determine the copy number of ura4+ relative to that of a single-copy gene, cam1+. Transformants produced up to 22 times the amount of ura4+ signal as a control (single-copy) strain (Table 6), demonstrating the presence of multiple, tandem copies of ura4+ DNA at the individual transgene loci.
Table 6. Copy number of single-locus ura4 transgenes.
| Strain | ura4+ copy numbera |
|---|---|
| WSP 0002 (ura4+)b | 1.0 |
| WSP 0571 (ura4-D18)c | 0.0 |
| WSP 1694 (T1)d | 1.0 |
| WSP 1695 (T2) | 1.6 |
| WSP 1696 (T3) | 1.8 |
| WSP 1697 (T4/scm1) | 2.4 |
| WSP 1698 (T5) | 17.9 |
| WSP 1699 (T6) | 5.2 |
| WSP 1700 (T7) | 5.0 |
| WSP 1701 (T8/scm2) | 6.2 |
| WSP 1702 (T9) | 2.5 |
| WSP 1703 (T10) | 10.3 |
| WSP 1704 (T11/scm3) | 2.8 |
| WSP 1705 (T12) | 5.4 |
| WSP 1706 (T13/scm4) | 3.1 |
| WSP 1707 (T14) | 21.6 |
| WSP 1708 (T15) | 9.3 |
| WSP 1709 (T16/scm5) | 4.9 |
| WSP 1710 (T17) | 10.4 |
| WSP 1711 (T18) | 3.0 |
| WSP 1712 (T19) | 1.2 |
| WSP 1713 (T20) | 5.7 |
| WSP 1714 (T21) | 1.3 |
| WSP 1715 (T22) | 1.6 |
| Average | 5.6 |
aReal-time PCR was used to determine ura4+ copy number relative to single-copy, internal control gene cam1+. Data were normalized to those for control strain WSP 0002.
bControl strain harboring a single copy of ura4+ at its endogenous locus.
cControl strain harboring deletion (ura4-D18) of the ura4+ locus.
dTransformant (T) number and scm gene names used in the text and figure labels.
For about half of the mutants, Southern blot analysis (Figure 5) revealed a DNA fragment of inadequate size to harbor the number of ura4+ transgenes detected by real-time PCR (Table 6), if each of those transgenes was full-length. For example, the T4/scm1, T6 and T8/scm2 mutants contained as many as six copies of the internal DNA sequence from ura4+, but produced hybridization signals in the 2–4 kbp range. (The ura4+ DNA fragment used for transformation was 1.7 kbp in length, so the expected minimum size for six copies of ura4+ would be ∼10 kbp.) Furthermore, DNA from those three mutants failed to generate PCR products using terminal outward-pointing and inward-pointing primers (data not shown). Those transformants contained multiple copies of internal ura4+ DNA (Table 6), but apparently lacked primer binding sites located toward the ends of the ura4+ DNA. We infer that the individual ura4+ repeats within those three tandem arrays had each lost 275 bp or more of terminal DNA.
Variable-length resection of DNA ends prior to concatomerization was also evident in all other transformants based on two criteria. First, for the diagnostic junction fragments, many transformants produced inverse-PCR products shorter than would be generated by templates with simple head-to-tail, head-to-head and tail-to-tail fusions (Figure 6B and C). Second, for all transformants that produced inverse-PCR products larger than those produced by junctions, most of the products deviated in size from the expected sizes of integer-length concatomers (Figure 6B and C).
To further characterize the variable-length resection of DNA ends, we purified individual DNA bands from ligation inverse-PCR reactions, reamplified them by PCR, and used nested oligoncleotide primers for DNA sequence analysis. Upon DNA sequencing, each of the products yielded information from the position of the sequencing primer to a variable distance toward the end of the ura4+ DNA fragment used for transformation (data not shown). This confirmed variable-length truncation of the ends of the individual ura4+ fragments in concatomeric arrays. After that point, the DNA sequence either became degenerate or jumped to a terminal or internal position of another copy of ura4+. This outcome, which is the expected result of concatomerization of ura4+ fragments with variably truncated ends, precluded the identification of the target genomic DNA sequences into which the ura4+ transgenes had integrated.
DISCUSSION
The goal of this study was to develop a novel type of genetic screen to identify mutations that affect meiotic chromosome dynamics. As part of that goal, we sought improved methods for insertional mutagenesis. We report that electroporation of linear ura4+ DNA into S.pombe in which the endogenous ura4+ locus has been deleted generates stable transformants (Figure 1C) in which the transgenes are integrated at a single locus in each transformant (Figure 5). This is in good agreement with studies that used chemical transformation in the presence of carrier DNA (17,18).
Individual transformants were subjected to a two-step screen for phenotypes associated with missegregation of chromosomes in meiosis (Figure 2) (6,8,9). Self-mating was used to render mutations homozygous, thereby permitting identification of recessive mutations. The primary screen sought transformants that produced either a decreased frequency of viable spores or an elevated frequency of diploid spores. Candidates that passed the primary screen were subjected to a secondary, cytological screen for aberrant partitioning of chromosomes in meiosis.
About half of all Ura+ transformants (i.e. prior to screening) were unstable for the Ura+ phenotype (Figure 1C), presumably because the ura4+ DNA fragments did not become incorporated into the genome and were diluted out by successive rounds of cell division. Because it was not practical to test thousands of colonies for stability prior to screening for meiotic phenotypes, individual transformants were subjected to the primary screen without prior knowledge of whether the Ura+ phenotype was due to extrachromosomal or intrachromosomal copies of ura4+ DNA. Candidates passing the primary screen were then tested for stability. As was the case for transformants prior to screening, about half (23 of 45) of the candidates from the primary screen were unstable transformants. This outcome was expected because the primary screen allowed analysis of many transformants, but (as such screens often do) it had an intrinsic false-positive rate. The false positives were identified, and eliminated from further consideration, by secondary screening.
Secondary screening and subsequent analysis of additional phenotypes placed the 22 stable candidates from the primary screen into four classes, each of which was expected to be found. Class I had two members (T6 and T19) which were proficient for mating and meiosis and had no obvious defects in chromosome segregation but produced a high frequency of inviable spores. The four members of class II (T1, T14, T15 and T18) exhibited a significant decrease in meiotic proficiency and hence a lower spore yield, but among those asci that were formed, chromosome partitioning was apparently normal. Transformants of class I and class II were expected because the primary screen sought candidates which produced a lower titer of viable spores (per unit volume) than wild-type cells. There were five class III transformants (T4/scm1, T8/scm2, T11/scm3, T13/scm4 and T16/scm5) which displayed the desired mutant phenotype. These mutants were proficient for mating and meiosis but exhibited significant defects in meiotic chromosome segregation and slightly reduced spore viability. Class IV transformants (11 isolates) were ‘weeds’ which failed to pass any of the secondary screens. These were expected based upon the normal distribution (Poisson) because the primary screen was a pseudo-quantitative assessment that scored candidates as ‘positive’ if they differed a few-fold in value from wild-type cells (see Materials and Methods).
Previous screens for meiotic hyporecombination mutants of fission yeast led to the discovery of meiotic cohesins (e.g. Rec8) and the key meiotic recombinase Rec12 (Spo11) (6–9,19,29,30). For this reason, and because crossover recombination structures (chiasmata) have an important role in aligning bivalents on the metaphase plate of meiosis I (1), most mutations known to affect meiotic chromosome segregation also affect recombination (31). We reasoned that there are likely many processes other than pairing and recombination that are required for proper segregation of chromosomes in meiosis. Our approach therefore sought missegregation mutants without regard to recombination status, and the five scm mutants obtained have wild-type (or higher) levels of recombination, suggesting that they define a largely unexplored class of genes required for meiotic chromosome dynamics.
For this study our goal was to analyze a number of mutations that would correspond to hits at 10% of the coding regions in the genome. The S.pombe genome has an average gene density of 0.56 and contains 4824 coding regions (32). We therefore set a lower limit for our analysis at 861 stable transformants. Because some of the transformants were unstable, we increased proportionately the total number of Ura+ colonies to be screened. Five scm mutants were obtained and their respective recessive mutations defined five complementation groups. Based upon the numbers of transformants screened and number of mutants obtained, a saturating screen could identify ∼50 genes of this class and, as described above, many of those genes would likely affect processes other than cohesion, pairing and recombination.
Because the ura4+ DNA was located at a single locus in each transformant, we expected that we could easily use inverse-PCR and DNA sequencing to identify the mutated loci (Figure 6A) (18,26). However, none of the mutants yielded data on DNA sequence flanking the integrated transgene, despite extensive efforts to optimize conditions for inverse-PCR. We therefore applied a variety of techniques to reveal the molecular basis for this finding.
Of the 22 ura4+ transgene loci that we examined 19 harbored concatomeric arrays with variable-length truncations of repeats and the remaining 3 loci were refractory to inverse-PCR (Figure 6). At least two of those three refractory loci also harbored concatomers, because there were multiple copies of internal ura4+ DNA (Table 6) at a single site in the genome of each respective mutant (Figure 5). We conclude that electroporation-mediated, nonhomologous integration of DNA in fission yeast is accompanied by exonucleolytic processing and end-joining.
Transformant T1 was unique in that it had experienced end-joining (Figure 6) but harbored only a single copy of the small region of ura4+ (within the open reading frame) that was used for quantitative PCR (Table 6). This suggested that intramolecular ligation might have generated a circular DNA molecule that integrated by a single-exchange, nonhomologous recombination mechanism. If so, transformation with nonreplicating circular DNA might prove useful to generate single- copy insertion events. However, when we tested this hypothesis, we found that circular DNA molecules transformed with <1% of the efficiency of linear DNA molecules (data not shown). Thus, while we cannot disprove that T1 arose by integration of circular DNA, the efficiency of such transformation is too low to be of utility for insertional mutagenesis.
Insertion points of fission yeast transgenes generated by chemical transformation have been identified by inverse-PCR (18). This would seem to differ from our results following electroporation, where all informative transformants harbored concatomeric transgenes that are refractory to inverse-PCR. However, in the chemical method, transformants that failed to generate inverse-PCR products or that gave complicated patterns of inverse-PCR products were also observed, but their frequencies were not reported and those transformants were set aside without further characterization (18) (G. Chua and P. Young, personal communication). Others studies have also reported that transformants generated by the chemical method are refractory to analysis by inverse-PCR (33), but such failures are, understandably, seldom reported in the literature. We therefore used email to contact members of the fission yeast community in order to share our findings, to ask whether anyone had similar difficulties, and to seek advice. All respondents reported encountering difficulty when attempting to use inverse-PCR to identify transgene integration sites. Thus, available evidence suggests that transgenic loci of fission yeast are often refractory to analysis by inverse-PCR, regardless of the method used for transformation. Our finding that the transforming DNA forms concatomers explains why inverse-PCR often fails. This urges caution in the design of experiments using insertional mutagenesis—alternative approaches might be configured in such a way as to reduce concatomerization during transformation or to permit recovery of unique DNA flanking concatomeric transgenes (17).
CONCLUSION
Mutagenesis of S.pombe by electroporation-mediated, nonhomologous integration of a dominant, selectable marker gene is a useful tool for generating single-locus mutations linked to the transforming DNA. A direct, two-step screen revealed five complementation groups of mutations (scm1–scm5) that affect segregation of chromosomes in meiosis, suggesting that a saturating screen of this type could identify ∼50 scm loci. The utility of the mutagenesis approach is mitigated by the formation of concatomers of the transforming DNA, which renders mutant loci refractory to inverse-PCR to identify the genes of interest.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Charla Wiley and Audrey Stone for laboratory assistance; Charlie Hoffman, Paul Young, Gordon Chau, Gerry Smith and other members of the fission yeast community for sharing unpublished data; and our colleagues for helpful discussions. Work in our laboratory is supported by funds from the National Institute of General Medical Sciences at the National Institutes of Health (GM62244 and GM62801).
REFERENCES
- 1.Page S.L. and Hawley,R.S. (2003) Chromosome choreography: the meiotic ballet. Science, 301, 785–789. [DOI] [PubMed] [Google Scholar]
- 2.Buonomo S.B., Clyne,R.K., Fuchs,J., Loidl,J., Uhlmann,F. and Nasmyth,K. (2000) Disjunction of homologous chromosomes in meiosis I depends on proteolytic cleavage of the meiotic cohesin Rec8 by separin. Cell, 103, 387–398. [DOI] [PubMed] [Google Scholar]
- 3.Rabitsch K.P., Gregan,J., Schleiffer,A., Javerzat,J.P., Eisenhaber,F. and Nasmyth,K. (2004) Two fission yeast homologs of Drosophila Mei-S332 are required for chromosome segregation during meiosis I and II. Curr. Biol., 14, 287–301. [DOI] [PubMed] [Google Scholar]
- 4.Kitajima T.S., Kawashima,S.A. and Watanabe,Y. (2004) The conserved kinetochore protein shugoshin protects centromeric cohesion during meiosis. Nature, 427, 510–517. [DOI] [PubMed] [Google Scholar]
- 5.Davis L. and Smith,G.R. (2003) Nonrandom homolog segregation at meiosis I in Schizosaccharomyces pombe mutants lacking recombination. Genetics, 163, 857–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sharif W.D., Glick,G.G., Davidson,M.K. and Wahls,W.P. (2002) Distinct functions of S.pombe Rec12 (Spo11) protein and Rec12-dependent crossover recombination (chiasmata) in meiosis I; and a requirement for Rec12 in meiosis II. Cell Chromosome, 1, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Watanabe Y. and Nurse,P. (1999) Cohesin Rec8 is required for reductional chromosome segregation at meiosis. Nature, 400, 461–464. [DOI] [PubMed] [Google Scholar]
- 8.Molnar M., Bahler,J., Sipiczki,M. and Kohli,J. (1995) The rec8 gene of Schizosaccharomyces pombe is involved in linear element formation, chromosome pairing and sister-chromatid cohesion during meiosis. Genetics, 141, 61–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Krawchuk M.D., DeVeaux,L.C. and Wahls,W.P. (1999) Meiotic chromosome dynamics dependent upon the rec8+, rec10+, and rec11+ genes of the fission yeast Schizosaccharomyces pombe. Genetics, 153, 57–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Parisi S., McKay,M.J., Molnar,M., Thompson,M.A., van der Spek,P.J., van Drunen-Schoenmaker,E., Kanaar,R., Lehmann,E., Hoeijmakers,J.H.J. and Kohli,J. (1999) Rec8p, a meiotic recombination and sister chromatid cohesion phosphoprotein of the Rad21p family, conserved from fission yeast to humans. Mol. Cell. Biol., 19, 3515–3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bhatt A.M., Lister,C., Page,T., Fransz,P., Findlay,K., Jones,G.H., Dickinson,H.G. and Dean,C. (1999) The DIF1 gene of Arabidopsis is required for meiotic chromosome segregation and belongs to the REC8/RAD21 cohesin gene family. Plant J., 19, 463–472. [DOI] [PubMed] [Google Scholar]
- 12.Klein F., Mahr,P., Galova,M., Buonomo,S.B., Michaelis,C., Nairz,K. and Nasmyth,K. (1999) A central role for cohesins in sister chromatid cohesion, formation of axial elements, and recombination during yeast meiosis. Cell, 98, 91–103. [DOI] [PubMed] [Google Scholar]
- 13.Klapholz S. and Esposito,R.E. (1982) A new mapping method employing a meiotic rec-mutant of yeast. Genetics, 100, 387–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Molnar M., Parisi,S., Kakihara,Y., Nojima,H., Yamamoto,A., Hiraoka,Y., Bozsik,A., Sipiczki,M. and Kohli,J. (2001) Characterization of rec7, an early meiotic recombination gene in Schizosaccharomyces pombe. Genetics, 157, 519–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grimm C., Kohli,J., Murray,J. and Maundrell,K. (1988) Genetic engineering of Schizosaccharomyces pombe: a system for gene disruption and replacement using the ura4 gene as a selectable marker. Mol. Gen. Genet., 215, 81–86. [DOI] [PubMed] [Google Scholar]
- 16.Grallert B., Nurse,P. and Patterson,T.E. (1993) A study of integrative transformation in Schizosaccharomyces pombe. Mol. Gen. Genet., 238, 26–32. [DOI] [PubMed] [Google Scholar]
- 17.Hoffman C.S. and Welton,R. (2000) Mutagenesis and gene cloning in Schizosaccharomyces pombe using nonhomologous plasmid integration and rescue. Biotechniques, 28, 532–536. [DOI] [PubMed] [Google Scholar]
- 18.Chua G., Taricani,L., Stangle,W. and Young,P.G. (2000) Insertional mutagenesis based on illegitimate recombination in Schizosaccharomyces pombe. Nucleic Acids Res., 28, e53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ponticelli A.S. and Smith,G.R. (1989) Meiotic recombination-deficient mutants of Schizosaccharomyces pombe. Genetics, 123, 45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gutz H., Heslot,H., Leupold,U. and Loprieno,N. (1974) Schizosaccharomyces pombe. In King,R.C. (ed.), Handbook of Genetics. Plenum Press, New York, Vol. 1, pp. 395–446. [Google Scholar]
- 21.Kon N., Schroeder,S.C., Krawchuk,M.D. and Wahls,W.P. (1998) Regulation of the Mts1-Mts2-dependent ade6-M26 meiotic recombination hotspot and developmental decisions by the Spc1 mitogen-activated protein kinase of fission yeast. Mol. Cell. Biol., 18, 7575–7583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Okazaki K., Okazaki,N., Kume,K., Jinno,S., Tanaka,K. and Okayama,H. (1990) High-frequency transformation method and library transducing vectors for cloning mammalian cDNAs by trans-complementation of Schizosaccharomyces pombe. Nucleic Acids Res., 18, 6485–6489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Suga M. and Hatakeyama,T. (2003) High-efficiency electroporation by freezing intact yeast cells with addition of calcium. Curr. Genet., 43, 206–211. [DOI] [PubMed] [Google Scholar]
- 24.Hood M.T. and Stachow,C.S. (1995) Electroporation of Schizosaccharomyces pombe. Methods Mol. Biol., 47, 273–278. [DOI] [PubMed] [Google Scholar]
- 25.Innis M.R. and Gelfand,D.H. (1990) Optimization of PCRs. In Innis,M.A., Gelfand,D.H., Sninsky,J.J., and White,T.J. (eds), PCR Protocols: A Guide to Methods and Applications. Academic Press, Inc., San Diego, pp. 3–12. [Google Scholar]
- 26.Ochman H., Medhora,M., Garza,D. and Hartl,D.L. (1990) Amplification of flanking sequences by inverse PCR. In Innis,M.A., Gelfand,D.H., Sninsky,J.J., and White,T.J. (eds), PCR Protocols: A Guide to Methods and Applications. Academic Press, San Diego, pp. 219–227. [Google Scholar]
- 27.Giulietti A., Overbergh,L., Valckx,D., Decallonne,B., Bouillon,R. and Mathieu,C. (2001) An overview of real-time quantitative PCR: applications to quantify cytokine gene expression. Methods, 25, 386–401. [DOI] [PubMed] [Google Scholar]
- 28.Haldane J.B.S. (1919) The combination of linkage values, and the calculation of distances between loci of linked factors. J. Genet., 8, 299–309. [Google Scholar]
- 29.DeVeaux L.C., Hoagland,N.A. and Smith,G.R. (1992) Seventeen complementation groups of mutations decreasing meiotic recombination in Schizosaccharomyces pombe. Genetics, 130, 251–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krawchuk M.D. and Wahls,W.P. (1999) Centromere mapping functions for aneuploid meiotic products: analysis of rec8, rec10, and rec11 mutants of the fission yeast Schizosaccharomyces pombe. Genetics, 153, 49–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Davis L. and Smith,G.R. (2001) Meiotic recombination and chromosome segregation in Schizosaccharomyces pombe. Proc. Natl Acad. Sci. USA, 98, 8395–8402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wood V., Gwilliam,R., Rajandream,M.A., Lyne,M., Lyne,R., Stewart,A., Sgouros,J., Peat,N., Hayles,J., Baker,S. et al. (2002) The genome sequence of Schizosaccharomyces pombe. Nature, 415, 871–880. [DOI] [PubMed] [Google Scholar]
- 33.Rodriguez-Gabriel M.A., Burns,G., McDonald,W.H., Martin,V., Yates,J.R., 3rd, Bahler,J. and Russell,P. (2003) RNA-binding protein Csx1 mediates global control of gene expression in response to oxidative stress. EMBO J., 22, 6256–6266. [DOI] [PMC free article] [PubMed] [Google Scholar]