


Abstract
The Saccharomyces cerevisiae U6 RNA gene, SNR6, is transcribed by RNA polymerase III (Pol III), but lacks the intragenic B block promoter element found in most other Pol III transcription units. Rather, the SNR6 B block element is located 120 bp downstream of the terminator. In contrast, the Schizosaccharomyces pombe U6 RNA gene has an intragenic B block sequence in a short intron. We show that the S.pombe U6 intron, when inserted into SNR6, can functionally replace the downstream B block in vitro but not in vivo. The in vivo expression defect is caused by at least three different effects of the insertion: (i) the S.pombe intron is inefficiently spliced in S.cerevisiae due to the short distance between the 5′ splice site and branchpoint; (ii) the S.pombe B block sequence is suboptimal for S.cerevisiae; and (iii) a B block does not function well within the context of the SNR6 intron, especially when the gene is present at its normal chromosomal locus rather than on a plasmid. This last observation suggests that the chromatin structure of the SNR6 locus favors utilization of a downstream B block element. We also provide evidence that splicing of U6 RNA reduces its activity, presumably due to alterations in U6 RNA structure, localization and/or assembly into the spliceosome.
INTRODUCTION
Eukaryotic RNA polymerase III (Pol III) transcribes genes that code for short, untranslated RNAs, including transfer RNAs (tRNAs), the 5S ribosomal RNA and U6 spliceosomal RNA. All genes transcribed by Pol III appear to require binding of transcription factor (TF) IIIB upstream of the coding region for the initiation of RNA synthesis (1,2). A variety of promoter structures have evolved to accomplish the task of recruiting TFIIIB and, strikingly, many of these include intragenic elements. For example, promoters of tRNA genes consist of A and B block intragenic elements, which are 11 or 12 bp sequences that bind the six-subunit assembly factor TFIIIC. The A block corresponds to the D loop of the tRNA, and lies ∼20 bp downstream of the transcription start site. The B block corresponds to the T loop of the tRNA and is located 30–90 bp downstream of the A block, depending upon the length of the extra loop and the presence or absence of an intron. The B block is the high affinity binding site for TFIIIC, while the A block positions the upstream portion of TFIIIC properly to direct the placement of TFIIIB upstream of the transcription start site via protein–protein interactions (2,3). The optimal distance between the A and B blocks for TFIIIC binding and in vitro transcription is 30–60 bp (4,5).
Several other types of Pol-III-transcribed genes contain A and B block elements in their RNA coding regions, but these sequences are sometimes removed from the transcript by RNA processing. For example, in the Saccharomyces cerevisiae RNase P RNA (RPR1) gene, the A and B blocks lie in a leader region that is cleaved from the primary transcript (6). The snR52 small nucleolar RNA gene appears to also have the A and B block promoter elements in a 5′ leader (7). In the U6 RNA gene from Schizosaccharomyces pombe and related fission yeasts (8–10), a sequence that matches the B block consensus is found in a pre-mRNA-type intron (Figure 1). The intron is spliced out of the primary transcript via the normal pre-mRNA splicing pathway (11) to give mature U6 RNA (8,9). Presumably, these mechanisms have evolved to allow the utilization of intragenic promoter sequences that are incompatible with efficient function of the mature RNA.
Figure 1.
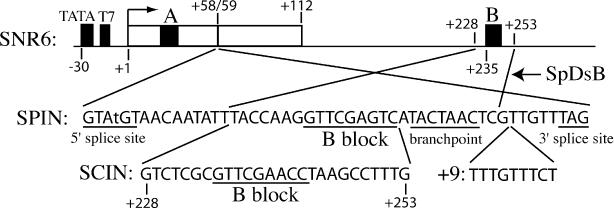
Promoter structure of wild type and mutant alleles of SNR6. The wild-type S.cerevisiae U6 RNA gene, SNR6, is shown schematically at the top. The open box represents U6 RNA coding sequence (+1 to +112) and black boxes represent promoter elements. The SPIN+B allele was created by inserting the 50 bp S.pombe U6 intron, the non-template strand sequence of which is shown, between positions +58 and +59 of SNR6. The consensus splice elements and core B block motif of the intron are underlined. (Note that the fourth intron nucleotide was mutated from A to T to match the S.cerevisiae 5′ splice site consensus.) The SPIN−B allele is truncated at position +235 (12). The SPIN alleles were modified by replacing 16 bp of the S.pombe U6 intron with 26 bp (+228 to +253) encompassing the SNR6 downstream B block (SCIN), or by addition of 9 bp between the branchpoint and 3′ splice site (+9). In a separate construct (SpDsB), SNR6 sequences +228 to +253 were replaced with the indicated sequence from the S.pombe intron.
The S.cerevisiae U6 RNA gene, SNR6, has a unique promoter structure that includes an extragenic B block located 120 bp downstream of the transcription termination signal and 200 bp downstream of the A block [(12); Figure 1]. In addition, SNR6 contains two upstream promoter elements: a consensus TATA box 30 bp upstream of the start site and a stretch of seven T residues between the TATA box and start site. Although the TATA box strongly influences start site selection, it is dispensable for transcription in vivo, whereas the A and B blocks are required for SNR6 expression in vivo (13,14). The T7 stretch is also dispensable, except in the presence of additional promoter mutations or in the absence of the non-histone chromatin protein Nhp6 (15). Thus, SNR6 has a promoter structure more similar to that of tRNA genes than to the entirely upstream promoters of the metazoan U6 RNA genes (1). The large distance between the SNR6 A and B blocks is surprising given that artificially increasing the spacing between the A and B blocks of the tRNA-like adenovirus VARNA1 gene beyond 100 bp is known to greatly diminish in vitro transcription activity and result in utilization of aberrant start sites (5). The steady-state level of U6 RNA in an actively growing yeast cell is approximately 1000 to 2000 molecules per cell (16), which predicts that SNR6 is transcribed at a high rate of at least once every 5 s during log phase growth.
A previous attempt to functionally replace the SNR6 downstream B block with an intragenic B block promoter element via site-directed mutagenesis had failed. The intragenic B block was functional in vitro but not in vivo; only in the presence of the downstream B block was transcript from the mutant allele detected in cells (13). It is not clear whether the intragenic B block failed to function in vivo because of suboptimal sequences flanking the core consensus sequence, or because of positional constraints on B block function in the context of SNR6, perhaps due to chromatin structure, or both. Furthermore, quantitation of the effects of the intragenic B block mutation on promoter strength is complicated by the fact that the product U6 RNA is apparently destabilized by the mutations. Here we have further examined positional constraints on SNR6 B block function by utilizing the S.pombe U6 RNA gene intron as a donor of an intragenic B block element. The S.pombe U6 intron contains consensus S.cerevisiae splicing signals, and so is expected to be efficiently removed from S.cerevisiae U6 RNA, as it is from S.pombe U6 RNA. If this is the case, then the amount of mature U6 RNA produced from the chimeric gene should accurately reflect the transcriptional activity of the gene.
We show that when the S.pombe U6 RNA gene intron is inserted into the analogous position of SNR6, it fully restores in vitro transcription of an SNR6 allele lacking the downstream B block, indicating that the S.pombe intron provides B block function in S.cerevisiae extracts. However, the SNR6 allele with only an intronic B block does not support viability in vivo, both because the expression of pre-U6 RNA is very low and because the S.pombe intron is not spliced out. The expression defect is in part due to suboptimal recognition of the S.pombe B block sequence, which we show is ∼3-fold less efficient at promoting SNR6 transcription when present in the downstream location. The splicing defect is due to inadequate spacing between the 5′ splice site and branchpoint. Substitution of the S.pombe B block sequence with the S.cerevisiae SNR6 B block sequence and a consequent increase in the 5′ splice site to branchpoint spacing rescues the lethality of the intron insertion when the allele is on a low-copy plasmid, but not when it is integrated into the chromosome at the SNR6 locus. These results suggest that the in vivo chromatin structure of SNR6 favors recognition of a downstream B block element relative to an intragenic B block element. Furthermore, we provide evidence that properly spliced U6 RNA is not as functional as wild-type U6 RNA that has not been spliced, which implies that acting as a splicing substrate alters the U6 RNA or U6 snRNP in some way.
MATERIALS AND METHODS
Plasmids
A DNA fragment containing the intron from the S.pombe U6 RNA gene was generated by PCR amplification of plasmid pDW19, a kind gift from Claudia Reich and Jo Ann Wise (9). The primer oligonucleotides SPIN–PCR 1 and SPIN–PCR 2 (see below) created MboI and AluI restriction sites on the 5′ and 3′ ends of the intron fragment, respectively. In addition, SPIN–PCR 1 also changed the fourth position of the intron to a T to better match the S.cerevisiae consensus 5′ splice site. PCR products were digested with MboI and AluI and purified on a 6% polyacrylamide gel. Approximately 20 ng of digested intron fragment was ligated with 150 ng of BclI-digested plasmid pEP6 [contains wild-type SNR6 gene; (17)] using 5 U of T4 DNA ligase (USB) for 4 h at 23°C. At this point, only the MboI site on the 5′ end of the intron fragment should have ligated to the BclI site of SNR6. The remaining BclI-generated 5′ overhang was removed by incubation of the DNA with 5 U of S1 nuclease for 20 min or filled in by incubation with 4 U of the Klenow fragment of DNA polymerase I and the four deoxynucleotides for 1 h to generate a blunt end for ligation to the AluI-generated blunt end on the 3′ end of the intron. The blunted DNA fragment was then ligated overnight using 10 U of T4 DNA ligase at 14°C. The Klenow treatment should result in a duplication of 4 bp corresponding to the overhang of the BclI site. However, clones obtained in this manner did not contain the 4 bp duplication, but rather contained the precise intron insertion between positions 58 and 59 of the U6 RNA gene. The resulting plasmid is called pEP6-SPIN.
The intron was transferred to SNR6 plasmid pCCs6 (12), which is truncated at its 3′ end in the middle of the B block consensus sequence and thus lacks a functional B block. The NruI–EcoNI fragment from pEP6-SPIN was ligated into NruI–EcoNI-digested pCCs6 to generate pCCs6-SPIN. To create the SPIN+9 intron, the sequence TTTGTTTCT was inserted between positions 42 and 43 of the intron by site-directed mutagenesis of pCCs6-SPIN using oligonucleotide SPIN+9 as described previously (13). The SPIN+9 intron was transferred into an SNR6 allele with the downstream B block by ligating the EcoRI–EcoNI fragment from the appropriate plasmid into EcoRI–EcoNI-digested pEP6-SPIN. The resulting plasmids have 120 bp of upstream SNR6 sequence instead of the 539 bp of the pEP6 plasmid. SNR6 constructs containing 120 bp of upstream sequence are transcribed with the same efficiency in vitro and in vivo as constructs containing 539 bp (12). For testing the in vivo functionality of the intron containing constructs, the EcoRI–SphI fragments from SNR6 plasmids were ligated into EcoRI–Sph I digested pSE358 [TRP1 CEN4 ARS1; (18)]. High-copy derivatives of the SPIN+B and SPIN+B+9 alleles of SNR6 were made by ligating the appropriate EcoRI–PstI fragments into EcoRI–PstI-digested pRS424 [TRP1 2μ ORI; (19)]. Several alleles were created which contained insertions of DNA between the 5′ splice site and branch point consensus sequences by digesting pRS424-EP6SPIN with StyI, filling in the 5′ overhang with T4 DNA polymerase and the four deoxynucleotides and religation with or without BamHI linkers (New England Biolabs).
The downstream B block of the S.cerevisiae SNR6 gene was substituted with the S.pombe B block and 10 bp of flanking sequence at either side by PCR, using pRS314-539H6 (15) as the template and oligos U6-5′EcoRI and U6-SpDsB/Bam as primers. The EcoRI/BamHI-digested PCR fragment was ligated to EcoRI/BamHI-digested pRS314 [TRP1 CEN6 ARSH4; (20)] to create pRS314-SpDsB. pRS314-SCIN−B (Figure 1) was constructed by ‘inverse’ PCR using pCCs6-SPIN as the template and oligos ScB-SPIN1 and ScB-SPIN2 as primers. The PCR product was digested by BstBI and self-ligated to give the pCCs6-SCIN plasmid. The U6 allele in pCCs6-SCIN was amplified by PCR using U6-5′EcoRI and U6-3′PstI-B as primers and ligated to EcoRI–PstI-digested pRS314. pRS314-SCIN+B was constructed by the same procedure, except pEP6-SPIN was used as the initial PCR template. Integrating plasmids pRS306-SCIN−B and pRS306-SCIN+B were made by ligating the appropriate BamHI–EcoRI-digested fragments from pRS314-SCIN−B and pRS314-SCIN+B into BamHI–EcoRI-digested pRS306 (20).
Oligonucleotides
The following oligonucleotides were used: SPIN–PCR 1, 5′-GAGAAGATCGTATGTAACAAT; SPIN–PCR 2, 5′-CGAGCTAAACAACGAGTTAG, SPIN3; 5′-GTATGACTCGAACC, SPIN+9, 5′-GCTCTAAACAAAGAAACAAACGAGTTAGTATG; 6D, 5′-AAAACGAAATAAATCTCTTTG; U6-5′EcoR I, 5′-CGGAATTCTTCGGCTACTATAAATAAA;U6-SpDsB/Bam, 5′-CGGGATCCGATAGGAGTTAGTATGACTCGAACCTTGGTAAATTTTCTATTCGAGATGTTA;ScB-SPIN1, 5′-TCGCGTTCGAACCTAAGCCTTTGATACTAACTCGTTGTTTAGAGCA; ScB-SPIN2, 5′-TTAGGTTCGAACGCGAGACAATATTGTTACATACGATCATCTC; U6-3′PstI-B, 5′-AACTGCAGCGAGACAATTTTCTATTCGA; U1-SH, 5′-CCGTATGTGTGTGTGACC.
In vitro transcription
In vitro transcription was carried out as described in (13) using subcellular extract (21) and 100 ng of CsCl-gradient-purified (22) plasmid DNA. The RNA products were run on 6% polyacrylamide, 8.3 M urea gels and the gels were exposed to Kodak XAR-5 film with or without Cronex Lightning Plus intensifying screens (DuPont). For primer extension analysis of the in vitro synthesized pre-U6 RNA, transcription was carried out as normal except that 50 mM GTP was added and 32P-labeled GTP was omitted. The RNA products were treated as usual except the final nucleic acid pellet was resuspended in 10 μl of 10 mM Tris–HCl, pH 8.0, 1 mM EDTA. One microliter of the RNA was sequenced as described (13) using a 32P-labeled oligonucleotide SPIN3 (complementary to positions 22 to 35 of the intron) and the cDNA products were run on a 6% polyacrylamide, 8.3 M gel. The gel was exposed to Kodak XRP-1 film for 16 h without an intensifying screen.
In vivo expression analysis
For the in vivo analysis, SNR6 alleles carried on pSE358 or pRS314 were transformed into yeast strain DAB016 (12), MWK003 or MWK027 (23) by the lithium acetate procedure (24). All three strains have a LEU2-marked disruption of the SNR6 locus, complemented by either a wild-type (DAB016) or pseudo-wild-type [MWK003, MWK027; (25)] allele of SNR6 carried on the URA3-marked centromere plasmid YCp50. The ability of the intron-containing alleles to function as the sole copy of SNR6 in the cell was determined by the plasmid shuffle assay (26), i.e. scoring for growth on medium containing 0.75 mg/ml of 5-fluoroorotic acid (5-FOA), which selects for loss of the URA3-marked plasmid.
The SCIN+B and SCIN−B alleles were integrated at the SNR6 locus by the ‘pop-in/pop-out’ procedure (27). A URA3-marked integrating plasmid pRS306 (20) containing the SCIN+B allele of SNR6 was linearized at the NruI site and transformed into a wild-type haploid yeast strain PJ43-2b (MATα trp1-1 ura3-52 can1-100 leu2-3,112 his3-11,15 ade2-1 met2-Δ1 lys2-Δ2; kindly provided by Phil James), while NruI-cut pRS306-SCIN−B was transformed into strain AKA001. AKA001 is isogenic to PJ43-2b, except that it is MATa and has the lethal ‘C4G’ mutation integrated at the SNR6 downstream B block (23), which is complemented with a pseudo-wild-type copy of SNR6 carried on the LYS2-marked plasmid pRS317 [LYS2 CEN6 ARSH4; (28)]. After SCIN+B ‘pop-in’, the strain was transformed with pRS317-SNR6, followed by plating of both strains on medium containing 5-FOA to select for loss of the URA3 gene along with one of the two copies of chromosomal SNR6 by intra-chromosomal homologous recombination (‘pop-out’). After the ‘pop-out’, the LYS2-marked SNR6 plasmid was replaced with a URA3-marked plasmid pRS316 [URA3 CEN6 ARSH4; (20)] that contains a pseudo-wild-type copy of SNR6 with minimal upstream and downstream sequences. The presence of the mutant SNR6 alleles was confirmed by PCR amplification of the genomic locus and direct sequencing of the PCR product.
Total cellular RNA was isolated using the guanidinium thiocyanate method (29), including a 65°C phenol extraction. Primer extension analysis (13) of 4 μg of total cellular RNA was carried out using 32P-labeled oligonucleotide 6D (complementary to the 3′ end of U6 RNA) or SPIN3 (see above). In the experiment shown in Figure 5, oligonucleotide U1-SH was also included to measure the U1 RNA level for normalization. The cDNA products were electrophoresed on 6% polyacrylamide, 8.3 M urea gels. The gels were exposed to film as described for in vitro transcription, or visualized with a PhosphorImager (Molecular Dynamics). Data were quantitated with the Molecular Dynamics ImageQuant software.
Figure 5.
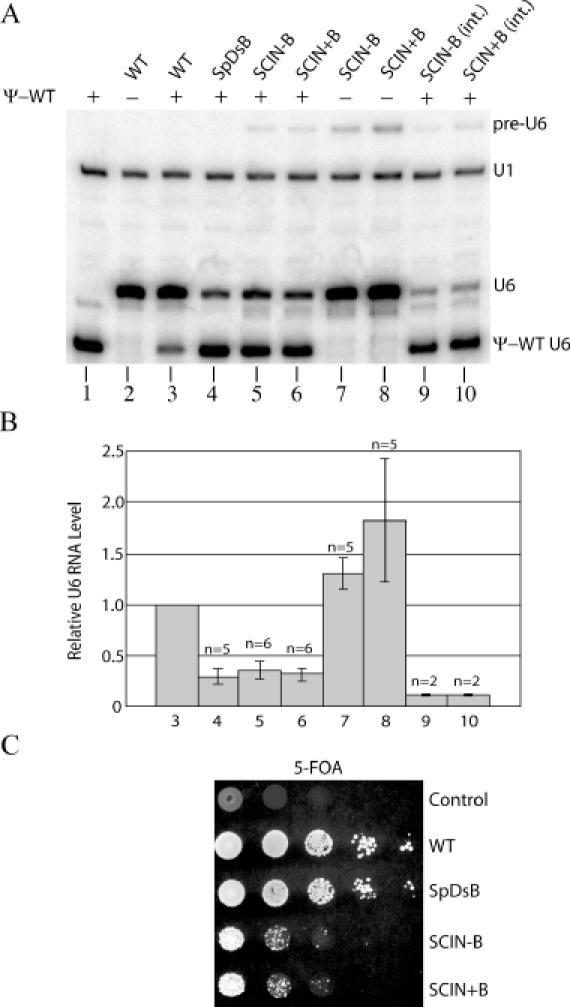
In vivo expression levels and function of plasmid-borne and integrated SNR6–SCIN alleles. (A) Primer extension analysis of U6 and U1 (internal control) RNA levels in strains transformed with SNR6–SpDsB and SNR6–SCIN constructs. A URA3-marked centromere plasmid bearing the pseudo-wild-type (Ψ-WT) SNR6 allele was present (+) or absent (−) as indicated. The allele of SNR6 present on a TRP1-marked centromere plasmid (lanes 2–8) or integrated (int.) at the SNR6 locus (lanes 9 and 10) is listed above each lane. The position of pre-U6, U6, Ψ-WT U6 and U1 cDNAs are indicated on the right. (B). Relative U6 RNA levels in the strains described in (A). The sample numbers below each bar correspond to the lanes of the gel in (A). The number of determinations for each sample is indicated. Error bars denote standard deviation, except for samples 9 and 10, where they denote the range. Pre-U6 RNA was not included in the quantitation, as it accounted for <5% of the total U6 RNA. The ratio of U6 to U1 in each sample was normalized to the U6/U1 ratio in sample #3 (WT + Ψ-WT). (C) Growth phenotypes of strains bearing mutant SNR6 alleles on a TRP1-marked centromere plasmid. Cells were grown overnight in trp media before spotting on medium containing 5-FOA to select for loss of the Ψ-WT U6 gene. Spotted left to right are 10 μl aliquots of successive 10-fold dilutions of an A600 = 10 suspension of cells. Plates were incubated at 30°C for 3 days. The TRP1 plasmid-borne alleles of SNR6 are indicated on the right. The control strain has a TRP1 plasmid without a SNR6 allele.
RESULTS
The 50 bp S.pombe U6 RNA intron was inserted into SNR6 alleles that include or lack the downstream B block (Figure 1). The resulting alleles are named SPIN+B (S.pombe intron + downstream B block) and SPIN−B, respectively. The intron position is identical in the S.pombe U6 gene and the SNR6–SPIN alleles, relative to the conserved central core of the U6 RNA (30). The fourth position of the intron was changed from an A to a T to create a perfect match to the S.cerevisiae 5′ splice site consensus (Figure 1).
In vitro transcription of intron-containing alleles of SNR6
The promoter activity of the SPIN±B alleles was tested in vitro with yeast subcellular extract. Wild-type SNR6 (Figure 2, lanes 3 and 5) directs synthesis of two sets of transcripts: full-length U6 RNA (112–115 nt) and downstream transcripts ∼240 nt in length, which initiate downstream of the SNR6 terminator and terminate at position +420 (13). The downstream transcripts use the SNR6 B block as an intragenic element, in conjunction with A-block-like sequences downstream of the U6 RNA coding region. These transcripts are not detected in vivo, presumably because the native chromatin represses their formation. As expected, removal of most of the downstream B block by 3′ truncation abolishes synthesis of both U6 RNA and the downstream transcripts (Figure 2, lane 4). Both the SPIN−B (lane 6) and SPIN+B (lane 7) alleles produce three major sets of transcripts that are ∼160–165, 150–155 and 140 nt long. The 160–165 nt transcripts correspond in size to full-length pre-U6 RNA (intron-containing U6 RNA), which is expected to be 162–166 nt long if the normal SNR6 initiation and termination sites are utilized. Primer extension analysis with an intron-specific oligonucleotide revealed that the transcripts produced in vitro all initiate at the normal +1 position (data not shown), indicating that the bands below the 160–165 nt band are probably 3′ degradation products of full-length pre-U6 RNA. The 3′ degradation products of wild-type U6 RNA are also observed when long incubation times are used (13). No transcripts corresponding in length to properly spliced product were detected in vitro, but the subcellular extract is not known to be active for pre-mRNA splicing.
Figure 2.
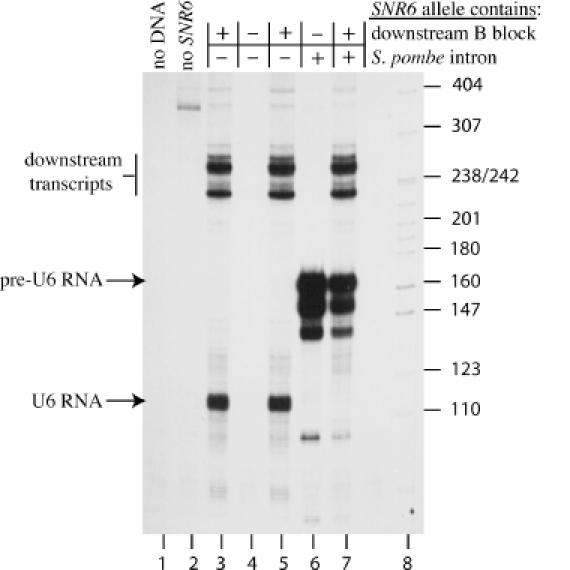
In vitro transcription of SNR6–SPIN alleles. Transcription in yeast subcellular extract was carried out as described in Materials and Methods using 100 ng of plasmid DNA per reaction, and the products were resolved on a denaturing polyacrylamide gel. Positions of U6 RNA, pre-U6 RNA and downstream transcripts are indicated on the left. The presence (+) or absence (−) of the intact SNR6 downstream B block and the S.pombe U6 RNA gene intron (SPIN) is indicated above each lane. Lane 1 contains the products of a reaction with no added plasmid DNA. The following plasmids were used: lane 2, pUC118; lane 3, p-539H6; lane 4, pCCs6; lane 5, pEP6; lane 6, pCCs6-SPIN; lane 7, pEP6-SPIN. Lane 8 contains 32P-labeled MspI cut pBR322 markers. The size of the markers (in nucleotides) is indicated on the right.
The SPIN−B allele produces no downstream transcripts, and makes several times more pre-U6 RNA than the SPIN+Ballele (compare lanes 6 and 7). The decreased synthesis of pre-U6 RNA in the presence of the downstream B block is almost certainly a result of competition between the two B blocks for TFIIIC. The fact that approximately equal amounts of downstream transcripts are produced in the presence and absence of the S.pombe intron suggests that the downstream SNR6 B block binds TFIIIC more strongly than the intron B block. This notion is supported by in vitro DNase I footprinting data, which show that the SPIN−B allele is a poor competitor for the downstream B block footprint on SNR6 [(13); V. L. Gerlach and D. A. Brow, unpublished data]. Thus, the S.pombe U6 intron provides strong B block function in vitro, but may compete poorly with the downstream B block for TFIIIC.
In vivo expression of intron-containing SNR6 alleles
We tested if the intron-containing alleles of SNR6 are also well transcribed in vivo, and if so, whether the S.pombe intron is spliced out of pre-U6 RNA in S.cerevisiae. The SPIN−B and SPIN+B alleles on yeast centromere plasmids were introduced into a yeast strain in which the chromosomal SNR6 locus is disrupted and wild-type SNR6 is carried on a URA3-marked plasmid. Transformants were plated on medium containing 5-FOA to select for loss of the URA3 plasmid. After 4 days of incubation at 30°C no colonies arose in the SPIN−B and SPIN+B sectors, indicating that neither of the SPIN alleles is able to function as the sole copy of SNR6. RNA was examined from the strains containing both wild-type SNR6 and either of the SPIN alleles by primer extension using a 32P-labeled oligonucleotide complementary to the 3′ end of U6 RNA. This analysis revealed that total cellular RNA from SPIN+B and SPIN−B strains contains low amounts of a transcript of the expected size of pre-U6 RNA (Figure 3A, lanes 6 and 7). Interestingly, the SPIN+B allele makes much more pre-U6 RNA than the SPIN−B allele, in contrast to what was seen in vitro. This indicates that the intron B block is poorly utilized in vivo. To verify the identity of pre-U6 RNA, primer extension sequencing was carried out using an oligonucleotide complementary to positions 22 to 35 of the intron sequence. The pre-U6 RNA transcript produced by the SPIN−B and SPIN+B alleles initiates properly at +1 of SNR6 (Figure 3B).
Figure 3.
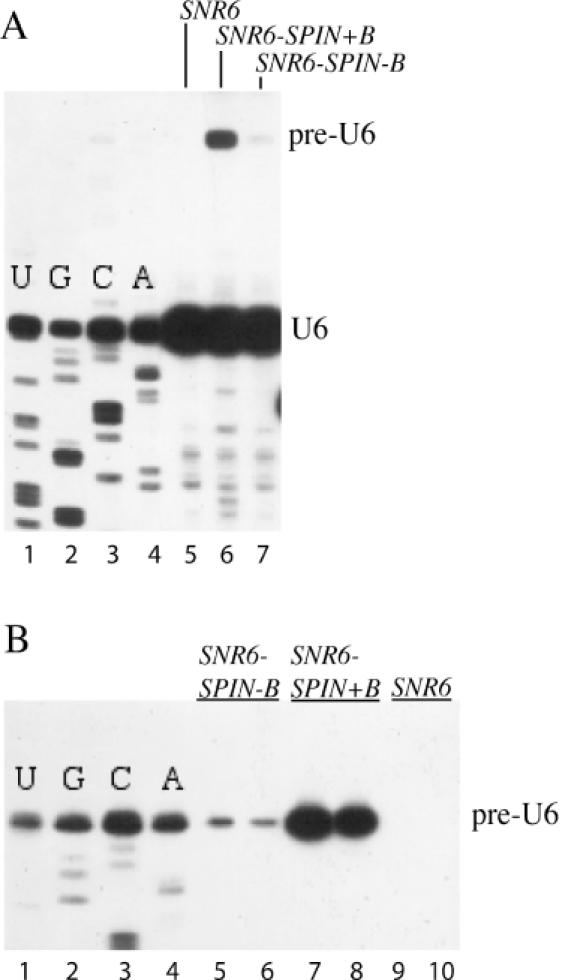
In vivo expression of SNR6-SPIN alleles. Primer extension analysis of total cellular RNA from strains containing SNR6–SPIN alleles (or a wild-type control) on a TRP1-marked centromere plasmid and wild-type SNR6 on a URA3-marked centromere plasmid. (A) Primer extension was done using 32P-labeled oligonucleotide 6D (complementary to the 3′ end of U6 RNA) and the resulting cDNA products were run on a denaturing 6% polyacrylamide gel. Lanes 1–4 (U, G, C, A) represent a sequencing ladder generated from the RNA used in lane 6. Lanes 5–7 show cDNAs from strains with the indicated alleles on the TRP1 plasmid. The positions of pre-U6 and U6 cDNAs are indicated on the right. (B) Primer extension was done using 32P-labeled oligonucleotide SPIN3 (complementary to positions 22 to 35 of the S.pombe U6 intron) and the resulting cDNA products were run on a denaturing 6% polyacrylamide gel. Lanes 1–4 (U, G, C, A) represent a sequencing ladder generated from the RNA used in lane 7. Lanes 5–10 show cDNAs from duplicate RNA preparations from strains containing the indicated alleles on the TRP1 centromere plasmid. The position of pre-U6 cDNA is indicated on the right.
To determine if any of the pre-U6 RNA that is made is properly spliced, the SPIN+B allele on either a low-copy centromere plasmid or a high-copy plasmid with a 2 μm origin was introduced into a strain that carries a pseudo-wild-type U6 RNA gene as its sole functional copy of SNR6. The pseudo-wild-type U6 gene makes an RNA that is 13 nt shorter at its 5′ end than wild-type U6 RNA (25). This RNA can be distinguished from full-length spliced U6 RNA in a primer extension reaction when an oligonucleotide complementary to the 3′ end of U6 RNA is used. Little or no spliced U6 RNA is detected in cells carrying the SPIN+B allele on a low-copy centromere plasmid (Figure 4, lane 3). Expression of pre-U6 RNA from a 2 μm plasmid is ∼15-fold higher than from a centromere plasmid, yet only a faint band corresponding in size to mature U6 RNA can be seen (Figure 4, lane 5). Thus, it appears that the pre-U6 RNA produced from the SPIN+B allele is spliced very inefficiently, if at all, in vivo. Consequently, the low level of pre-U6 RNA that accumulates from the SPIN+B allele must reflect either decreased transcription due to the intron insertion or rapid degradation of the unspliced pre-U6RNA (or some combination thereof).
Figure 4.
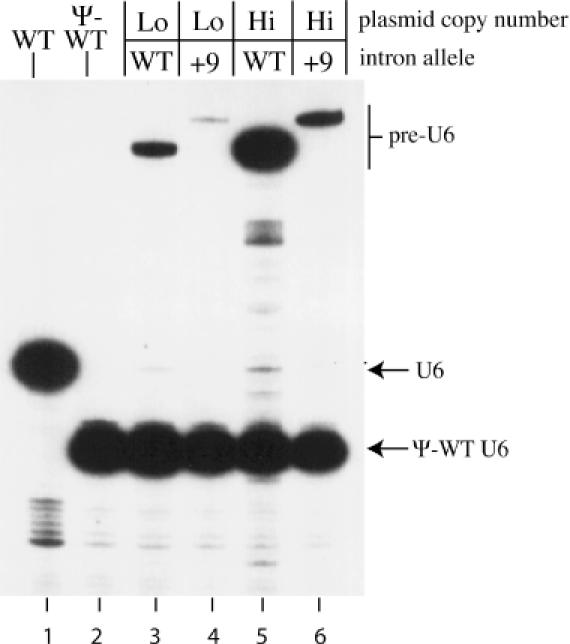
Inefficient splicing of SPIN pre-U6 RNA in vivo. Primer extension analysis of total cellular RNA from strains containing SNR6–SPIN+B alleles on low-copy-number or high-copy-number plasmids. Primer extension was done using 32P-labeled oligonucleotide 6D (complementary to the 3′ end of U6 RNA) and the resulting cDNA products were run on a denaturing 6% polyacrylamide gel. The positions of pre-U6, U6 and Ψ-WT U6 cDNAs are indicated on the right. Lane 1, RNA from a strain containing only wild-type (WT) SNR6 on a centromere plasmid. Lane 2, RNA from a strain containing only pseudo-wild-type (Ψ-WT) SNR6 on a centromere plasmid. Lanes 3–6, RNA from strains containing Ψ-WT SNR6 on a centromere plasmid and SNR6–SPIN+B (lanes 3 and 5) or SNR6–SPIN+B+9 (lanes 4 and 6) on a TRP1-marked centromere (lanes 3 and 4) or 2 μm (lanes 5 and 6) plasmid.
Increasing the distance between the 5′ splice site and branchpoint sequences facilitates splicing of pre-U6 RNA in S.cerevisiae
Given that the S.pombe U6 intron contains consensus S.cerevisiae splice signals, we hypothesized that the lack of splicing of the SPIN+B transcript in S.cerevisiae is due to the close spacing of these elements. The shortest known S.cerevisiae intron is the 52 nt intron 2 of MATa1 (31), which has 42 nt from the 5′ splice site through branchpoint A residue (lariat length) and 10 nt between the branchpoint A and the 3′ splice site (tail length). Reduction of the lariat length to 37 nt has been shown to inactivate splicing of MATa1 intron 1 (32). The S.pombe U6 intron has a 38 nt lariat length and 12 nt tail length. Therefore, the lack of splicing of this intron is most likely due to inadequate spacing between the 5′ splice site and branchpoint. Consistent with this expectation, insertion of the sequence TTTGTTTCT between the branchpoint and 3′ splice site of the U6 intron (SPIN+9 allele) did not improve splicing (Figure 4, lanes 4 and 6). Unexpectedly, this mutation resulted in a greatly decreased level of pre-U6 RNA.
To determine if increasing the lariat length would increase the splicing efficiency, insertions of 4 or 12 bp were made at a unique StyI restriction site immediately upstream of the intron B block in the SPIN+B allele. The 4 nt insertion strongly enhanced splicing of the pre-U6 RNA, resulting in ∼40% spliced U6 RNA, while the 12 nt insertion resulted in >90% splicing efficiency (data not shown, but see below). Thus, the splicing defect of the S.pombe U6 gene intron in S.cerevisiae can be corrected by increasing the 5′ splice site to branchpoint spacing.
Sequence dependence and position dependence ofSNR6 B block function
The fact that the S.pombe B block competes poorly for TFIIIC in vitro (see above) suggests that it may not have the optimal sequence for binding to S.cerevisiae TFIIIC. Indeed, when we replaced 26 bp encompassing the SNR6 downstream B block (+228 to +253) with the equivalent 26 bp from the S.pombe U6 intron (‘SpDsB’ in Figure 1), the transcription efficiency of SNR6 decreased ∼3-fold in vivo (Figure 5A and B, lane 4). In an attempt to increase both the splicing efficiency of the S.pombe intron in S.cerevisiae and its affinity for S.cerevisiae TFIIIC, we replaced the 9 bp B block core and 7 bp of upstream sequence with base pairs +228 to +253 of SNR6 (‘SCIN’ in Figure 1). This substitution increases the distance between the 5′ splice site and branchpoint sequence by 10 bp, and so should result in efficient splicing. Furthermore, it contains the S.cerevisiae B block and flanking sequences protected from DNase I by TFIIIC binding (13), and so should bind TFIIIC as efficiently as the downstream B block, if accessible. We call this mutant form of the S.pombe U6 intron ‘SCIN’, which stands for S.cerevisiae intron, and we generated both SCIN+B and SCIN−B alleles, which include or lack the wild-type downstream B block, respectively.
Plasmid shuffle analysis showed that both the SCIN+B and SCIN−B alleles complement the SNR6 disruption when carried on a centromere plasmid, indicating that they produce sufficient spliced U6 RNA to support life (Figure 5C). However, the viability of SCIN+B and SCIN−B strains is low relative to strains with wild-type SNR6, or with the S.pombe B block in a downstream location (Figure 5C). We performed primer extension analysis of total cellular RNA from strains that contain both a pseudo-wild-type U6 gene and either a SCIN+B or SCIN−B allele on low-copy plasmids, as well as strains picked from 5-FOA plates that contain the SCIN+B or SCIN−B allele as their sole source of U6 RNA. In the heterozygous strains, both the SCIN+B and SCIN−B alleles are expressed at a 3-fold lower level than wild-type U6 RNA (Figure 5A and B, lanes 5 and 6). The very low level of pre-U6 RNA that is observed (<5% of total U6) suggests that the expression defect is due to decreased transcription rather than inefficient splicing. Interestingly, the downstream B block appears to be completely inactive in this context, as its presence has no effect on the U6 RNA level.
Surprisingly, when the SCIN+B or SCIN−B allele is the only SNR6 allele in the cell, the level of U6 RNA is ∼1.5-fold higher than wild type. This finding suggests that there is strong selection for increased copy number of the SCIN plasmids when pseudo-wild-type U6 RNA is not present, a conclusion consistent with the slow growth of the strains bearing a SCIN allele as their only copy of SNR6 (Figure 5C). However, our past experience with B block point mutants (23) indicates that such selection normally takes place only when U6 RNA levels fall below ∼25% of wild-type levels. Therefore, the high level of SCIN+B- and SCIN−B-derived U6 RNA in the strains lacking pseudo-wild-type U6 suggests that spliced U6 RNA has significantly reduced function relative to wild-type U6 RNA.
Integration of the SCIN alleles into the SNR6 locus impairs their expression
To prevent changes in gene dosage from influencing the level of U6 RNA expression from the SCIN alleles, and to assure that the alleles are present in the context of native SNR6 chromatin, we integrated the SCIN alleles at the SNR6 locus on chromosome XII. To generate the integrated SCIN−B allele, we used a yeast strain with a single lethal point mutation in the chromosomal SNR6 downstream B block as the recipient. This point mutation, C4G, reduces B block function ∼50-fold in vivo (23). Neither the SCIN+B nor SCIN−B integrant strain is viable in the absence of a functional SNR6 allele on a plasmid, consistent with our conclusion that increased gene dosage is required to make adequate amounts of U6 RNA from the SCIN alleles. Surprisingly, the integrated SCIN alleles produce only 11–12% of the wild-type level of U6 RNA (Figure 5A and B, lanes 9 and 10). These results indicate that the SNR6 downstream B block element cannot function efficiently from an intronic location in the context of native chromatin, and that promoter activity cannot be rescued by an additional B block in the conventional downstream location, perhaps because of the increased distance from the A block caused by the intron insertion. The 3-fold lower level of SNR6–SCIN expression from the chromosome than from a centromere plasmid in the presence of pseudo-wild-type U6 RNA suggests either that there is some selection for increased copy number even when pseudo-wild-type U6 is available, or that the chromatin structure on the plasmid is more permissive to intronic B block function than is the chromatin structure at the chromosomal SNR6 locus.
DISCUSSION
Our impetus for inserting the S.pombe intron into the S.cerevisiae U6 RNA gene, SNR6, was to determine the effect of moving the SNR6 B block from its unique downstream location to a conventional intragenic location. Several modifications of our initial constructs were required before we could address this issue. First, the 5′ splice site to branchpoint distance had to be increased to allow efficient removal of the intron from pre-U6 RNA. This was necessary to assure that the steady-state level of U6 RNA accurately reflects the rate of transcription of SNR6. Second, the S.pombe B block element had to be replaced with the S.cerevisiae SNR6 B block sequence, so that B block function would be altered only by the change in location, and not the sequence specificity of TFIIIC binding. Third, it was necessary to integrate the mutant alleles at the SNR6 locus to control copy number and provide a native chromatin environment. When all of these modifications are made, we find that an intronic B block is utilized ∼9-fold less efficiently than a downstream B block in the context of the SNR6 locus. Furthermore, while the addition of a downstream B block did stimulate transcription of a plasmid-borne construct with the S.pombe intronic B block sequence (SPIN+B; Figure 3), it did not do so either for plasmid-borne or integrated alleles containing the S.cerevisiae B block sequence in the intron (SCIN+B; Figure 5).
The most likely explanation for decreased SNR6 B block function in the intronic location is the presence of a chromatin structure that prevents access of TFIIIC to the promoter element. Chromatin footprinting studies detected a protected region of ∼90 bp centered on the terminator of SNR6 (33,34). This protection is presumably due to a protein complex that may also bind to the SCIN alleles and occlude the intronic B block. While the identity of the factor(s) that protect the DNA from nuclease cleavage in this region is unknown, it is known that the non-histone chromatin protein Nhp6 is important for efficient SNR6 transcription (15,35,36). As an HMG-box protein, Nhp6 may either bind directly to the SNR6 gene in vivo, or it may recruit or modify the binding of other chromatin proteins (37). This same chromatin structure may be the reason that addition of the downstream B block does not increase expression of the SCIN allele. We have proposed that the chromatin structure evolved to bring the SNR6 A and B blocks into optimal spatial arrangement for binding of TFIIIC. An increase of 60 bp between the A and B blocks due to insertion of the modified U6 gene intron (SCIN) may place these elements in an unfavorable geometry for binding of TFIIIC, when constrained by the chromatin structure. A strong prediction of this hypothesis is that deletion of 60 bp between the terminator and B block of the SCIN+B allele would greatly increase transcription of this allele at the SNR6 locus.
An alternative possibility is that the SNR6 B block element impedes elongation by Pol III when present within the transcribed region. Since most B blocks are intragenic, this must not be a general property of B block elements. Yet, it could be that the SNR6 downstream B block has sequence characteristics that result in stable retention of TFIIIC. Chromatin footprinting of SNR6 gives a protection pattern consistent with a high level of occupancy of TFIIIC at the downstream B block in vivo (33). However, this could simply be a consequence of the lack of transcription through the downstream element, rather than indicative of an unusually stable association with TFIIIC.
The low viability of strains that carry only the SCIN+B or SCIN−B allele on a plasmid despite the relatively high levels of mature U6 RNA produced in these strains (Figure 5) suggests that the spliced U6 RNA is not fully functional. An alternative explanation, that pre-U6 has a dominant inhibitory effect, seems unlikely since heterozygous strains that produce high levels of pre-U6 (e.g. Figure 4, lane 5) grow well. Decreased function of spliced U6 RNA could be due to as yet undetected changes in the covalent or non-covalent (secondary or tertiary) structure of the spliced RNA, although intron removal appears to occur correctly. Another possibility is that a protein that is normally recruited to spliced mRNAs, such as Yra1 (38), associates with U6 RNA during splicing and either blocks U6 snRNP assembly, or results in export of U6 RNA to the cytoplasm. Indeed, we cannot exclude the possibility that part or all of the decreased expression of U6 RNA from the SCIN alleles is due to increased turnover as a consequence of traversing the splicing pathway. Further experiments will be required to fully understand the cellular adaptations that have evolved to accommodate the unusual promoter architectures of both the S.cerevisiae and S.pombe U6 RNA genes.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Claudia Reich and Jo Ann Wise for providing the S.pombe U6 RNA gene plasmid, Valerie Gerlach, Michael Martin and Anisa Kaenjak-Angeletti for providing yeast strains, and Eric Steinmetz and Jason Kuehner for comments on the manuscript. This work was supported by grant GM44665 from the NIH. M.W.K. was a trainee of NIH training grant T32 GM07215.
REFERENCES
- 1.Schramm L. and Hernandez,N. (2002) Recruitment of RNA polymerase III to its target promoters. Genes Develop., 16, 2593–2620. [DOI] [PubMed] [Google Scholar]
- 2.Geiduschek E.P. and Kassavetis,G.A. (2001) The RNA polymerase III transcription apparatus. J. Mol. Biol., 310, 1–26. [DOI] [PubMed] [Google Scholar]
- 3.Chaussivert N., Conesa,C., Shaaban,S. and Sentenac,A. (1995) Complex interactions between yeast TFIIIB and TFIIIIC. J. Biol. Chem., 270, 15353–15358. [DOI] [PubMed] [Google Scholar]
- 4.Baker R.E., Camier,S., Sentenac,A. and Hall,B.D. (1987) Gene size differentially affects the binding of yeast transcription factor tau to two intragenic regions. Proc. Natl Acad. Sci. USA, 84, 8768–8772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cannon R.E., Wu,G.-J. and Railey,J.F. (1986) Functions of and interactions between the A and B blocks in adenovirus type 2-specific VARNA1 gene. Proc. Natl Acad. Sci. USA, 83, 1285–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee J.-Y., Evans,C.F. and Engelke,D.R. (1991) Expression of RNase P RNA in Saccharomyces cerevisiae is controlled by an unusual RNA polymerase III promoter. Proc. Natl Acad. Sci. USA, 88, 6986–6990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harismendy O., Gendrel,C.-G., Soularue,P., Gidrol,X., Sentenac,A., Werner,M. and Lefebvre,O. (2003) Genome-wide location of yeast RNA polymerase III transcription machinery. EMBO J., 22, 4738–4747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tani T. and Ohshima,Y. (1989) The gene for the U6 small nuclear RNA in fission yeast has an intron. Nature, 337, 87–90. [DOI] [PubMed] [Google Scholar]
- 9.Reich C. and Wise,J.A. (1990) Evolutionary origin of the U6 small nuclear RNA intron. Mol. Cell. Biol., 10, 5548–5552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Frendewey D., Barta,I., Gillespie,M. and Potashkin,J. (1990) Schizosaccharomyces U6 genes have a sequence within their introns that matches the B box consensus of tRNA internal promoters. Nucleic Acids Res., 18, 2025–2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Potashkin J. and Frendewey,D. (1989) Splicing of the U6 RNA precursor is impaired in fission yeast pre-mRNA splicing mutants. Nucleic Acids Res., 17, 7821–7831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brow D.A. and Guthrie,C. (1990) Transcription of a yeast U6 snRNA gene requires a polymerase III promoter element in a novel position. Genes Develop., 4, 1345–1356. [DOI] [PubMed] [Google Scholar]
- 13.Eschenlauer J.B., Kaiser,M.W., Gerlach,V.L. and Brow,D.A. (1993) Architecture of a yeast U6 RNA gene promoter. Mol. Cell. Biol., 13, 3015–3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burnol A.-F., Margottin,F., Schultz,P., Marsolier,M.-C., Oudet,P. and Sentenac,A. (1993) Basal promoter and enhancer element of yeast U6 snRNA gene. J. Mol. Biol., 233, 644–658. [DOI] [PubMed] [Google Scholar]
- 15.Martin M.P., Gerlach,V.G. and Brow,D.A. (2001) A novel upstream RNA polymerase III promoter element becomes essential when the chromatin structure of the yeast U6 RNA gene is altered. Mol. Cell. Biol., 21, 6429–6439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li Z. and Brow,D.A. (1993) A rapid assay for quantitative detection of specific RNAs. Nucleic Acids Res., 21, 4645–4646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brow D.A. and Guthrie,C. (1988) Spliceosomal RNA U6 is remarkably conserved from yeast to mammals. Nature, 334, 213–218. [DOI] [PubMed] [Google Scholar]
- 18.Elledge S.J. and Davis,R.W. (1988) A family of versatile centromeric vectors designed for use in the sectoring-shuffle mutagenesis assay in Saccharomyces cerevisiae. Gene, 70, 303–312. [DOI] [PubMed] [Google Scholar]
- 19.Christianson T.W., Sikorski,R.S., Dante,M., Shero,J.H. and Hieter,P. (1992) Multifunctional yeast high-copy-number shuttle vectors. Gene, 110, 119–122. [DOI] [PubMed] [Google Scholar]
- 20.Sikorski R.S. and Hieter,P. (1989) A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics, 122, 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Evans C.F. and Engelke,D.R. (1990) Yeast extracts for transfer RNA gene transcription and processing. Methods Enzymol., 181, 439–450. [DOI] [PubMed] [Google Scholar]
- 22.Maniatis T., Fritsch,E.F. and Sambrook,J. (1982) Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 23.Kaiser M.W. and Brow,D.A. (1995) Lethal mutations in a yeast U6 RNA gene B block promoter element identify essential contacts with transcription factor-IIIC. J. Biol. Chem., 270, 11398–11405. [DOI] [PubMed] [Google Scholar]
- 24.Ito H., Fukuda,Y., Murata,K. and Kimura,A. (1983) Transformation of intact yeast cells treated with alkali cations. J. Bacteriol., 153, 163–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Madhani H.D., Bordonné,R. and Guthrie,C. (1990) Multiple roles for U6 snRNA in the splicing pathway. Genes Dev., 4, 2264–2277. [DOI] [PubMed] [Google Scholar]
- 26.Boeke J.D., Truehart,J., Natsoulis,G. and Fink,G.R. (1987) 5-Fluoroorotic acid as a selective agent in yeast molecular genetics. Methods Enzymol., 154, 164–175. [DOI] [PubMed] [Google Scholar]
- 27.Scherer S. and Davis,R.W. (1979) Replacement of chromosome segments with altered DNA sequences constructed in vitro. Proc. Natl Acad. Sci. USA, 76, 4951–4955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sikorski R.S. and Boeke,J.D. (1991) In vitro mutagenesis and plasmid shuffling: from cloned gene to mutant yeast. Methods Enzymol., 194, 302–318. [DOI] [PubMed] [Google Scholar]
- 29.Wise J.A., Tollervey,D., Maloney,D., Swerdlow,H., Dunn,E.J. and Guthrie,C. (1983) Yeast contains small nuclear RNAs encoded by single copy genes. Cell, 35, 743–751. [DOI] [PubMed] [Google Scholar]
- 30.Fortner D.M., Troy,R.G. and Brow,D.A. (1994) A stem/loop in U6 RNA defines a conformational switch required for pre-mRNA splicing. Genes Develop., 8, 221–233. [DOI] [PubMed] [Google Scholar]
- 31.Spingola M., Grate,L., Haussler,D. and Ares,M.,Jr (1999) Genome-wide bioinformatic and molecular analysis of introns in Saccharomyces cerevisiae. RNA, 5, 221–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Köhrer K. and Domdey,H. (1988) Splicing and spliceosome formation of the yeast MATa1 transcript require a minimum distance from the 5′ splice site to the internal branch acceptor site. Nucleic Acids Res., 16, 9457–9475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gerlach V.L., Whitehall,S.K., Geiduschek,E.P. and Brow,D.A. (1995) TFIIIB placement on a yeast U6 RNA gene in vivo is directed primarily by TFIIIC rather than by sequence-specific DNA contacts. Mol. Cell. Biol., 15, 1455–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marsolier M.C., Tanaka,S., Livingstone-Zatchej,M., Grunstein,M., Thoma,F. and Sentenac,A. (1995) Reciprocal interferences between nucleosomal organization and transcriptional activity of the yeast SNR6 gene. Genes Develop., 9, 410–422. [DOI] [PubMed] [Google Scholar]
- 35.Kruppa M., Moir,R.D., Kolodrubetz,D. and Willis,I.M. (2001) Nhp6, an HMG1 protein, functions in SNR6 transcription by RNA Polymerase III in S. cerevisiae. Mol. Cell, 7, 309–318. [DOI] [PubMed] [Google Scholar]
- 36.Lopez S., Livingstone-Zatchej,M., Jourdain,S., Thoma,F., Sentenac,A. and Marsolier,M.C. (2001) High-mobility-group proteins NHP6A and NHP6B participate in activation of the RNA polymerase III SNR6 gene. Mol. Cell. Biol., 21, 3096–3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thomas J.O. and Travers,A.A. (2001) HMG1 and 2, and related ‘architectural’ DNA-binding proteins. Trends Biochem. Sci., 26, 167–174. [DOI] [PubMed] [Google Scholar]
- 38.Preker P.J., Kim,K.S. and Guthrie,C. (2002) Expression of the essential mRNA export factor Yra1p is autoregulated by a splicing-dependent mechanism. RNA, 8, 969–980. [DOI] [PMC free article] [PubMed] [Google Scholar]