


Abstract
One of the largest families of small RNAs in eukaryotes is the H/ACA small nucleolar RNAs (snoRNAs), most of which guide RNA pseudouridine formation. So far, an effective computational method specifically for identifying H/ACA snoRNA gene sequences has not been established. We have developed snoGPS, a program for computationally screening genomic sequences for H/ACA guide snoRNAs. The program implements a deterministic screening algorithm combined with a probabilistic model to score gene candidates. We report here the results of testing snoGPS on the budding yeast Saccharomyces cerevisiae. Six candidate snoRNAs were verified as novel RNA transcripts, and five of these were verified as guides for pseudouridine formation at specific sites in ribosomal RNA. We also predicted 14 new base-pairings between snoRNAs and known pseudouridine sites in S.cerevisiae rRNA, 12 of which were verified by gene disruption and loss of the cognate pseudouridine site. Our findings include the first prediction and verification of snoRNAs that guide pseudouridine modification at more than two sites. With this work, 41 of the 44 known pseudouridine modifications in S.cerevisiae rRNA have been linked with a verified snoRNA, providing the most complete accounting of the H/ACA snoRNAs that guide pseudouridylation in any species.
INTRODUCTION
The small nucleolar RNAs (snoRNAs) define one of the largest families of small non-coding RNAs known in eukaryotes. A homologous class of RNAs (sRNAs) exists in archaeal organisms as well [see reviews (1–3)]. Divided by sequence and secondary structure motifs into the box C/D and box H/ACA families, most snoRNAs serve as guide RNAs in 2′-O-ribose methylation or pseudouridylation of specific nucleotides of ribosomal RNA (rRNA) and other RNAs, including spliceosomal small nuclear RNAs (snRNAs). A small number of C/D and H/ACA snoRNAs also play essential roles in the cleavage of precursor rRNA (1,2). In all cases, the snoRNAs function as part of a snoRNA–ribonucleoprotein complex (snoRNP) and are associated with four core proteins.
For the C/D guide snoRNAs, the presence of relatively well-conserved box motifs and 10–21 nt complementary guide sequences has enabled the development of a successful computational screen (4). As a result, it has been possible to determine the nearly complete complement of C/D guide snoRNAs in the budding yeast Saccharomyces cerevisiae, as well as hundreds of C/D snoRNA-like genes in archaeal species (5,6). Moreover, it has been possible to associate all but four ribose methylations in ribosomal RNA in S.cerevisiae with a guide snoRNA, and to show that, with one possible exception, each C/D box snoRNA targets at most two sites of ribosomal methylation; yeast U24 may guide three sites of which two are adjacent (7).
In contrast to the C/D guide snoRNAs, the H/ACA guide snoRNAs have shorter and less well-conserved primary sequence motifs, making the development of an effective computational screen for H/ACA snoRNAs and their associated pseudouridylation (Ψ) sites significantly more difficult. One screening strategy has been reported, although its candidates have not been tested experimentally (8). Because simple, comprehensive experimental means for identifying these snoRNAs are also lacking, H/ACA guide snoRNAs remain hidden in most genomes, even where complete genomic sequences and modification maps exist. In order to address this, we have developed a computational screen for this class of snoRNAs and have demonstrated its effectiveness on the yeast genome.
In S.cerevisiae, there are 44 known rRNA Ψ sites (9,10). Although pseudouridylation can occur by RNA-independent mechanisms, we hypothesize that, like ribose methylation of rRNA (4), most or all Ψ modifications require snoRNA cofactors as guides. When we began our studies, only 27 sites had been linked to 22 known S.cerevisiae H/ACA snoRNAs (1,9). If all 44 Ψs in yeast rRNA were guided by H/ACA snoRNAs, as many as 17 additional guide snoRNAs would remain to be identified. The suggestion that numerous H/ACA snoRNAs remained to be detected in S.cerevisiae was supported by the observation that fractionation of total small RNA from this yeast revealed upwards of 50–60 species in the size range of ∼70–330 nt (11). Moreover, nearly all of the 33 species examined by sequencing in this study were snoRNAs, including 20 H/ACA species (11). Thus, despite the availability of the complete genome sequence for yeast over the last eight years, it appeared likely at the beginning of this investigation that numerous snoRNAs remained to be discovered.
A number of H/ACA snoRNAs have also been identified in other eukaryotes by experimental screens (12). Homologous sRNAs have been discovered in archaeal organisms as well (13). Such experimental screens are labor-intensive and costly, and consequently are likely to be performed only for a limited number of model organisms. These experimental approaches also tend to favor discovery of the most abundant RNAs so that species of lower abundance may not be detected. Another experimental challenge is defining Ψ modifications in RNA transcripts. Information about the sites of Ψ modification in rRNA and other RNAs is only available for a few organisms (9), and mapping new sites biochemically is time-consuming (14). Identifying new guide snoRNAs greatly facilitates the identification of previously unknown Ψ sites with which they interact. For these reasons, there is an important need for computational screening methods to guide experimental efforts to identify H/ACA guide RNAs and their sites of Ψ modification.
Here, we report the computational identification and experimental verification of H/ACA snoRNAs that guide pseudouridylation in S.cerevisiae. We have developed a program that screens genomes for candidate guide snoRNAs called ‘snoGPS’ (for ‘snoRNAs-Guiding-Pseudouridylation Scanner’). The program implements a combination of a deterministic search algorithm and a probabilistic gene model of H/ACA snoRNA primary sequence and secondary structure, trained on known H/ACA guide snoRNAs. Primary sequence motifs, rRNA guide sequences, stem–loop structures, and interval spacing between the different motifs are all used within the model. In the present work, we used snoGPS, in conjunction with comparative sequence analysis and Gibbs free-energy-minimization calculations to select 17 candidate genomic regions for experimental study. From our computationally identified candidates, we confirmed six new members of the H/ACA class of RNAs, including five that were experimentally shown to guide pseudouridylation at specific sites in rRNA. We also established assignments of guide snoRNAs to 14 of the known Ψs in S.cerevisiae rRNA. With this work, guide RNAs have been assigned to 41 of the 44 known pseudouridylation modifications in yeast rRNA.
MATERIALS AND METHODS
Data sources
Saccharomyces cerevisiae sequence data were taken from the Stanford Saccharomyces Genome Database (SGD) (15). Searches were performed against either the entire S.cerevisiae genome or against the SGD ‘Not-Feature’ component created by removing all open reading frames, known RNAs and other annotated sequences from the genome. Specific searches of S.cerevisiae intron sequences were also performed using sequence data from the yeast intron database (16). Sequence data for five other Saccharomyces genomes (S.bayanus, S.castellii, S.kluyveri, S.kudriavzevii and S.mikatae) were taken from unannotated genomic-sequence files provided by Cliften et al. (17). Sequence and annotation data for the known snoRNAs (which were used in program training) were initially taken from the University of Massachusetts snoRNA database (18). Data on additionally verified H/ACA snoRNAs was added to the training data during the course of the project, as they became available, either from the literature (19,20) or from newly verified genes in the present study.
Algorithm description
The snoGPS program employs a deterministic search algorithm and a probabilistic gene model to search for RNA genes with weakly conserved primary and secondary structure motifs [see (21) for a review of deterministic and probabilistic sequence models]. The hybrid nature of the program is intended to combine the efficiency of deterministic algorithms with the sensitivity of probabilistic models to detect multiple weakly conserved motifs. In practice, the program generally executes in two stages. An initial series of deterministic tests limits the potential search space, enumerating all possible features for a given candidate. The second phase consists of scoring routines that measure how similar the identified features are to those of a training set of known RNAs. Summing of the component scores in the framework of a probabilistic model gives a final bit score used to rank candidates.
snoGPS can be configured by means of user-specified ‘descriptor files’. The descriptor files are similar in spirit to those found in RNAMOT (22) and several similar RNA-motif-searching programs (23,24). However, in contrast to most of these programs, snoGPS is designed to facilitate the incorporation of probabilistic scoring matrices for any of the specified motifs. The user can also specify a target file consisting of short ‘target sequences’ to be used by the program (e.g. when searching for Ψ guide snoRNAs, these are the sequences immediately flanking the Ψ in the target RNA). In this way, a single invocation of the program can search for guide snoRNAs for multiple Ψ sites.
The model of H/ACA snoRNA genes used by snoGPS is based on the canonical H/ACA structure shown in Figure 1A. The actual tests performed by snoGPS are shown in the schematic diagram in Figure 1B. Most of our genomic screens utilized a descriptor file based on the entire two-hairpin molecule shown in Figure 1A and including all tests shown in Figure 1B (the ‘two-stem scanner’). An alternative descriptor file implementing a search for one half of the snoRNA molecule in Figure 1A and tests 1 through 9 of Figure 1B (the ‘one-stem scanner’) was used during the initial screen and for sites where the two-stem scanner did not score any candidates above our threshold. (A variant of the one-stem scanner specific for the 3′ half of the snoRNA structure and executing tests 1 though 7 followed by tests 11, 22 and 9 was also implemented. However, it was rarely used since it has low sensitivity for snoRNAs with guide sequences in 5′ hairpins.)
Figure 1.
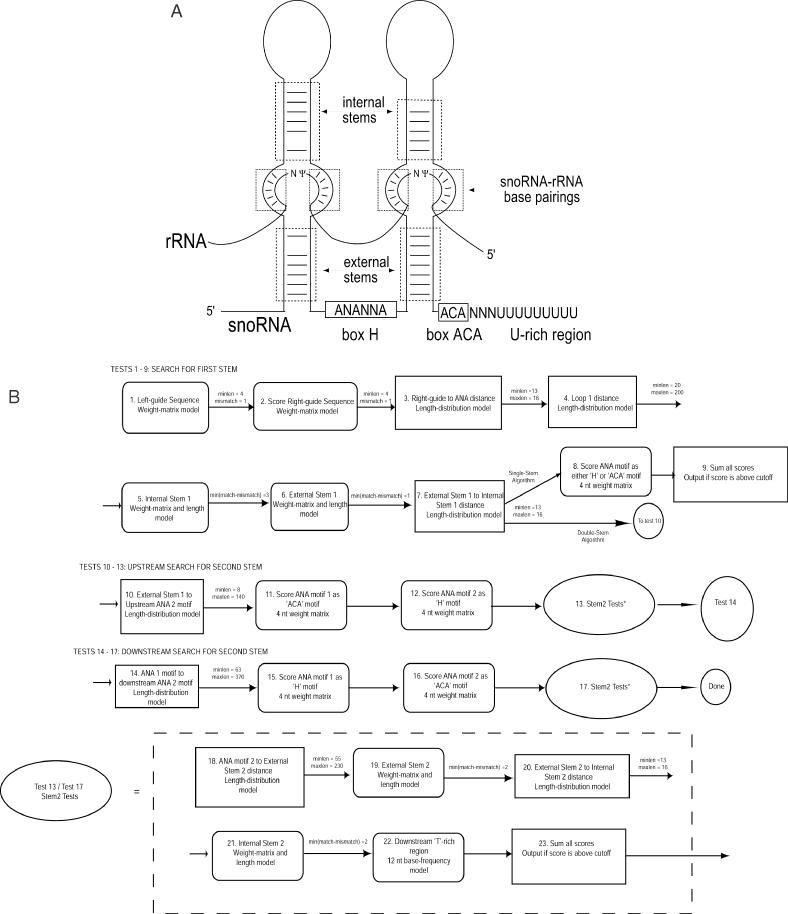
H/ACA model and snoGPS algorithm. (A) Schematic diagram of a consensus H/ACA snoRNA. A snoRNA in which both guide regions interact with a substrate RNA is shown. The classic H/ACA snoRNA sequence motifs are indicated, including left and right guide sequences, ‘H’ and ‘ACA’ boxes, 5′ and 3′ stems and the downstream U-rich region. The U-rich region is not part of the fully processed snoRNA. The model on which the ‘one-stem’ version of snoGPS is based consists of a single helix–bulge–helix stem with one pair of guide elements. (B) Schematic representation of the one-stem and two-stem snoGPS algorithms. Rectangles indicate length distribution tests. Rounded rectangles indicate nucleotide weight-matrix tests. For computational efficiency, tests are not necessarily computed in the order shown. The one-stem algorithm consists of tests 1–9. For the two-stem algorithm, snoGPS first searches for the hairpin structure containing the rRNA-guide sequences (tests 1–7). Then it searches both upstream (tests 10–13) and downstream (tests 14–17) for the second hairpin structure. To speed up the search, some tests have ranges on the allowable test results (shown to the right of the corresponding test). ‘minlen’ and ‘maxlen’ indicate the minimum and maximum allowed values for a length test. ‘Mismatch’ is the maximum allowed number of mismatches for a guide-region matching test and ‘min(match-mismatch)’ is the minimum excess of matches (Watson–Crick or G–U) over mismatches for a stem. If the score for a candidate is outside the allowed range, the sequence is rejected and the remaining tests are not executed.
snoGPS also utilizes a set of score tables created by a separate program that uses sequences of the known H/ACA snoRNAs as training data. For each feature or motif, a corresponding score table is created by calculating a log-odds ratio of scores from two models: one that describes H/ACA snoRNAs and a null model that uses the background nucleotide composition of the S.cerevisiae genome (21).
Comparative genome analyses
As the study progressed, all high-scoring candidates were further screened using a BLASTn (NCBI Stand-Alone BLAST, version 2.2.6) (25) similarity search of the other five Saccharomyces genomes. For these BLASTn searches, default parameters were used except for wordsize, ‘W = 8’ and expected value, ‘e = (1 × 10−4)’. In addition to screening on the basis of simple overall sequence similarity, the strongest BLAST hits for each candidate were aligned using T-Coffee (version 1.37) (26), and the alignments were annotated using the snoGPS output. The annotated alignments were manually evaluated and compensatory changes that preserved the H/ACA secondary structure (as occurs in the known snoRNAs) were taken as additional evidence that a candidate snoRNA was a true positive.
Free-energy calculations
Minimum Gibb's free-energy values were calculated for candidate hits using the mfold program (version 3.1) with default program values (27). Minimum free-energy (MFE) values were divided by the length of the candidate sequence to remove biases toward longer candidates. A cutoff score was determined by comparing the MFEs of known snoRNAs with those of the top 100 snoGPS ‘hits’ found in 36 million base pairs (36 Mb) of random sequence. Candidates with MFEs-per-nucleotide significantly outside the range of values from the known H/ACA snoRNAs were removed from the candidate list.
Algorithm assessment
Algorithm sensitivity was initially assessed by testing the program on all of the sites targeted by the previously known snoRNAs. We performed cross-validation tests of the probabilistic parameters used in the score tables, by means of separate searches for each known snoRNA, using training data derived from all other snoRNA sequences [i.e. ‘leave-one-out’ testing (28,29)]. However, since the overall algorithm utilized information from all the known snoRNAs, this approach cannot be considered as a genuine cross-validation test. Ultimately, we assessed the sensitivity of snoGPS on the basis of our experimental tests and none of our conclusions hinge on cross-validation. Program specificity was assessed by searching random sequence generated with a fifth-order Markov model (21) of S.cerevisiae, which uses hexanucleotide frequencies in the genome. The random data experiments were carried out using 36 Mb of sequence (corresponding to three complete S.cerevisiae genomes), and the experiments were repeated using all 44 Saccharomyces Ψ sites to determine whether the random-sequence test scores were target-dependent.
Experimental verification
Initial experimental verification of candidate RNAs was performed with northern analysis, by probing total small RNA. Total RNA was isolated from strains grown to mid-log phase in rich media using a hot phenol method as described previously (30). Total RNA (10 μg) was separated on a denaturing polyacrylamide gel (7 M urea, 1× TBE), electrophoretically transferred onto a nylon membrane (Hybond-N; Amersham) and cross-linked to the membranes with ultraviolet light. The membranes were treated for 1 h at 37°C in the formamide buffer [50% (w/v) formamide, 5× SSC, 5× Denhardts, 5 mM disodium EDTA, 0.5% SDS, 250 μg/ml herring sperm DNA and 50 mM Tris–HCl, pH 8.0]. Membranes were probed overnight in formamide buffer at 42°C with internally labeled PCR products corresponding to the genome regions as follows: snR80(chrV:52057-52775), snR81(chrXV:233743-234566), snR82(chrVII:316607-317187), snR83(chrXIII:626422-626743), snR84(chrIV:1492605-1492976) and snR85(chrXIII:67699-68228). Each membrane was also incubated with 5′ end-labeled snR64 oligo 5′-ATGTTCCTCGTCACTTGAGAATCTGTTGTC. Post-hybridization washes were done at 42°C; twice with 2× SSC and once with 0.1× SSC/0.1% SDS. Hybridization patterns were determined using a PhosphorImager (Molecular Dynamics). For 5′ end mapping, total RNA (20 μg) was precipitated with 106 c.p.m. of each 5′ end-labeled oligo. Samples were dissolved in distilled water and heat-denatured. Reverse transcription reactions were carried out in a final volume of 20 μl for 45 min at 42°C with the following components: 1× First-strand buffer, 0.01 M DTT, 2.5 mM dNTPs, 20 U RNaseIN (Promega) and 100 U Superscript RT (Invitrogen). Samples were treated with RNAseA, phenol:chloroform:isoamyl alcohol extracted, ethanol precipitated and half of the reaction mixture loaded onto a 6% sequencing gel (7 M urea, 1× TBE). A mapping ladder was generated with dideoxy-sequencing reactions using the primers indicated above.
Candidate sequences were evaluated for Ψ modification activity by disrupting the RNA coding sequence and screening for target rRNA modification using a primer extension assay. Strains corresponding to knockouts of the known snoRNAs were described previously (31). The strain YRP1145 for RNA161, which was formerly suggested to be an H/ACA snoRNA (20), was kindly provided by Roy Parker. All other strains were produced for the present work by PCR-mediated gene disruption (32) in the yeast strain YS602 (MAT:α ade2-101 his3-11,15 trp1Δ901 ura3-52 leu2-3,112). In each case, the PCR product used in the gene disruption was the Kluyveromyces lactis TRP1-marker gene flanked by ∼50 bp upstream and downstream of the genomic region of interest. To produce the PCR products, the template was pBS1479 (33) and two primers of ∼70 nt each were used. The 3′ ends of each set of primers were constant: 5′-TGATATCGAATTCCTGC-3′ for the upstream primer and 5′-TACGACTCACTATAGGG-3′ for the downstream primer.
Further information including source code, a sample descriptor file and details of the snoGPS implementation, experimental procedures and primer sequences can be found in the Supplementary Material.
RESULTS
A computational screen can detect H/ACA snoRNAs
We sought to determine the sensitivity of snoGPS by searching for known H/ACA snoRNAs with the algorithm depicted in Figure 1B. This algorithm takes into account the following features: the ability of the rRNA guide sequence(s) to base pair with target RNAs; the stem–loop structure of the 5′ and 3′ hairpins; similarity of the H and ACA box motifs to those in known H/ACA snoRNAs; nucleotide distances between features; and the number of thymine residues in the 12 nt region immediately 3′ to the candidate sequence (11). All of the 22 known snoRNAs except snR30 were included in the training set for these tests (snR30 was excluded because, at the time, it had no known or predicted Ψ target).
snoGPS ranked 10 of the training set snoRNA sequences as having the highest score in the entire S.cerevisiae genome for at least one of its target Ψ sites (Table 1). Six others had among the four highest scores in the genome for at least one site (Table 1). Two more (snR10 and snR34) had scores among the top six in the intergenic region of the genome (data not shown), so that, in all, 18 of the 21 training set snoRNAs scored among the top six hits in the intergenic region for at least one target.
Table 1. Cross-validated snoGPS scores of all H/ACA snoRNAs using the two-stem model.
| snoRNA | Target site with highest rank | Scorea | Rank | BLAST homologyb | E/base |
|---|---|---|---|---|---|
| (A) Previously known | |||||
| snR3 | LSU-2263 | 62.5 | 1 | 2 | −0.28 |
| snR5 | LSU-1123 | 44.0 | 1 | 3 | −0.18 |
| snR8 | LSU-959 | 29.6 | >20 | 5 | −0.32 |
| snR9 | LSU-2339 | 23.1 | >20 | 3 | −0.21 |
| snR10 | LSU-2919 | 36.5 | 10 | 5 | −0.28 |
| snR11 | LSU-2415 | 41.0 | 1 | 4 | −0.26 |
| snR30c | LSU-1109 | 24.5 | >20 | 5 | −0.32 |
| snR31 | SSU-1000 | 51.2 | 1 | 4 | −0.24 |
| snR32 | LSU-2190 | 38.9 | 2 | 4 | −0.25 |
| snR33 | LSU-1041 | 38.0 | 2 | 3 | −0.26 |
| snR34 | LSU-2876 | 34.9 | 16 | 5 | −0.26 |
| snR35 | SSU-1189 | 46.4 | 1 | 3 | −0.31 |
| snR36 | SSU-1185 | 22.6 | >20 | 4 | −0.22 |
| snR37 | LSU-2940 | 37.6 | 3 | 4 | −0.28 |
| snR42 | LSU-2971 | 49.5 | 1 | 5 | −0.31 |
| snR43 | LSU-965 | 40.0 | 2 | 3 | −0.23 |
| snR44 | SSU-106 | 36.2 | 4 | 5 | −0.25 |
| snR46 | LSU-2861 | 52.8 | 2 | 3 | −0.24 |
| snR49 | LSU-989 | 39.2 | 1 | 4 | −0.28 |
| snR161c | SSU-766 | 48.8 | 1 | 3 | −0.30 |
| snR189 | LSU-2730 | 48.5 | 1 | 3 | −0.21 |
| snR191c | LSU-2257 | 49.8 | 1 | 3 | −0.27 |
| (B) Newly identified | |||||
| snR80 | LSU-775 | 37.9 | 3 | 4 | −0.15 |
| snR81 | LSU-1051 | 49.3 | 1 | 2 | −0.28 |
| snR82 | LSU-2350 | 39.8 | 2 | 3 | −0.19 |
| snR83 | LSU-1289 | 38.0 | 1 | 4 | −0.23 |
| snR84 | LSU-2265 | 42.7 | 1 | 3 | −0.23 |
| snR85 | SSU-1179 | 28.8 | >20 | 3 | −0.21 |
For each snoRNA, the Ψ site which ranked the highest (by score) in the entire S.cerevisiae genome is indicated, as well as the score and the ranking. The table also lists the number of other Saccharomyces species with putative homologs to the snoRNA as well as the calculated minimum Gibbs-free energy of the snoRNA in kcal/mol/nt, denoted as ‘E/base’.
aCross-validation score.
bNumber of other Saccharomyces species with BLAST hit to snoRNA with BLAST E-score < 10−4.
csnoRNAs not included in initial training set. snR30 was not included in the training set because no target site was known until the present work was almost complete. snR161 and snR191 were not initially in the training set because they are not included in the snoRNA database (18). After they were independently identified in the present study, they were added to the training set.
Only snR8, snR9 and snR36 received lower rankings, in each case because of the presence of an insertion in one hairpin. Interestingly, two of these (snR36 and snR8) were found to have homologous sequences in S.castellii that received snoGPS scores of 36.8 and 33.7, respectively (data not shown), significantly higher than their S.cerevisiae scores of 22.6 and 29.6. This observation led us to expand our direct screening for new snoRNAs from S.cerevisiae to all six of the Saccharomyces genomes.
We tested the specificity of snoGPS using randomly generated sequence as a negative dataset. These experiments allowed us to develop general expectations for the rate of false positives at various score thresholds (Figure 2A). From the figure one sees that for a typical randomly generated genome sequence and Ψ target site, the highest two-stem model snoGPS score was ∼36.5. Very little target dependence was observed among these scores which ranged from 35.5 to 37.5. This result, combined with the observation that most of the training set snoRNAs had scores greater than 36 bits, suggested using a threshold of 36 bits when searching for new snoRNAs.
Figure 2.
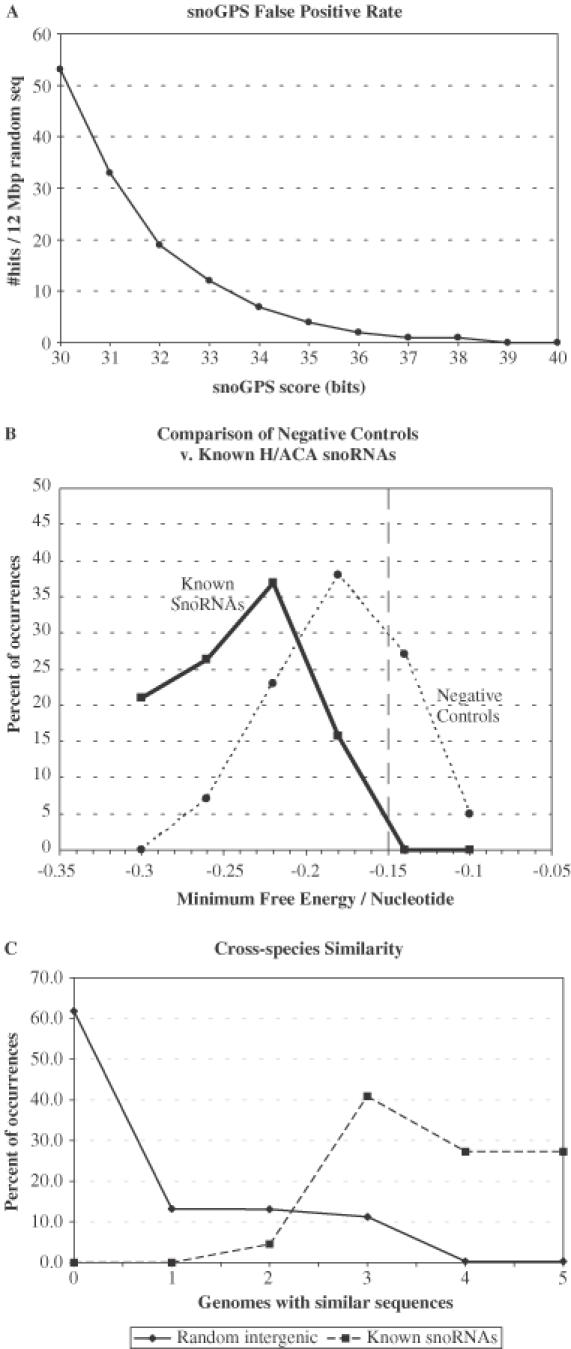
Scores of randomized and background sequences. (A) Number of snoGPS hits per 12 Mb random genome sequence, when averaged over three 12 Mb random genomes and all 44 Ψ target sites. The graph indicates that a threshold score of 36 would be expected to include approximately two false positives/12 Mb. (B) mfold MFE-per-base scores for the known snoRNAs compared with those of the top 100 highest-scoring snoGPS hits found in one of the random sequence runs. Solid line is for known snoRNAs; dashed line for random snoGPS hits. Although there is considerable overlap between the two curves, the figure suggests that candidates with MFE-per-base scores greater than −0.15 kcal/mol/nt are probably false positives. (C) Percentage of sequences in the Not-Feature part of the S.cerevisiae genome with putative homologs in other Saccharomyces genomes. The dashed line shows that three to five homologs in other Saccharomyces genomes were found essentially for all known snoRNAs. The solid line is for an average of one-thousand 200 nt sequences randomly selected from the S.cerevisiae Not-Feature genome. The data suggest that Not-Feature sequences with fewer than three homologs outside S.cerevisiae are probably not H/ACA snoRNAs.
Comparative genomics and free-energy calculations improve detection specificity
The results of MFE computations using the mfold program indicated that in general, known snoRNAs have slightly lower (i.e. more stable) MFEs than randomly generated sequences (Figure 2B). However, as seen in Figure 2B, there is a considerable overlap between the free energy values of known snoRNAs and the strongest snoGPS hits to randomized sequences. Consequently, a conservative free-energy cutoff score of less than −0.15 kcal/mol/nt was used to avoid inadvertently missing an snoRNA with relatively high (less stable) MFE.
BLASTn was used to detect possible orthologous snoRNA gene sequences in other Saccharomyces genomes. BLASTn similarity scores of known snoRNAs were found to be well separated from those of randomly selected 200 nt intergenic subsequences (Figure 2C). Approximately 75% of the randomly selected sequences have only one similar sequence (13%) in another Saccharomyces genome or none at all (62%). In contrast, all but one of the 22 known snoRNAs has apparent homologs in at least three genomes as seen in Table 1 [snR3 has only two putative homologs among the other yeast genomes; however, this is probably because the other yeast genome sequences are incomplete (17)]. This suggested that a candidate sequence with only one homolog in the other genomes, or none at all, is probably a false positive and this cutoff was also incorporated into the screen for new H/ACA snoRNAs.
In addition, alignments of known H/ACA snoRNAs with corresponding other yeast homologs confirmed that most of the sequence variations are in the loop and hinge regions (data not shown). The few differences that do occur in the stem segments almost always conserve snoGPS-predicted base pairings. This is illustrated for two of the newly identified snoRNAs in Figure 3. Consequently, visual inspection of cross-species alignments of candidate sequences also served to eliminate probable false positives.
Figure 3.
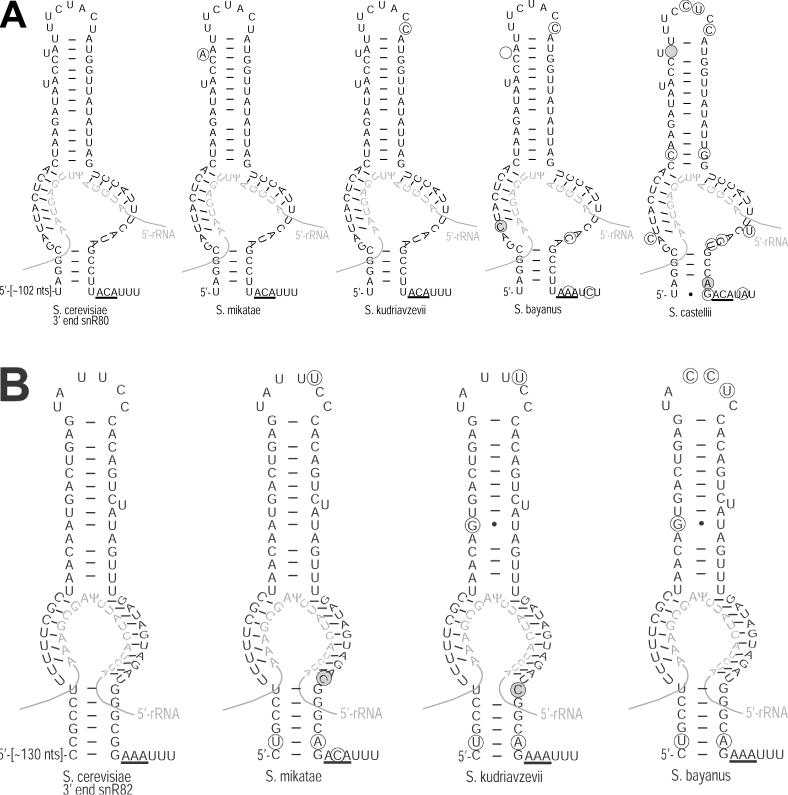
Phylogenetic comparison is consistent with the presumed secondary structure of newly discovered snoRNAs. The proposed secondary structures of the guide regions of the newly discovered S.cerevisiae (A) snR80 and (B) snR82 snoRNAs are shown along with the homologous sequences from several other Saccharomyces species. Nucleotides differing from S.cerevisiae are encircled. Those changes that presumably disrupt base pairing within the snoRNA or between the snoRNA and the rRNA are also shaded. The rRNA sequences are shown in light gray. The box ACA element at the 3′ end of each RNA is underlined. The Ψ at LSU-2350 is shown for the snR82 guide region. For clarity, only one of the two H/ACA hairpins for each of the snoRNA homologs is shown.
These results suggested that by post-processing candidate sequences we would eliminate many false positives. This would enable us to consider candidates with snoGPS scores lower than the threshold of 36 bits. Consequently, in practice, candidate sequences were filtered by mfold scores, presence of likely orthologs in other Saccharomyces genomes, and by knowledge of whether the sequence is intergenic, intronic or overlapping a known gene. With these additional filters, candidate sequences with snoGPS scores as low as 32 bits were considered for further testing.
New H/ACA snoRNAs detected by snoGPS are verified with gene disruption experiments
The searches for new H/ACA snoRNAs were carried out with three separate computational screens. The initial screen used the one-stem algorithm and the S.cerevisiae genome, only. In this phase, we also specifically searched for single-stem hits near one another that could form a double-guide molecule. In the second screen, both the one-stem and two-stem algorithms were used and the mfold and BLASTn homology post-processing filters were included. In the third screen, we modified the descriptor file to allow more G–U base pairings in the guide region and we analyzed all six Saccharomyces genomes directly with snoGPS.
The searches for single-stem hits near one another that could form a double-guide molecule resulted in two candidates that turned out to be previously identified RNAs. One of these was identified as snR191 (19). The other was a small RNA called RNA161 (20) which for consistency we refer to as snR161. This RNA had been suggested to be an H/ACA RNA but no target for it had been identified (20). These two RNAs were not in the University of Massachusetts snoRNA database (18) and had not been included in our initial training set (they were subsequently added to the training set). Identification of these snoRNAs indicated that the program was performing well in searching for snoRNAs in which both guide domains have a known target.
The screens for snoRNAs resulted in the selection of 17 candidates (Tables 2 and 3) for northern analysis (Figure 4) and gene disruption tests (Figure 5). All candidates were included in the genetic disruption experiments on the chance that those with negative northern signals may still be expressed at very low levels. Of the 17 candidates, 6 were observed to have positive northern signals (Figure 4). Putative base pairings of these snoRNAs and the cognate target regions are shown in Figure 6C. The results from the genetic disruption analyses showed that five of the six (snR80, snR81, snR82, snR83 and snR84) are required for Ψ formation at specific positions in rRNA (Figure 5). Unexpectedly, one snoRNA (snR82) was found to be required for Ψ synthesis at both a predicted site (LSU-2350) and a nearby site that was not predicted (LSU-2348).
Table 2. Predicted and experimentally determined genomic coordinates of candidates that were verified as snoRNAs.
| snoRNA | Ch | St | Predicted location | Predicted length | Coordinates from primer extensions | Northern size (nt) | Comp. screena | GenBank accession no. |
|---|---|---|---|---|---|---|---|---|
| snR80 | V | C | 52143–52315 | 173 | 52150–52320 | 170 ± 3 | 1 | AY679768 |
| snR81 | XV | W | 234350–234531 | 182 | 234344–234544 | 200 ± 3 | 1 | AY679769 |
| snR82 (RUF2) | VII | W | 316857–317056 | 200 | 316756–317056 | 300 ± 10 | 1 | AY679770 |
| snR83 (RUF3) | XIII | W | 626392–626653 | 262 | 626312–626657 | 345 ± 10 | 2 | AY679771 |
| snR84 (RUF1) | IV | C | 1492684–1493013 | 330 | 1492480–1493020 | 540 ± 20 | 2 | AY679772 |
| snR85 | XIII | C | 67763–67932 | 170 | 67768–67938 | 170 ± 3 | 3 | AY679773 |
‘Ch’ and ‘St’ indicate chromosome and strand, respectively.
aPhase of investigation in which snoRNA was selected for experimental testing.
Table 3. Predicted coordinates, snoGPS scores, number of BLASTn homologs and mfold scores of candidates with negative northern results.
| Predicted location | Comp. screena | Target site | snoGPS score | BLAST homologyb | E/base |
|---|---|---|---|---|---|
| chrIV:52259-52334_C | 2 (1-stem scan) | LSU-1109 | 23.3 | 3 | −0.28 |
| chrVII:93143-93309_C | 2 | LSU-1179 | 35.1 | 4 | −0.21 |
| chrIV:1156504-1156702_C | 3 | LSU-1179 | 40.60 | 3 | −0.25 |
| chrVIII:48383-48701_Wc | 2 | LSU-2265 | 37.8 | 3 | −0.28 |
| chrII:326304-326546_W | 2 | LSU-2313 | 36.1 | 2 | −0.28 |
| chrXI:430576-430759_W | 2 | LSU-2348 | 36.7 | 3 | −0.22 |
| chrXVI:753114-753200_W | 2 (1-stem scan) | SSU-1414 | 22.8 | 2 | −0.18 |
| chrXVI:860687-860758_C | 2 (1-stem scan) | SSU-1414 | 21.2 | 2 | −0.31 |
| chrXIII:223196-223414_W | 3 | SSU-1414 | 31.6 | 5d | −0.20 |
| chrX:277746-277979_Cc | 2 | SSU-759 | 33.4 | 3 | −0.18 |
| chrXIII:661806-661928_W | 2 (1-stem scan) | SSU-759 | 23.2 | 3 | −0.18 |
‘E/base’ indicates the calculated minimum Gibbs-free energy of the candidate sequence in kcal/mole/nt (scores for verified candidates are shown in Table 1). Note that snoGPS scores with the one-stem-scanner are generally lower than those from two-stem-scanner. A threshold of 20 bits was used with the one-stem-scanner.
aPhase of investigation in which snoRNA was selected for experimental testing. ‘1-stem scan’ indicates that candidate was identified with the one-stem-scanner descriptor file.
bNumber of other Saccharomyces species with BLAST hit to snoRNA with BLAST E-score < 10−4.
cLoci for which knock-out strains showed significantly slower growth rates.
dOverlaps known protein-coding gene RPS18B.
Figure 4.
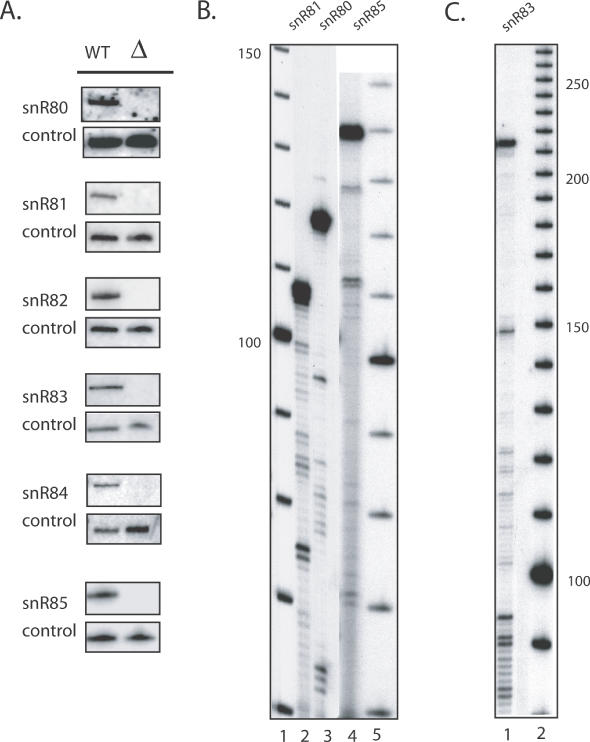
Experimental verification of predicted guide RNAs by northern hybridizations and primer extensions. (A) Northern-blot analysis of snoGPS snoRNA predictions. Total RNA was isolated from a wild-type (WT) and a strain in which snoGPS predicted snoRNA regions were chromosomally deleted. snR64 served as a loading control. (B) Primer extension 5′ end mapping of snR80, snR81 and snR85 from WT RNA relative to a 10 bp marker. (C) Primer extension 5′ end mapping of snR83 from WT RNA relative to a 10 bp marker. End-mapping of snR82, snR83 and snR84 has been reported previously (40). For snR82 and snR84, our results are very similar to those of (40) (data not shown). For snR83 our results show a strong stop signal corresponding to coordinates in reasonable agreement with those in the corrected version of (40) (see http://www.genetics.wustl.edu/eddy/publications/#McCutcheonEddy03).
Figure 5.
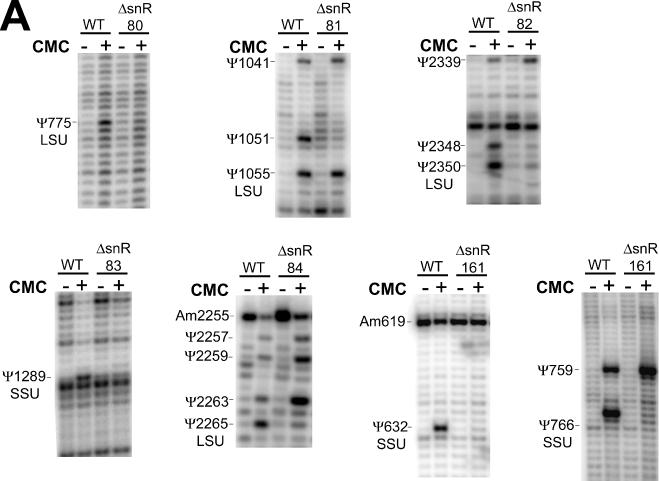
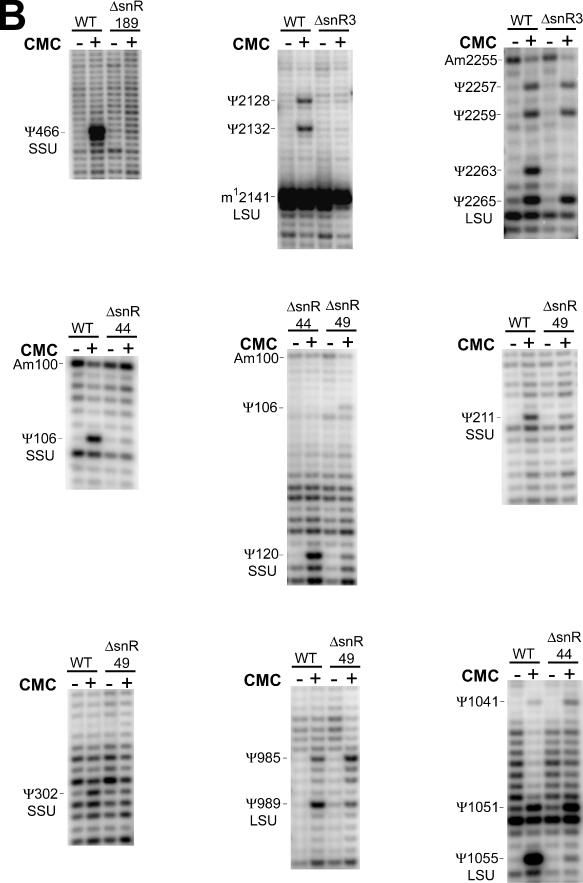
The results of gene disruption experiments. Experimental verification of the loss of specific rRNA pseudouridines in (A) strains with candidate loci and an unconfirmed snoRNA gene disrupted and (B) strains with experimentally identified-snoRNA genes disrupted. Following treatment of RNA from several strains with or without CMC, primer extension was performed with various primers to check positions in the 18S (SSU) or 25S (LSU) subunit ribosomal RNAs. ‘WT’ represents RNA from yeast where the locus under question has not been disrupted.
Figure 6.
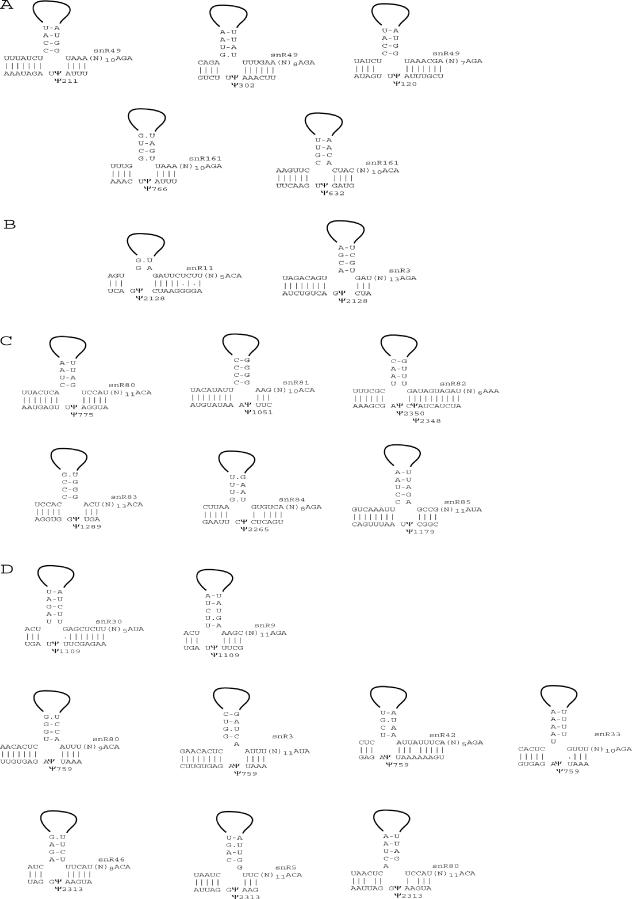
Putative base pairings between Ψ target regions and known snoRNAs. Pairings are shown for (A) snR49 and snR161 and experimentally verified Ψ targets. (B) The putative snR3 and snR11 guide snoRNAs and LSU-2128; snR3 was experimentally shown to be the actual guide RNA. The association predicted for snR11, though plausible, was demonstrated experimentally to be incorrect. (C) The newly identified Ψ guide snoRNAs. (D) Potentially redundant guide snoRNAs.
Unfortunately, we were unable to determine whether the sixth verified RNA (snR85) is required for guiding its predicted Ψ modification. This was because, despite repeated efforts, we were unable to detect the Ψ at SSU-1179 even in the S.cerevisiae wild-type strain. The reason for this result is not clear, but may be related to the presence of two other Ψ sites, SSU-1185 and the hypermodified SSU-1189, in the same region as SSU-1179. Interestingly, snR85 has an snoGPS score of only 28.8 in S.cerevisiae, below the threshold of 32 bits. However, the BLASTn homologous sequence of snR85 in S.kudriavzevii received a snoGPS score of 33.6 bits (data not shown). Consequently, snR85 was included for experimental testing.
The gene disruption analyses also showed that knock-out strains for two loci that had given negative northern and modification results (those at locations chrVIII:48383-48701 and chrX:277746-277979) grew at significantly slower rates than those of the other 15 gene disruption strains (data not shown). This suggests that these two sequences—while apparently not H/ACA guide snoRNAs for ribosomal RNA—may either exert regulatory control on other loci of biological importance or may be expressed at very low levels with the laboratory yeast strains and standard growth conditions used.
Forty-one of forty-four S.cerevisiae rRNA Ψs correlate with a known snoRNA
In addition to searching for new snoRNAs, we applied snoGPS to search for additional targets of the known snoRNAs. From an analysis of known snoRNAs with rRNA guide sequences where the Ψ guide function has been experimentally verified, we observed that the ‘match score’ (total number of Watson–Crick or G–U base pairings minus the number of mismatches in the rRNA guide-sequence region) ranged from 8 to 16 (Table 4). In addition, we observed that the rRNA guide sequences were generally well conserved among the other Saccharomyces species (e.g. see Figure 3). We consequently set our main criteria in searching for unassigned Ψs that might be guided by already-known snoRNAs to be a minimum match score of 7 in the rRNA guide sequence and strong cross-species conservation of the rRNA guide sequence.
Table 4. Predictions and experimental verifications of H/ACA snoRNA guide associations for all S.cerevisiae rRNA Ψ sites.
| Target site | snoRNA | Hairpin | Scorea | Genome rank | Match/mismatch | Expb |
|---|---|---|---|---|---|---|
| SSU-106 | snR44 | 5 | 36.2 | 4 | 12/0 | 4 |
| SSU-120c | snR49 | 5 | 38.9 | 1 | 11/1 | 4 |
| SSU-211c | snR49 | 5 | 41.9 | 2 | 11/0 | 4 |
| SSU-302c | snR49 | 3 | 41.7 | 1 | 10/0 | 4 |
| SSU-466 | snR189 | 5 | 39.4 | 2 | 11/0 | 4 |
| SSU-632c | snR161 | 3 | 41.7 | 2 | 10/0 | 4 |
| SSU-759 | * | — | ||||
| SSU-766c | snR161 | 5 | 48.8 | 1 | 8/0 | 4 |
| SSU-1000 | snR31 | 3 | 51.2 | 1 | 10/0 | 3 |
| SSU-1179c | snR85 | 5 | 28.8 | >20 | 12/0 | — |
| SSU-1185 | snR36 | 3 | 22.6 | >20 | 11/0 | 2 |
| SSU-1189 | snR35 | 5 | 46.4 | 1 | 12/0 | — |
| SSU-1414 | Unknown | — | ||||
| LSU-775c | snR80 | 3 | 37.9 | 3 | 12/0 | 4 |
| LSU-959 | snR8 | 5 | 29.6 | >20 | 9/0 | 3 |
| LSU-965 | snR43 | 5 | 40.0 | 2 | 10/0 | — |
| LSU-985 | snR8 | 3 | 17.0 | >20 | 10/1 | 3 |
| LSU-989 | snR49 | 5 | 39.2 | 1 | 11/0 | 4 |
| LSU-1003 | snR5 | 3 | 34.1 | >20 | 14/1 | 2 |
| LSU-1041 | snR33 | 3 | 38.0 | 2 | 10/0 | 3 |
| LSU-1051c | snR81 | 3 | 49.3 | 1 | 11/0 | 4 |
| LSU-1055 | snR44 | 3 | 31.0 | >20 | 12/0 | 4 |
| LSU-1109c | snR30 | 5 | 24.5 | >20 | 11/0 | — |
| LSU-1123 | snR5 | 5 | 44.0 | 1 | 13/0 | 2 |
| LSU-1289c | snR83 | 3 | 38.0 | 1 | 8/0 | 4 |
| LSU-2128c | snR3 | 5 | 55.5 | 1 | 11/0 | 4 |
| LSU-2132 | snR3 | 3 | 62.4 | 1 | 11/1 | 4 |
| LSU-2190 | snR32 | 3 | 38.9 | 2 | 12/0 | 3 |
| LSU-2257 | snR191 | 5 | 49.8 | 1 | 10/0 | 1 |
| LSU-2259 | snR191 | 3 | 32.1 | >20 | 8/0 | 1 |
| LSU-2263 | snR3 | 3 | 62.5 | 1 | 9/0 | 3 |
| LSU-2265c | snR84 | 5 | 42.7 | 1 | 10/1 | 4 |
| LSU-2313 | * | — | ||||
| LSU-2339 | snR9 | 5 | 23.1 | >20 | 9/1 | 5 |
| LSU-2348c | snR82 | 3 | 0 | >20 | 14/2 | 4 |
| LSU-2350c | snR82 | 3 | 39.8 | 2 | 16/0 | 4 |
| LSU-2415 | snR11 | 5 | 41.0 | 1 | 10/0 | 5 |
| LSU-2730 | snR189 | 3 | 48.5 | 1 | 11/0 | 5 |
| LSU-2822 | snR34 | 5 | 34.8 | >20 | 12/0 | 5 |
| LSU-2861 | snR46 | 5 | 52.8 | 2 | 11/0 | 3 |
| LSU-2876 | snR34 | 3 | 34.9 | 16 | 13/0 | 3 |
| LSU-2919 | snR10 | 3 | 36.5 | 10 | 10/0 | 3 |
| LSU-2940 | snR37 | 3 | 37.6 | 3 | 10/1 | 2 |
| LSU-2971 | snR42 | 3 | 49.5 | 1 | 11/0 | 3 |
For each Ψ, the experimentally verified or predicted associated guide H/ACA snoRNA is listed, as well as the snoRNA hairpin (5′ or 3′) that includes the putative guide sequence. The snoGPS score for the associated guide snoRNA is shown as well as the rank of that score within the entire S cerevisiae genome for the site. The total number of matches and mismatches in the rRNA guide sequence is also shown. The final column lists references for the experimental verification of the guide activity of the snoRNAs. Asterisks denote sites with multiple unverified predictions.
asnoGPS cross-validation score.
bExperimental verification of pseudouridylation: 1, (19); 2, (35); 3, (31); 4, present work; 5, Ni et al. (unpublished) cited in (9).
cNot previously predicted.
Using these criteria, we used snoGPS to analyze the known snoRNAs (including those newly identified in the present study) to determine whether they might guide any additional pseudouridylations for which a guide-snoRNA had not been experimentally confirmed. This resulted in new predictions of guides for nine Ψs with no experimentally confirmed, associated snoRNA (Table 5 and Figure 6). Interestingly, in four cases, more than one potential guide snoRNA was found with match scores above the cutoff (SSU-759, LSU-1109, LSU-2128 and LSU-2313).
Table 5. Potential Ψ guide associations to known snoRNAs.
| SnoRNA | Target site | Match/mismatch | Scorea | rRNA guide sequence conservation | Verification |
|---|---|---|---|---|---|
| snR44 | SSU-106* | 12/0 | 36.2 | Good | + |
| snR49 | SSU-120 | 11/1 | 38.9 | Good | + |
| snR49 | SSU-211 | 11/0 | 41.9 | Good | + |
| snR49 | SSU-302 | 10/0 | 41.7 | Good | + |
| snR189 | SSU-466* | 11/0 | 39.4 | Good | + |
| snR161 | SSU-632 | 10/0 | 41.7 | Good | + |
| snR42 | SSU-759 | 11/1 | 34.6 | Poor | − |
| snR80 | SSU-759 | 11/0 | 32.6 | Good | − |
| snR3 | SSU-759 | 12/0 | 26.7 | Good | − |
| snR33 | SSU-759 | 9/0 | 26.5 | Fair | nab |
| snR161 | SSU-766 | 8/0 | 48.8 | Fair | + |
| snR49 | LSU-989* | 11/0 | 39.2 | Good | + |
| snR44 | LSU-1055* | 12/0 | 31.0 | Good | + |
| snR9 | LSU-1109 | 7/0 | 26.6 | Fair | − |
| snR30 | LSU-1109 | 11/0 | 24.5 | Good | nab |
| snR3 | LSU-2128 | 11/0 | 55.5 | Good | + |
| snR11 | LSU-2128* | 12/0 | 40.2 | Poor | − |
| snR3 | LSU-2132* | 11/1 | 62.4 | Good | + |
| snR46 | LSU-2313 | 8/0 | 32.1 | Poor | − |
| snR5 | LSU-2313 | 8/0 | 24.4 | Good | − |
| snR80 | LSU-2313 | 9/2 | 23.1 | Good | − |
Six of these associations (indicated by asterisks) had been predicted previously. For sites SSU-759, LSU-2128, LSU-1109 and LSU-2313 more than one known snoRNA could plausibly guide the reaction as shown in (Figure 6B and D). Guide-sequence matches and mismatches are listed in addition to the overall snoGPS score. The table also includes a qualitative assessment of the conservation of the guide sequence in the other Saccharomyces species.
aCross-validation scores are shown for snoRNAs that were included in the training set.
bGene disruption experiment not attempted.
To remove ambiguity from these often conflicting snoRNA guide/target predictions, we performed gene disruption experiments. We also tested previously proposed, but unverified guide functions for snR3 (for LSU-2132), snR11 (LSU-2128), snR44 (SSU-106 and LSU-1055), snR49 (LSU-989) and snR189 (SSU-466). The results of these experiments confirmed 11 pairings of snoRNAs with Ψ modifications (Table 5). The target sites include SSU-106, SSU-120, SSU-211, SSU-302, SSU-466, SSU-632, SSU-766, LSU-989, LSU-1055, LSU-2128 and LSU-2132.
For LSU-2128, the Ψ modification had previously been assigned to snR11 (34). However, the putative guide sequences in snR3 are better conserved in other yeast genomes than those of snR11 (data not shown). This suggested that LSU-2128 may be guided by snR3, and, in fact, this assignment was confirmed experimentally (Figure 5). Interestingly, single-gene disruptions of four different snoRNAs predicted to target site SSU-759 (snR3, snR33, snR42 and snR80) and three others predicted to target LSU-2313 (snR5, snR46 and snR80) did not alter the Ψ phenotype.
Thus, with seven Ψ sites guided by new snoRNAs, seven sites guided by known snoRNAs with newly determined associations, and 27 sites with previously known associations, we get 41 total sites with associations (Table 4).
Two snoRNAs are required for the modification of three or more non-adjacent Ψ sites
Among the newly predicted target-guide associations, those for snR49 and snR3 are of particular interest. Our results demonstrate that snR49 is required for four separate, non-adjacent Ψ modifications. Three of these sites (SSU-120, SSU-211 and LSU-989) are predicted to be guided by a single guide sequence within the 5′ hairpin that is complementary to all three rRNA target sites. We have also shown that snR3 is required for the formation of three non-adjacent Ψs. These are the first verified cases of a single snoRNA specifying three or more Ψs in any species. In contrast, only one S.cerevisiae methylation guide snoRNA (U24) has been shown to be required for more than two modifications, and two of the three sites that it guides are adjacent (7). These findings were quite unexpected as it has been widely assumed that no snoRNA could guide more than two non-adjacent sites (1,3).
DISCUSSION
This study yielded two important advances. First, we have demonstrated that a computational screen is capable of detecting H/ACA snoRNAs in Saccharomyces genomes. Second, we have assembled the first nearly complete set of experimentally verified associations between guide snoRNAs and Ψ sites in a single RNA type, in this case pre-ribosomal RNA. In particular, we have experimentally verified 17 H/ACA snoRNA Ψ guide assignments, confirming that for S.cerevisiae, at least 37 of 44 Ψs in ribosomal RNA are guided by a snoRNA (Table 4).
The study also identified areas where snoGPS could be improved in the future. In six cases, experimentally verified snoRNA target site associations received low scores. One site (LSU-2348) was missed entirely, presumably because it is only two nucleotides from another Ψ site (LSU-2350) and the same snoRNA guides both pseudouridylations (although it is formally possible that the first Ψ is needed for the second modification). Strikingly, the complementarity between the 3′ region at LSU-2350 and the corresponding 5′ guide region in snR82 is 10 bp in length, which is the longest base pairing potential of any Ψ guide snoRNA known to us (Figure 6C). In matching snR82 to the nearby LSU-2348 site, the base pairing potential is still eight nucleotides. A similar phenomenon was previously observed for two yeast C/D box methylation guide snoRNAs (U24 and snR13), where methylation of two adjacent ribose sites is guided by a single snoRNA (4,7).
Aside from adjacent modified sites, low snoGPS scores are generally caused by some unusual feature in the snoRNA that has not been incorporated into the model underlying the snoGPS algorithm. An example is that of snR36 [see Figure 4C of (34)], which appears to have a large insertion in the 5′ hairpin, and does not conform well to the consensus snoRNA model used by snoGPS. In principle, it should be possible to modify the descriptor file to allow such structures to be detected. Interestingly, another snoRNA (snR42) with a large insertion in the 5′ hairpin (34) received a high score from snoGPS because snoGPS identified an alternative secondary structure scheme that is more similar to the canonical one. It would be interesting to determine which of these predicted structures is closer to the natural secondary structure of snR42 or if both forms occur in the cell. While allowing flexibility for these extra structures may be necessary for searching in other yeasts, we have observed yeast non-coding RNAs to be, by far, the most problematic in terms of insertions of stem–loops in the canonical structures relative to orthologs found in other species (unpublished data). Thus, we anticipate yeast to be among the most difficult organisms in terms of flexibility required in the gene models, implying that our sensitivity measures presented here are likely to be a lower bound compared to other sequenced organisms.
Of the seven Ψ sites in S.cerevisiae rRNA for which no guide snoRNA has been experimentally confirmed, four are predicted to be guided by a unique known H/ACA snoRNA. Sites LSU-965 and SSU-1189 were predicted previously to be served by the snR43 and snR35 snoRNAs, respectively (31,35), and our analysis supports these assignments. The snoGPS scores for these pairs are 40.0 and 46.4, respectively, and taken with perfect complementarities of 10 and 12 nt for the target-guide sequences, it seems very likely that these associations are correct. Site LSU-1179 has a perfect 12 bp match to the target-guide sequence of snR85; however, as noted above, we were unable to detect the Ψ at LSU-1179 in our primer extension experiments even for wild-type S.cerevisiae. Similarly, site LSU-1109 has a perfect 11 bp match to the target-guide sequence of snR30 (Table 5). Testing this prediction is also not straightforward, however, as snR30 is an essential snoRNA required for nucleolytic processing of pre-rRNA (36). Separating these functions genetically may be possible, however, based on success with another dual function H/ACA snoRNA (snR10). In that study, modification was blocked with a point mutation that did not affect the nucleolytic cleavage function (37).
This leaves just three sites without clear assignments (SSU-1414, SSU-759 and LSU-2313). Two of these Ψ sites (SSU-759 and LSU-2313) have high target-guide match scores to multiple snoRNAs (Table 5). However, disrupting the coding sequences for these snoRNAs individually did not block modification. A formal possibility is that either or both modifications could be formed by classic Ψ synthases that are not part of snoRNP complexes, and do not require snoRNA cofactors. Another possibility is that the modifications may be mediated by redundant mechanisms. Such redundancies might involve multiple snoRNAs acting with one or more snoRNA-dependent Ψ synthases. Alternatively, both snoRNA-dependent and snoRNA-independent pathways may exist as was recently discovered for the 2′-O-methylation of LSU-U2918 in S.cerevisiae (38). These issues could be addressed by examining the modification states of LSU-2313 and SSU-759, after disrupting all of the candidate guide snoRNAs in the same yeast strain.
For SSU-1414, our computational screen did not yield a likely candidate. Potential explanations here include one or more of the following possibilities: (i) the guide snoRNA may have some unusual structural feature(s) that causes it to elude the snoGPS program, as occurs with snR36; (ii) the site may not occur in the other Saccharomyces species analyzed, in which case a guide snoRNA in S.cerevisiae might not have recognizable orthologs and hence would be rejected by our candidate-filtering procedure; and (iii) this modification may be catalyzed without the use of a guide snoRNA, as appears to be the case for the pseudouridylation of S.cerevisiae tRNAs. These possibilities might apply to the negative experimental results obtained for Ψ sites LSU-2313 and SSU-759 as well. In fact, the Ψ corresponding to LSU-2313 is conserved in Escherichia coli (and in several other prokaryotes and eukaryotes) and is known to be formed by a protein-only Ψ synthase in E.coli (9). However, the Ψ modification at SSU-759 is not conserved in E.coli and the modification at SSU-1414 is not known to occur anywhere other than in Saccharomyces (9).
With the completion of the present work, it is possible to begin to make systematic comparisons of the nearly complete sets of snoRNAs that guide rRNA ribose-methylations with those that guide rRNA pseudouridylations in a single species. One noticeable difference is in gene organization. Among the 42 C/D box methylation-guide snoRNAs, six occur in introns and 17 occur in one of five polycistronic transcripts. In contrast, only two of the 28 H/ACA snoRNAs are intronic. Moreover, the distance of each of the H/ACA snoRNA genes from its neighboring genes in the S.cerevisiae genome suggests that no H/ACA snoRNA genes occur in a polycistronic transcript. Another difference is that with the possible exception of one or two C/D box snoRNAs that may guide methylations at adjacent sites, each C/D guide snoRNA targets at most two sites of ribosomal methylation (4,18). In contrast, we have seen that at least two of the H/ACA snoRNAs guide Ψ formation at three or four non-adjacent sites.
Comparison with other computational methods for H/ACA snoRNA detection
It is interesting to compare our method to two other recent approaches to H/ACA snoRNA genefinding in S.cerevisiae: the MFE method (8) and a method known as QRNA (39,40). The MFE approach seeks to identify H/ACA snoRNAs using a probabilistic search algorithm based on H/ACA snoRNA structure and free-energy minimization (41). A detailed comparison of the present results with those in the MFE study is not possible because that report does not include experimental verification, nor does it list the genomic coordinates of the 50 candidate sequences identified. For three of the top candidate sequences, approximate locations are indicated. Two of these are within an intron of a known gene (RPS11A and RPL43A) and one overlaps a known protein gene (MPP10). We have tested these three regions with snoGPS using as targets all of the S.cerevisiae Ψ sites for which there is as yet no experimentally verified guide snoRNA. The highest snoGPS scores for these candidates are 23.1, 18.1 and 24.2, respectively, all of which are well below our usual snoGPS cutoff of approximately 36 (or 32 for highly conserved intergenic candidates); thus snoGPS would not select these sequences as probable H/ACA snoRNAs. Approximate chromosomal locations are provided for 37 other candidates (Figure 9 in that report) (8). Comparing these approximate locations with those for the six new RNAs identified here, it is clear that five of the six new RNAs are not among those listed. The DNA segment for one candidate is in the general region of snR80, however, it is not possible to determine from the report if the candidate is indeed snR80. We conclude that at most one of the six snoRNAs identified in the present study are among the top 40 candidates of the MFE approach.
In contrast to snoGPS and the MFE approach, QRNA (39) is designed to search for any well-structured small non-coding RNA (ncRNA) conserved in at least two species. QRNA does not use any sequence-specific training data or descriptor files. Instead, QRNA looks for covariation patterns found in alignments of putative homologs of the candidate RNA sequence. While the present study was in progress, McCutcheon and Eddy (40) applied QRNA to S.cerevisiae, and identified 92 candidate ncRNAs. Eight of these candidates were confirmed by northern analysis (40) including three of the H/ACA snoRNAs independently identified in the present study. The designations in that study (RUF1, RUF2 and RUF3) correspond to our RNA species snR84, snR82 and snR83, respectively. Interestingly, McCutcheon and Eddy (40) were able to classify only one of these three as a H/ACA snoRNA without the use of snoGPS. We believe that this example illustrates how QRNA and snoGPS can be powerful complementary tools in searches for ncRNAs; QRNA is able to screen a genome for potential ncRNAs while snoGPS can efficiently identify which ones are H/ACA snoRNAs and what Ψ modifications they guide.
Searching for Ψ guide snoRNAs in other species and for other classes of RNAs
By changing the target files, descriptor files and score tables used, snoGPS can also be adapted to search for Ψ guide snoRNAs in other organisms. Such searches will be much easier, of course, in cases where the locations of target Ψ sites are known precisely. However, in cases where Ψ sites are not known, snoGPS can still be applied by using all uridine sites as potential targets. An additional challenge is that in many species only a few H/ACA snoRNAs are currently known and available for use as training data. We are currently developing modified target files, descriptor files and score tables that address these issues in order to search for H/ACA snoRNAs and homologs in archaea, mammals and other model organisms.
In addition to searching for snoRNAs that guide Ψ formation in rRNA, snoGPS could be used to search for snoRNAs that guide pseudouridylation of other classes of RNAs. Based on the results with snoRNA-like RNAs that guide Ψ synthesis in non-rRNAs—including the related scaRNAs that act on spliceosomal snRNAs in vertebrate Cajal bodies (42), it seems likely that the key structural properties featured here will be present in other Ψ guide RNAs as well. In fact, in mammals, experimental screens have identified snoRNA-like RNAs with canonical H/ACA features and with guide-elements complementary to known Ψ sites in spliceosomal snRNA (12). We have not yet applied snoGPS to search for snoRNA variants that target snRNAs, in part because Ψ modifications in S.cerevisiae snRNAs may be mediated by a snoRNA-independent mechanism (43).
Beyond searching for Ψ guide snoRNAs, snoGPS can be reconfigured—through changes to its descriptor files and score tables—to search for other families of RNAs, provided that they have characteristic primary sequence and secondary structure motifs. Examples might include telomerase RNAs and microRNAs (44) and their precursors. The only requirement is the availability of a training set of known or related RNAs to aid in the creation of the necessary descriptor files and score tables.
In searching for classes of RNAs with few known examples for training, the issue of large numbers of false positives is likely to become significant. We anticipate that in such cases, snoGPS may be more effective when applied to sets of known RNAs of unknown structure and function than to genome-wide searches. In the last few years, hundreds of such RNAs of unknown function have been identified by experimental (12,13) and computational (41,45) means. The results from microarray experiments and cross-species homology analyses suggest that many more such RNAs will be found [e.g. (46–48)]. Classifying and establishing relationships among these newly identified RNAs will be an important challenge in the coming years. snoGPS, which combines much of the flexibility of RNA-motif searching programs with the detailed, probabilistic score tables typically found in customized programs, provides a promising approach for accomplishing this goal.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at NAR Online.
Acknowledgments
ACKNOWLEDGEMENTS
We are grateful to Paul Cliften and Mark Johnston for providing us with prepublication access to their sequence database for the five additional Saccharomyces genomes and to Roy Parker for providing the knockout strain for RNA161. We thank Karen Artiles for carrying out northern analyses during the initial phase of the study, Mark Diekhans for numerous helpful discussions and an anonymous reviewer for many thoughtful and constructive comments. This work was supported by NIH grants GM040478 (M.A.) and GM19351 (M.J.F.), an Alfred P. Sloan Research Fellowship (T.M.L.) and a grant from theWM Keck Foundation to the RNA Center at UCSC.
REFERENCES
- 1.Bachellerie J.P., Cavaille,J. and Huttenhofer,A. (2002) The expanding snoRNA world. Biochimie, 84, 775–790. [DOI] [PubMed] [Google Scholar]
- 2.Bertrand E. and Fournier,M.J. (2004) The snoRNPs and related machines: ancient devices that mediate maturation of rRNA and other RNAs. In Olson,M. (ed.), The Nucleolus. Landes Bioscience, Georgetown, TX, pp. 225–261. [Google Scholar]
- 3.Kiss T. (2002) Small nucleolar RNAs: an abundant group of noncoding RNAs with diverse cellular functions. Cell, 109, 145–148. [DOI] [PubMed] [Google Scholar]
- 4.Lowe T.M. and Eddy,S.R. (1999) A computational screen for methylation guide snoRNAs in yeast. Science, 283, 1168–1171. [DOI] [PubMed] [Google Scholar]
- 5.Omer A.D., Lowe,T.M., Russell,A.G., Ebhardt,H., Eddy,S.R. and Dennis,P.P. (2000) Homologs of small nucleolar RNAs in Archaea. Science, 288, 517–522. [DOI] [PubMed] [Google Scholar]
- 6.Gaspin C., Cavaille,J., Erauso,G. and Bachellerie,J.P. (2000) Archaeal homologs of eukaryotic methylation guide small nucleolar RNAs: lessons from the Pyrococcus genomes. J. Mol. Biol., 297, 895–906. [DOI] [PubMed] [Google Scholar]
- 7.Kiss-Laszlo Z., Henry,Y., Bachellerie,J.P., Caizergues-Ferrer,M. and Kiss,T. (1996) Site-specific ribose methylation of preribosomal RNA: a novel function for small nucleolar RNAs. Cell, 85, 1077–1088. [DOI] [PubMed] [Google Scholar]
- 8.Edvardsson S., Gardner,P.P., Poole,A.M., Hendy,M.D., Penny,D. and Moulton,V. (2003) A search for H/ACA snoRNAs in yeast using MFE secondary structure prediction. Bioinformatics, 19, 865–873. [DOI] [PubMed] [Google Scholar]
- 9.Ofengand J. and Fournier,M.J. (1998) The pseudouridine residues of rRNA: number, location, biosynthesis, and function. In Grosjean,H. and Benne,R. (eds), Modification and Editing of RNA. ASM Press, Washington DC, pp. 229–253. [Google Scholar]
- 10.Ofengand J. (2002) Ribosomal RNA pseudouridines and pseudouridine synthases. FEBS Lett., 514, 17–25. [DOI] [PubMed] [Google Scholar]
- 11.Balakin A.G., Smith,L. and Fournier,M.J. (1996) The RNA world of the nucleolus: two major families of small RNAs defined by different box elements with related functions. Cell, 86, 823–834. [DOI] [PubMed] [Google Scholar]
- 12.Huttenhofer A., Kiefmann,M., Meier-Ewert,S., O'Brien,J., Lehrach,H., Bachellerie,J.P. and Brosius,J. (2001) RNomics: an experimental approach that identifies 201 candidates for novel, small, non-messenger RNAs in mouse. EMBO J., 20, 2943–2953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tang T.H., Bachellerie,J.P., Rozhdestvensky,T., Bortolin,M.L., Huber,H., Drungowski,M., Elge,T., Brosius,J. and Huttenhofer,A. (2002) Identification of 86 candidates for small non-messenger RNAs from the archaeon Archaeoglobus fulgidus. Proc. Natl Acad. Sci. USA, 99, 7536–7541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ofengand J., Del Campo,M. and Kaya,Y. (2001) Mapping pseudouridines in RNA molecules. Methods, 25, 365–373. [DOI] [PubMed] [Google Scholar]
- 15.Cherry J.M., Adler,C., Ball,C., Chervitz,S.A., Dwight,S.S., Hester,E.T., Jia,Y., Juvik,G., Roe,T., Schroeder,M., Weng,S. and Botstein,D. (1998) SGD: Saccharomyces Genome Database. Nucleic Acids Res., 26, 73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grate L. and Ares,M. (2002) Searching yeast intron data at the Ares lab website. In Guthrie,C. and Fink,G. (eds), Guide to Yeast Genetics and Molecular and Cell Biology, Part B, Methods Enzymology. Vol. 350, Academic Press, San Diego, CA, pp. 380–392. [DOI] [PubMed] [Google Scholar]
- 17.Cliften P., Sudarsanam,P., Desikan,A., Fulton,L., Fulton,B., Majors,J., Waterston,R., Cohen,B.A. and Johnston,M. (2003) Finding functional features in Saccharomyces genomes by phylogenetic footprinting. Science, 301, 71–76. [DOI] [PubMed] [Google Scholar]
- 18.Samarsky D.A. and Fournier,M.J. (1999) A comprehensive database for the small nucleolar RNAs from Saccharomyces cerevisiae. Nucleic Acids Res., 27, 161–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Badis G., Fromont-Racine,M. and Jacquier,A. (2003) A snoRNA that guides the two most conserved pseudouridine modifications within rRNA confers a growth advantage in yeast. RNA, 9, 771–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Olivas W.M., Muhlrad,D. and Parker,R. (1997) Analysis of the yeast genome: identification of new non-coding and small ORF-containing RNAs. Nucleic Acids Res., 25, 4619–4625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Durbin R., Eddy,S.R., Krogh,A. and Mitchison,G. (1998) Biological Sequence Analysis. Cambridge Unversity Press, Cambridge, UK. [Google Scholar]
- 22.Gautheret D., Major,F. and Cedergren,R. (1990) Pattern searching/alignment with RNA primary and secondary structures: an effective descriptor for tRNA. Comput. Appl. Biosci., 64, 325–331. [DOI] [PubMed] [Google Scholar]
- 23.Macke T.J., Ecker,D.J., Gutell,R.R., Gautheret,D., Case,D.A. and Sampath,R. (2001) RNAMotif, an RNA secondary structure definition and search algorithm. Nucleic Acids Res., 29, 4724–4735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gautheret D. and Lambert,A. (2001) Direct RNA motif definition and identification from multiple sequence alignments using secondary structure profiles. J. Mol. Biol., 313, 1003–1011. [DOI] [PubMed] [Google Scholar]
- 25.Altschul S.F., Gish,W., Miller,W., Myers,E.W. and Lipman,D.J. (1990) Basic local alignment search tool. J. Mol. Biol., 215, 403–410. [DOI] [PubMed] [Google Scholar]
- 26.Notredame C., Higgins,D.G. and Heringa,J. (2000) T-Coffee: a novel method for fast and accurate multiple sequence alignment. J. Mol. Biol., 302, 205–217. [DOI] [PubMed] [Google Scholar]
- 27.Zuker M. and Stiegler,P. (1981) Optimal computer folding of large RNA sequences using thermodynamics and auxiliary information. Nucleic Acids Res., 9, 133–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mosteller F. and Tukey,J.W. (1977) Data Analysis and Regression: A Second Course in Statistics. Addison-Wesley, Reading, MA. [Google Scholar]
- 29.Efron B. and Gong,G. (1983) A leisurely look at the bootstrap, jackknife, and cross-validation. Am. Stat., 37, 36–48. [Google Scholar]
- 30.Zavanelli M.I. and Ares,M.,Jr (1991) Efficient association of U2 snRNPs with pre-mRNA requires an essential U2 RNA structural element. Genes Dev., 5, 2521–2533. [DOI] [PubMed] [Google Scholar]
- 31.Ni J., Tien,A.L. and Fournier,M.J. (1997) Small nucleolar RNAs direct site-specific synthesis of pseudouridine in ribosomal RNA. Cell, 89, 565–573. [DOI] [PubMed] [Google Scholar]
- 32.Baudin A., Ozier-Kalogeropoulos,O., Denouel,A., Lacroute,F. and Cullin,C. (1993) A simple and efficient method for direct gene deletion in Saccharomyces cerevisiae. Nucleic Acids Res., 21, 3329–3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Puig O., Caspary,F., Rigaut,G., Rutz,B., Bouveret,E., Bragado-Nilsson,E., Wilm,M. and Seraphin,B. (2001) The tandem affinity purification (TAP) method: a general procedure of protein complex purification. Methods, 24, 218–229. [DOI] [PubMed] [Google Scholar]
- 34.Ganot P., Caizergues-Ferrer,M. and Kiss,T. (1997) The family of box ACA small nucleolar RNAs is defined by an evolutionarily conserved secondary structure and ubiquitous sequence elements essential for RNA accumulation. Genes Dev., 11, 941–956. [DOI] [PubMed] [Google Scholar]
- 35.Ganot P., Bortolin,M.L. and Kiss,T. (1997) Site-specific pseudouridine formation in preribosomal RNA is guided by small nucleolar RNAs. Cell, 89, 799–809. [DOI] [PubMed] [Google Scholar]
- 36.Morrissey J.P. and Tollervey,D. (1993) Yeast snR30 is a small nucleolar RNA required for 18S rRNA synthesis. Mol. Cell. Biol., 4, 2469–2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.King T.H., Liu,B., McCully,R.R. and Fournier,M.J. (2003) Ribosome structure and activity are altered in cells lacking snoRNPs that form pseudouridines in the peptidyl transferase center. Mol. Cell, 11, 425–435. [DOI] [PubMed] [Google Scholar]
- 38.Bonnerot C., Pintard,L. and Lutfalla,G. (2003) Functional redundancy of Spb1p and a snR52-dependent mechanism for the 2′-O-ribose methylation of a conserved rRNA position in yeast. Mol. Cell, 12, 1309–1315. [DOI] [PubMed] [Google Scholar]
- 39.Rivas E., Klein,R.J., Jones,T.A. and Eddy,S.R. (2001) Computational identification of noncoding RNAs in E.coli by comparative genomics. Curr. Biol., 11, 1369–1373. [DOI] [PubMed] [Google Scholar]
- 40.McCutcheon J.P. and Eddy,S.R. (2003) Computational identification of non-coding RNAs in Saccharomyces cerevisiae by comparative genomics. Nucleic Acids Res., 31, 4119–4128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schuster P., Fontana,W., Stadler,P.F. and Hofacker,I.L. (1994) From sequences to shapes and back: a case study in RNA secondary structures. Proc. R. Soc. Lond. B Biol. Sci., 255, 279–284. [DOI] [PubMed] [Google Scholar]
- 42.Darzacq X., Jady,B.E., Verheggen,C., Kiss,A.M., Bertrand,E. and Kiss,T. (2002) Cajal body-specific small nuclear RNAs: a novel class of 2′-O-methylation and pseudouridylation guide RNAs. EMBO J., 21, 2746–2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ma X., Zhao,X. and Yu,Y.T. (2003) Pseudouridylation (Psi) of U2 snRNA in S.cerevisiae is catalyzed by an RNA-independent mechanism. EMBO J., 22, 1889–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ambros V. (2001) MicroRNAs: tiny regulators with great potential. Cell, 107, 823–826. [DOI] [PubMed] [Google Scholar]
- 45.Argaman L., Hershberg,R., Vogel,J., Bejerano,G., Wagner,E.G., Margalit,H. and Altuvia,S. (2001) Novel small RNA-encoding genes in the intergenic regions of Escherichia coli. Curr. Biol., 11, 941–950. [DOI] [PubMed] [Google Scholar]
- 46.Wassarman K.M., Repoila,F., Rosenow,C., Storz,G. and Gottesman,S. (2001) Identification of novel small RNAs using comparative genomics and microarrays. Genes Dev., 15, 1637–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kapranov P., Cawley,S.E., Drenkow,J., Bekiranov,S., Strausberg,R.L., Fodor,S.P. and Gingeras,T.R. (2002) Large-scale transcriptional activity in chromosomes 21 and 22. Science, 296, 916–919. [DOI] [PubMed] [Google Scholar]
- 48.Mural R., Adams,M., Myers,E., Smith,H.O., Miklos,G.L., Wides,R., Halpern,A., Li,P.W., Sutton,G.G., Nadeau,J. et al. (2002) A comparison of whole-genome shotgun-derived mouse chromosome 16 and the human genome. Science, 296, 1661–1671. [DOI] [PubMed] [Google Scholar]