


Abstract
Hexitol nucleic acids (HNAs) are nuclease resistant and provide strong hybridization to RNA. However, there is relatively little information on the biological properties of HNA antisense oligonucleotides. In this study, we compared the antisense effects of a chimeric HNA ‘gapmer’ oligonucleotide comprising a phosphorothioate central sequence flanked by 5′ and 3′ HNA sequences to conventional phosphorothioate oligonucleotides and to a 2′-O-methoxyethyl (2′-O-ME) phosphorothioate ‘gapmer’. The antisense oligomers each targeted a sequence bracketing the start codon of the message of MDR1, a gene involved in multi-drug resistance in cancer cells. Antisense and control oligonucleotides were delivered to MDR1-expressing cells using transfection with the cationic lipid Lipofectamine 2000. The anti-MDR1 HNA gapmer was substantially more potent than a phosphorothioate oligonucleotide of the same sequence in reducing expression of P-glycoprotein, the MDR1 gene product. HNA and 2′-O-ME gapmers displayed similar potency, but a pure HNA antisense oligonucleotide (lacking the phosphorothioate ‘gap’) was ineffective, indicating that RNase H activity was likely required. Treatment with anti-MDR1 HNA gapmer resulted in increased cellular accumulation of the drug surrogate Rhodamine 123 that correlated well with the reduced cell surface expression of P-glycoprotein. Thus, HNA gapmers may provide a valuable additional tool for antisense-based investigations and therapeutic approaches.
INTRODUCTION
The development of resistance to chemotherapeutic agents is a major obstacle in the treatment of cancer. P-glycoprotein, the product of MDR1 gene, is a transmembrane ATP-dependent drug efflux pump that belongs to ABC transporter family. P-glycoprotein is found in many types of cancer cells (1–4) as well as in normal tissues including liver, kidney, intestine, placenta and the blood brain barrier (5,6). Expression of MDR1 has been correlated with the increased drug resistance and decreased clinical response in human cancers (7–9). P-glycoprotein over-expression is also believed to be closely related to the development of cancer cells, and inhibition of the protein has been shown to inhibit the growth of various carcinomas (10,11). However, most small-molecule inhibitors of P-glycoprotein suffer from high toxicity, complicated drug–drug interactions and metabolic interference, most notably through P-450 enzymes (12,13).
Nucleic-acid-based drug design has provided therapeutic agents such as antisense oligonucleotides, hammerhead ribozymes and siRNA that provide an alternative to small molecule drugs. These oligonucleotides all act at the mRNA level in a sequence-specific manner (14). Antisense oligonucleotides usually work either by translation arrest or by target degradation through RNase H activation (15–20). Extensive modifications of antisense oligonucleotides have been developed to overcome the intrinsic instability of normal phosphodiester oligonucleotides in a cellular environment (21–26). However, most first- and second-generation modified antisense oligonucleotides suffer from undesirable effects due to non-specific binding to certain cell proteins (27), lack of RNase H activation (28,29), or poor cellular uptake (29). A chimeric or ‘gapmer’ approach has been employed to overcome these obstacles by combining stable modified residues at 5′ and 3′ positions with phosphodiester or phosphorothioate residues in a central segment; this has proved to be a worthwhile approach (30–35). The ribozyme approach has also encountered similar problems, and is in the process of optimization for therapeutic purposes (36). SiRNA is thought to work as an RNA–protein complex known as RNA-induced silencing complex, and its activity on target gene regulation has been widely demonstrated (37–41). Concerns about in vivo therapeutic application of siRNA include actions on non-targeted genes (42–44) and activation of the innate immune response (45). MDR1 gene expression has been successfully modulated by antisense oligonucleotides (34,35,46–51), hammerhead ribozymes (52–56) and siRNA (57,58).
Hexitol nucleic acids (HNAs; Figure 1) are oligonucleotide derivatives with a six-membered carbohydrate moiety that display strong hybridization affinity and resistance to nucleases (59). In solution, HNA–RNA hybrids form an A-type anti-parallel heteroduplex similar to dsRNA (60). Antisense effects of HNA oligonucleotides have been evaluated for mutated Ha-ras mRNA in a rabbit reticulocyte extract and for intracellular adhesion molecule-1 expression in a mammalian cell system (61). Also, the carbamoyl phosphate synthatase II gene of the human malaria parasite Plasmodium falciparum has been targeted (62). In the cellular study, HNAs were less effective than phosphorothioates; this was attributed to the lack of RNase H activation mechanism and low cellular uptake (59,61). However, this study did not make use of HNA gapmers capable of supporting RNaseH activity in the cell-based experiments.
Figure 1.
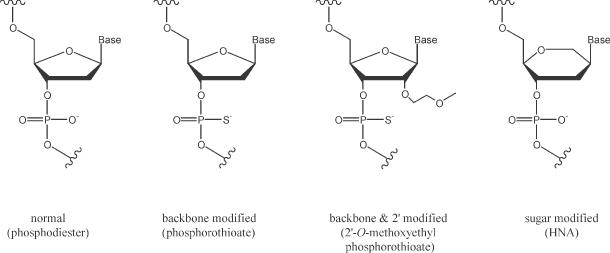
Chemical structures of oligonucleotides employed in this study as compared to phosphodiester oligonucleotides.
In the current study, chimeric HNA-phosphorothioate ‘gapmers’ targeting the AUG start codon of MDR1 gene were prepared and complexed with Lipofectamine 2000 for cellular delivery. The antisense effects of the HNA derivatives on multi-drug resistant (MDR) NIH 3T3 MDR cells and NCI/ADR-RES cells were compared to those of a pure phosphorothioate counterpart, as well as to a chimeric 2′-O-methoxyethyl (2′-O-ME) phosphorothioate oligonucleotide reported previously (34,35).
MATERIALS AND METHODS
Preparation of oligonucleotides
The HNA phosphoramidite building blocks were synthesized as described previously (63). The HNA and chimeric HNA-phosphorothioate derivatives were synthesized using the phophoramidites according to a previously reported procedure (64). Unmodified phosphorothioate oligonucleotide was purchased from Midland Certified Reagent Company (Midland, TX). Chimeric 2′-O-ME phosphorothioate derivatives were synthesized at ISIS Pharmaceticals using previously described procedures (65). Oligonucleotide sequences either targeting the AUG start codon of the MDR1 gene or scrambled/mismatched sequences are summarized in Table 1. All the oligonucleotides used in this study were purified by high-performance liquid chromatography (HPLC). Mass spectra were acquired for HNA derivatives on a quadrupole/orthogonal-acceleration time-of-flight tandem mass spectrometer equipped with a standard electrospray ionization interface: HNA antisense (GS1954) [calculated, 6350.3 mass units; found, 6350.7 mass units]; HNA mismatch (GS1955) [calculated, 6366.4 mass units; found, 6366.7 mass units]; HNA gapmer (GS1956) [calculated, 6366.0 mass units; found, 6366.7 mass units]; HNA gapmer mismatch (GS1957) [calculated, 6382.1 mass units; found, 6382.8 mass units].
Table 1. Oligonucleotides targeted to the MDR1 gene and scrambled/mismatched controls.
| Name | Target | Sequence | Chemical property |
|---|---|---|---|
| 5995 | AUG start codon of MDR1 | 5′-CCA TCC CGA CCT CGC GCT CC-3′ | PS |
| 10 221 | Scrambled control of 5995 | 5′-CAC CAC CCC CCT CGC TGG TC-3′ | PS |
| 13 758 | AUG start codon of MDR1 | 5′-CCA TCC CGA CCT CGC GCT CC-3′ | PS, 2′-O-ME and PS |
| 13 753 | Scrambled control of 13 758 | 5′-CAC CAC CCC CCT CGC TGG TC-3′ | PS, 2′-O-ME and PS |
| GS1954 | AUG start codon of MDR1 | 6′-CCA TCC CGA CCT CGC GCT CC-propanediol-4′ | HNA |
| GS1955 | Mismatched control of GS1954 | 6′-CCA TAC CAA CAT CAC GCT CC-propanediol-4′ | HNA |
| GS1956 | AUG start codon of MDR1 | 6′-CCA TCC CGA CCT CGC GCT CC-propanediol-4′ | PS, HNA |
| GS1957 | Mismatched control of GS1956 | 6′-CCA TAC CAA CAT CAC GCT CC-propanediol-4′ | PS, HNA |
| GS1962 | AUG start codon of MDR1 | 6′-fl-CCA TCC CGA CCT CGC GCT CC-propanediol-4′ | HNA |
| GS1964 | AUG start codon of MDR1 | 6′-fl-CCA TCC CGA CCT CGC GCT CC-propanediol-4′ | PS, HNA |
PS, phosphorothioate internucleotide linkage; 2′-O-ME, 2′-O-methoxyethylribonucleoside; HNA, HNA with phosphodiester internucleotide linkage; fl: fluorescein.
Non-italic, phosphodiester; italic, phosphorothioate; bold, 2'-O-ME; underline, HNA.
Cells
NIH 3T3 cells stably transfected with a plasmid containing the human MDR1 gene (pSKI MDR) were a gift from M. M. Gottesman (66). The NIH 3T3 cells expressing the human MDR1 gene (NIH 3T3 MDR cells) were grown in Dulbecco's Modified Eagle's Medium (DMEM) containing 10% fetal bovine serum (FBS) and 60 ng/ml of colchicine in a humidified atmosphere of 95% air and 5% CO2 at 37°C. MDR NCI/ADR-RES breast carcinoma cells were grown in minimum essential medium (MEM) containing 10% FBS under the same conditions. These cells have attained their MDR status via chronic exposure to doxorubicin.
Oligonucleotide treatment
Lipofectamine 2000 (Invitrogen, 2 μg/ml) complexes of an oligonucleotide in Opti-MEM were freshly prepared according to the manufacturer's recommendations. DMEM and MEM media were used for NIH 3T3 MDR cells and NCI/ADR-RES cells, respectively, throughout the experiments. The cells were seeded onto six-well plates in aliquots of 3 × 105 per well in the corresponding medium containing 10% FBS. After 24 h, cells were treated with the oligonucleotide Lipofectamine 2000 complex (2 μg/ml) in the corresponding fresh medium (2 ml) containing 10% FBS for 4 h at 37°C. The cells were then washed twice with 10% FBS/DMEM or 10% FBS/MEM and incubated in the corresponding medium at 37°C. After 1 h, cells were either washed again for oligonucleotide uptake analysis or plated on fibronectin-coated coverslips for confocal fluorescence microscopy analysis. For studies of cytotoxicity, expressed P-glycoprotein levels through immunostaining (by flow cytometry and western blotting) and Rhodamine 123 accumulation (by flow cytometry), cells were further incubated in the corresponding medium containing 2% FBS for 64 h.
Oligonucleotide uptake by cells
NIH 3T3 MDR cells were treated with fluorescently labeled HNA or HNA gapmer derivatives (1, 3, 9 and 27 nM) complexed with Lipofectamine 2000 (2 μg/ml) for 4 h in 10% FBS/DMEM. The cells were washed twice with 10% FBS/DMEM and incubated for 1 h in 10% FBS/DMEM. The cells were then washed with phosphate-buffered saline (PBS), detached and centrifuged at 1100 r.p.m. for 5 min. PBS was added to the sedimented cells for fluorescence uptake analysis using flow cytometry.
Western blotting for P-glycoprotein expression
After treating with an oligonucleotide as described above, and further incubation for 64 h, NIH 3T3 MDR cells were detached, counted for normalization and harvested for western analysis. The cells were lysed in a modified radioimmunoprecipitation buffer (150 mM NaCl, 50 mM Tris pH 7.4, 1% NP40, 0.5 mM deoxycholate, 5 mM EDTA, 1 mM dithiothreitol, 2 mM phenylmethylsulfonyl fluoride, 0.1% aprotinin and 0.1% SDS), and lysates were microfuged at 12 000 r.p.m. for 10 min at 4°C. Equal amounts of protein (20 μg) were mixed with 4× SDS sample buffer and were boiled for 5 min. The proteins were electrophoresed on a 7% SDS–polyacrylamide gel and the separated proteins were transferred onto polyvinylidene difluoride membranes (Millipore, Bedford, MA). The MDR1 expression was detected using monoclonal anti-P-glycoprotein C219 antibody (Signet Laboratory, Dedham, MA) at 2 μg/ml in 1% BSA. Peroxidase-conjugated rat anti-mouse immunoglobulin G (IgG) antibody (Calbiochem, San Diego, CA) at a dilution of 1:3000 was used as a secondary antibody in 3% BSA/1% Tween-20. Actin was detected by anti-actin primary antibody (Sigma-Aldrich) at a dilution of 1:6000. Signals were detected by enhanced chemiluminescence (ECL kit, Amersham Biosciences, Piscataway, NJ).
Confocal microscopy
Cellular uptake and subcellular distribution of HNA oligonucleotide derivatives were investigated by confocal fluorescence microscopy. NIH 3T3 MDR cells were treated with fluorescently labeled HNA derivative (10 nM) complexed with Lipofectamine 2000 (2 μg/ml) for 4 h in 10% FBS/DMEM. The cells were washed twice with 10% FBS/DMEM and incubated for 1 h in 10% FBS/DMEM. The cells were then plated on fibronectin (10 μg/ml) coated glass coverslips for 2 h in complete media at 37°C. The adherent cells were fixed in 4% formaldehyde for 10 min, rinsed twice with PBS, rinsed once with distilled water and then mounted onto glass slides with PermaFluor mounting medium (Shandon Immunon, Pittsburgh, PA). Images were taken using an Olympus confocal microscope with a 60× objective lens and processed using Fluoview software.
Cytotoxicity
NIH 3T3 MDR cells were treated with Lipofectamine 2000 complexes of oligonucleotides, washed and further incubated for 64 h as described above. The cells were then detached, washed twice with PBS and counted using an Elzone particle cell counter (Micromeritics, Norcross, GA) to measure the number of surviving cells. Longer-term studies of cytotoxicity were performed by assaying cell growth in complete medium over a period of five days after oligonucleotide treatment.
Immunostaining of P-glycoprotein
The P-glycoprotein expression on viable cell membrane surfaces was studied by immunostaining using a flow cytometry assay. After treating NIH 3T3 MDR cells or NCI/ADR-RES cells with oligonucleotide and further incubating them for 64 h, as described above, the cells were trypsinized, washed twice with PBS, counted for normalization and incubated with MRK16 (Kamiya, Seattle, WA) anti-P-glycoprotein primary antibody in PBS (20 μg/ml, 45 min) at 4°C. The cells were then washed with PBS three times, and treated with an anti-mouse IgG secondary antibody conjugated with R-phycoerythrin (Sigma, St Louis, MO) for 30 min in 10% FBS/PBS at 4°C and then washed with 10% FBS/PBS three times. The levels of immunostaining by R-phycoerythrin in viable cells (identified by light scattering) were then quantified on a Becton Dickinson flow cytometer using Cicero software (Cytomation, Fort Collins, CO).
Rhodamine 123 accumulation
The fluorophore Rhodamine 123 is a substrate for the P-glycoprotein efflux pump. Thus, the Rhodamine 123 accumulation is often used as a surrogate for drug uptake. NIH 3T3 MDR cells were treated with oligonucleotides complexed with Lipofectamine 2000 as described above. After 64 h, the cells were trypsinized and suspended in DMEM/10% FBS. The cells were washed once and resuspended in complete medium and warmed to 37°C before adding Rhodamine 123 (1 μg/ml). After 1 h at 37°C, cells were washed once with cold PBS and resuspended in PBS. The accumulation of Rhodamine 123 inside viable cells was measured by flow cytometery as described (35).
RESULTS
Description of the antisense oligonucleotides studied
Oligonucleotides prepared for this study are summarized in Table 1. Antisense activity against MDR1 has been previously described for the phosphorothioate oligonucleotides 5995 and 10 221, and for the chimeric 2′-O-ME phosphorothioate oligonucleotides 13 758 and 13 753 (34,35). Phosphorothioate oligonucleotide 5995, chimeric (gapmer) 2′-O-ME phosphorothioate oligonucleotide 13 758, HNA oligonucleotide GS 1954 and chimeric (gapmer) HNA phosphorothioate oligonucleotide GS1956 are directed to the AUG start codon region (positions 409–428) of MDR1 message, while phosphorothioate oligonucleotide 10 221, chimeric (gapmer) 2′-O-ME phosphorothioate oligonucleotide 13 753, HNA oligonucleotide GS1955 and chimeric (gapmer) HNA phosphorothioate oligonucleotide GS1957 are their scrambled and mismatched controls, respectively. HNA GS1962 and chimeric (gapmer) HNA phosphorothioate oligonucleotide GS1964 are fluorescently labeled sequences of GS1954 and GS1956, respectively.
Cellular uptake and subcellular distribution of HNA derivatives
Lipofectamine 2000 (2 μg/ml) complexes of either fluorescently labeled HNA (GS1962, 10 nM) or fluorescently labeled chimeric HNA phosphorothioate derivative (HNA gapmer, GS1964, 10 nM) were transfected into NIH 3T3 MDR cells for cellular uptake and distribution analyses. Confocal microscopy indicated that fluorescence was mostly in cell nuclei (Figure 2), suggesting that both HNA and HNA gapmer were taken by NIH 3T3 MDR cells and eventually redistributed to nuclei. A fraction of the cell population was not transfected as indicated by cells lacking any fluorescence (Figure 2B, panel c).
Figure 2.

Cellular uptake and distribution of HNA derivatives. NIH 3T3 MDR cells were treated with fluorescein-labeled HNA derivatives (10 nM) complexed with Lipofectamine 2000. (A) HNA (GS1954); (B) HNA gapmer (GS1956). (a) fluor-labeled oligonucleotide; (b) phase contrast image; (c) overlay image. Note that some of the cells lack fluorophore (arrow).
Cytotoxicity of oligonucleotide complexes
The concentration dependence of the cytotoxicities of HNA (GS1954), HNA gapmer (GS1956), phosphorothioate (5995) and 2′-O-ME gapmer oligonucleotides (13 758) were evaluated under the same conditions as those used for testing antisense effects. All four types of antisense derivatives were relatively non-toxic up to ∼10 nM with cell survival being >80% of control; however, HNA (GS1954) and HNA phosphorothioate gapmer (GS1956) reduced cell survival to 53 and 36% of control, respectively, at 27 nM, while phosphorothioate (5995) and chimeric 2′-O-ME gapmer (13 758) reduced cell survival to 72 and 69% of control, respectively (Figure 3). Thus, the HNA oligomers were non-toxic at low concentrations but were more toxic than phosphorothioate or 2′-O-ME gapmer when used at higher concentrations. Cells were also assayed for growth in complete medium over a period of five days after treatment. No differences were observed between cells treated with Lipofectamine 2000 alone or with Lipofectamine 2000 plus HNA gapmer at 9 nM (data not shown). Thus, at lower concentrations the HNA gapmer is only minimally toxic.
Figure 3.
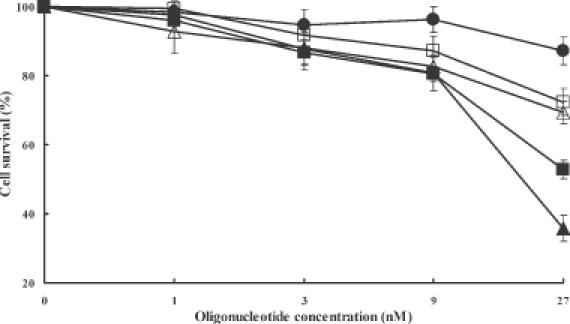
Cytotoxicity of oligonucleotides. The cytotoxicities of antisense oligonucleotides were determined after treatment of NIH 3T3 MDR cells with various concentrations of antisense/Lipofectamine 2000 complexes and further incubating them for 64 h as described. Closed circles, control cells treated with Lipofectamine 2000 only; closed squares, HNA (GS1954); closed triangles, HNA gapmer (GS1956); open squares, phosphorothioate (5995); open triangles, 2′-O-ME gapmer (13 158). Values represent cell population in percentage with 100% taken for untreated NIH 3T3 MDR cells. Means and standard errors of 3–6 determinations.
Cellular uptake of oligonucleotides
Fluorophore-labeled HNA and HNA gapmer exhibited similar uptake by NIH 3T3 MDR cells. Figure 4 shows uptake of the HNA gapmer. While the percentage of fluorescent cells increased linearly (up to 54% at 27 nM of HNA gapmer), the relative fluorescence strengths increased more sharply at higher oligonucleotide concentration (5.2 at 3 nM, 14.8 at 9 nM and 43.2 at 27 nM). The results imply that the amount of oligonucleotide per transfected cell is much greater at higher oligonucleotide concentration. This uptake result correlates with antisense effects on P-glycoprotein inhibition as well as the observed cytotoxicity at higher oligonucleotide concentrations.
Figure 4.
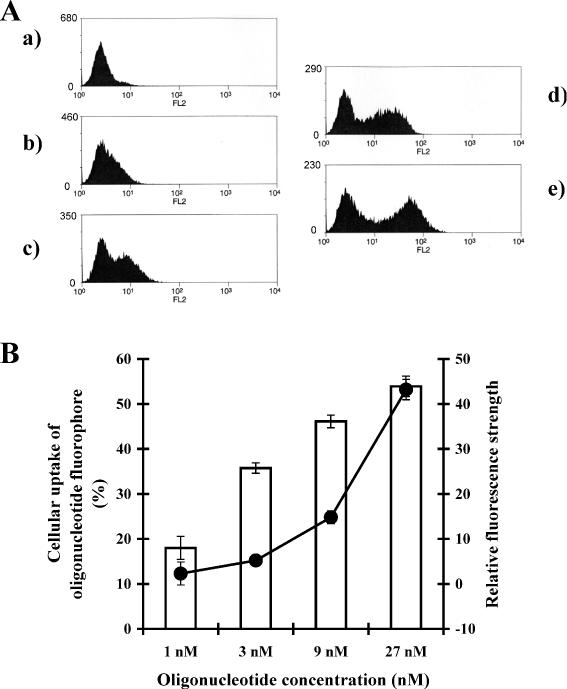
Oligonucleotide uptake by cells. NIH 3T3 MDR cells were treated with Lipofectamine 2000 complexes of fluorescein labeled HNA gapmer as described. The fluorescence of the cells was then measured by flow cytometry. (A) x-Axes represent fluorescence intensities and y-axes represent the cell number. (a) Untreated negative control cells; (b) 1 nM HNA gapmer; (c) 3 nM HNA gapmer; (d) 9 nM HNA gapmer; (e) 27 nM HNA gapmer. (B) x-Axis represents HNA gapmer concentrations, y-axis at left and open bars represent the percentage of the cell population that is transfected; y-axis at right and closed circles represent relative fluorescence strengths. Means and standard errors of three determinations.
Inhibition of P-glycoprotein expression measured by flow cytometry
Antisense effects of HNA (GS1954), HNA gapmer (GS1956), phosphorothioate (5995) and 2′-O-ME gapmer (13 758) derivatives on cell surface P-glycoprotein expression in viable NIH 3T3 MDR cells or NCI/ADR-RES cells were evaluated using immunofluorescence and flow cytometry. The control oligonucleotides 10 221, 13 753, GS1955 and GS1957 were also tested. The cells were treated with oligonucleotide complexed to Lipofectamine 2000 as described above. Figure 5 compares P-glycoprotein reductions in NIH 3T3 MDR cells caused by HNA gapmer, phosphorothioate and 2′-O-ME gapmer at 9 nM, while Figure 6 shows the effects on NCI/ADR-RES cells. As seen in both cell types, the HNA gapmer was substantially more effective than the phosphorothioate and was comparable to the 2′-O-ME gapmer derivative. Antisense effects could not be measured quantitatively at higher concentrations due to cytotoxicity (data not shown). Antisense effects on P-glycoprotein expression were concentration dependent (Figure 7). The HNA gapmer (GS1956) and 2′-O-ME gapmer (13 758) displayed a similar concentration–response profile, while over the concentration range tested, the phosphorothioate (5995) was less effective.
Figure 5.
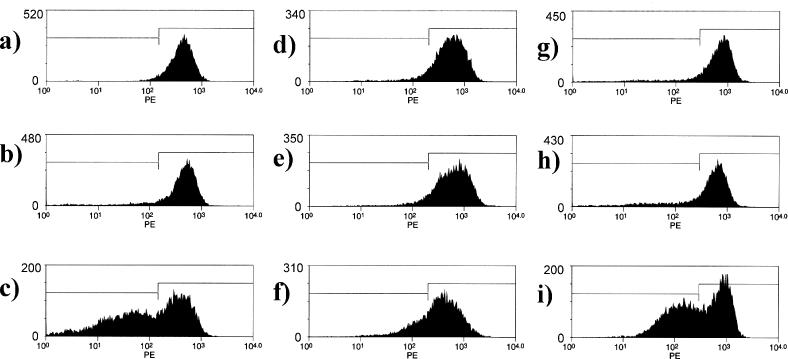
Antisense effects on P-glycoprotein expression in NIH 3T3 MDR cells. NIH 3T3 MDR cells were treated with Lipofectamine 2000 complexes of various oligonucleotides, and cell surface P-glycoprotein expression in the viable cells was evaluated as described in Materials and Methods. x-Axes represent fluorescence intensity and y-axes represent cell number. Panels (A, D and G), control (Lipofectamine 2000 only); (B) 9 nM HNA gapmer mismatched (GS1957); (C) 9 nM HNA gapmer (GS1956); (E) 9 nM phosphorothioate scrambled control (10 221); (F) 9 nM phosphorothioate (5995); (H) 9 nM 2′-O-ME gapmer scrambled (13 753) (I) 9 nM 2′-O-ME gapmer (13 758). The demarcations between the left and right boxes in the figures are set at one standard deviation below the mean of the untreated control.
Figure 6.
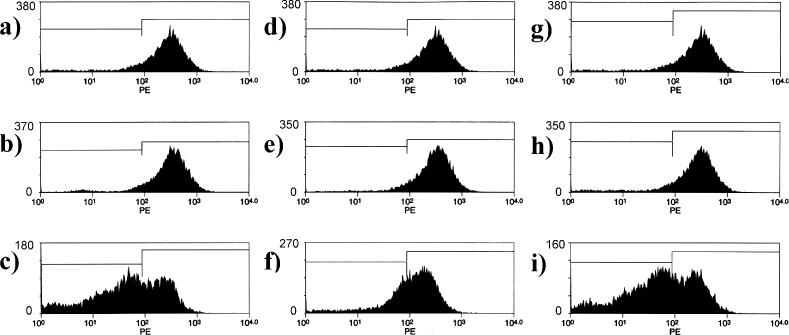
Antisense effects on P-glycoprotein expression in NCI/ADR-RES cells. NCI/ADR-RES cells were treated with Lipofectamine 2000 complexes of various oligonucleotides, and cell surface P-glycoprotein expression in the viable cells was evaluated as described in Materials and Methods. x-Axes represent fluorescence intensity and y-axes represent cell population. Panels (A, D and G), controls (Lipofectamine 2000 only); (B) 9 nM HNA gapmer mismatched (GS1957); (C) 9 nM HNA gapmer (GS1956); (E) 9 nM phosphorothioate scrambled control (10 221); (F) 9 nM phosphorothioate (5995); (H) 9 nM 2′-O-ME gapmer scrambled (13 753) (I) 9 nM 2′-O-ME gapmer (13 758). The demarcations between the left and right boxes in the figures are set at one standard deviation below the mean of the untreated control.
Figure 7.
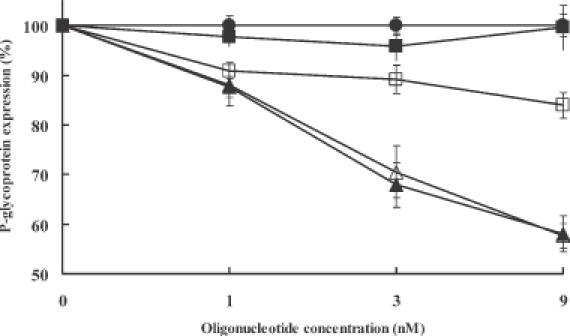
Antisense concentration dependence of P-glycoprotein expression. Cell surface P-glycoprotein expression in NIH 3T3 MDR cells was measured by immunostaining and flow cytometry as described in Materials and Methods. Closed circles, control cells treated with Lipofectamine 2000 only; closed squares, HNA (GS1954); closed triangles, HNA gapmer (GS1956); open squares, phosphorothioate (5995); open triangles, 2′-O-ME gapmer (13 158). Values are percentage of P-glycoprotein expression with 100% taken for NIH 3T3 MDR cells treated with corresponding mismatched or scrambled sequence. The percentage was calculated on the basis of the fraction of the cell population shifted to greater than one standard deviation below the mean of the untreated controls in terms of P-glycoprotein expression. Means and standard errors of 3–6 determinations.
Selective reduction of P-glycoprotein measured by western blot
The MDR1 gene expression in response to antisense and control oligonucleotides was evaluated by western blotting (Figure 8). At a concentration of 10 nM, the HNA gapmer (GS1956) and 2′-O-ME gapmer significantly reduced the P-glycoprotein expression, while the antisense phosphorothioate (5995) and the control oligonucleotides were less or not effective. Expression of actin was not affected by any of the oligomers.
Figure 8.
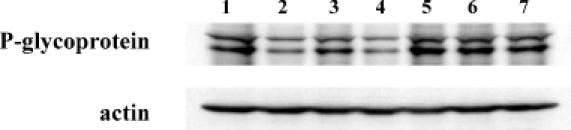
Western blots for P-glycoprotein expression. After treating NIH 3T3 MDR cells with various antisense/Lipofectamine 2000 complexes as described, cells were harvested for western analysis. Lane 1, Lipofectamine 2000 only control; lane 2, HNA gapmer (GS1956); lane 3, phosphorothioate (5995); lane 4, 2′-O-ME gapmer (13 758); lane 5, HNA gapmer mismatch (GS1957); lane 6, phosphorothioate scrambled (10 221); lane 7, 2′-O-ME gapmer scrambled (13 753). All oligonucleotides were used at a 10 nM concentration.
Accumulation of Rhodamine 123
Accumulation of fluorescent Rhodamine 123 by NIH 3T3 MDR cells was measured by flow cytometry after exposure to 9 nM oligonucleotide. Both the HNA (GS1956) and 2′-O-ME (13 758) gapmers enhanced Rhodamine 123 accumulation by ∼200%, while the control sequences GS1957 and 13 753 did not (Figure 9). Unmodified phosphorothioate (5995) slightly enhanced cellular accumulation of the drug, while the fully HNA oligonucleotide, GS1954, exhibited minimal effects.
Figure 9.
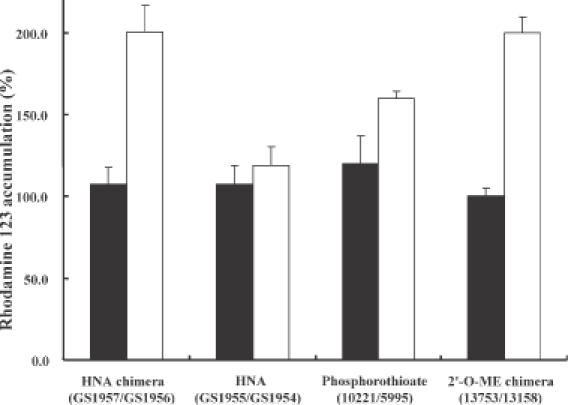
Effects of antisense oligonucleotides on Rhodamine 123 accumulation. NIH 3T3 MDR cells were treated with 9 nM of antisense oligonucleotides complexed with Lipofectamine 2000 as described. Closed bars, control oligonucleotides; open bars, oligonucleotides targeted to MDR1 gene. Values are Rhodamine 123 uptake increase, with the 100% level taken as that for untreated NIH3T3 MDR cells. Mean and standard errors of 3–6 determinations.
DISCUSSION
Selective inhibition of the P-glycoprotein efflux pump in MDR cancer cells is potentially an important objective in anti-cancer chemotherapeutics. This research demonstrates effective inhibition of MDR1 gene expression by a novel class of HNA antisense oligonucleotide derivatives. Both unmodified HNA and its phosphorothioate chimeric (gapmer) derivative were prepared; these targeted a sequence bracketing the AUG start codon of MDR1. The HNA oligonucleotides were compared to unmodified phosphorothioate and 2′-O-ME gapmer oligonucleotides with the same sequence.
Complexation with the cationic lipid Lipofectamine 2000 provided delivery of fluorophore-tagged HNA oligonucleotides to NIH 3T3 MDR cells, as evidenced by concentration of the fluorophore in the nucleus; however, some cells remained untransfected. HNA oligonucleotides were not toxic when used at low concentrations, but seemed to be somewhat more toxic to cells than the phosphorothioate or 2′-O-ME gapmer oligonucleotides when used at higher concentrations. The increased toxicity of HNA derivatives may be due to their stability and resistance to degradation (60–65).
The inhibition of MDR1 gene expression was assayed by examining levels of P-glycoprotein, the MDR1 gene product. The HNA gapmer proved to be extremely potent in these assays, displaying 50% inhibition of P-glycoprotein expression when used at ∼10 nM. This was equivalent to the potency of the 2′-O-ME gapmer which had provided by far the most effective inhibition of P-glycoprotein expression observed previously (34,35). The unmodified HNA oligomer was not effective in inhibiting P-glycoprotein expression, suggesting that RNase H activity is essential for this to occur. Reduced P-glycoprotein expression was reflected in a parallel increase in drug accumulation in the NIH 3T3 MDR cells, as evaluated using a flow cytometry assay for Rhodamine 123 accumulation. In these studies, we did not attempt to measure changes in resistance to anti-tumor drugs, since a fraction of the cell population did not take up the antisense oligonucleotides, and thus would be expected to remain resistant.
In summary, this research has demonstrated that chimeric HNA phosphorothioate oligonucleotide gapmers can effectively inhibit MDR1 gene expression. The great potency of the HNA gapmer allows its use at non-toxic concentrations. Further, although the HNA gapmer displayed some toxicity at higher concentrations, this might be attenuated by use of phosphodiester internucleotide linkages, rather than phosphorothioate, in the ‘gap’ segment. This approach may reduce the observed cytotoxicity while maintaining the required nuclease resistance and target binding, thus enhancing the desired gene regulation capabilities. It would seem worthwhile to pursue this approach in future studies, as well as seek more efficient methods for delivery of HNAs and other antisense oligonucleotides.
Acknowledgments
ACKNOWLEDGEMENTS
This work was supported by NIH grant PO1 GM59299 to R.L.J. The authors wish to thank Dr Anna Astriab-Fisher for helpful advice and David Rinker for editorial assistance.
REFERENCES
- 1.Ambudkar S.V., Kimchi-Sarfaty,C., Sauna,Z.E. and Gottesman,M.M. (2003) P-glycoprotein: from genomics to mechanism. Oncogene, 22, 7468–7485. [DOI] [PubMed] [Google Scholar]
- 2.Juliano R.L. and Ling,V. (1976) A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim. Biophys. Acta, 455, 152–162. [DOI] [PubMed] [Google Scholar]
- 3.Licht T., Pastan,I., Gottesman,M. and Herrmann,F. (1994) P-glycoprotein-mediated multidrug resistance in normal and neoplastic hematopoietic cells. Ann. Hematol., 69, 159–171. [DOI] [PubMed] [Google Scholar]
- 4.Bradley G. and Ling,V. (1994) P-glycoprotein, multidrug-resistance and tumor progression. Cancer Metastasis Rev., 13, 223–233. [DOI] [PubMed] [Google Scholar]
- 5.Ambudkar S.V., Dey,S., Hrycyna,C.A., Ramachandra,M., Pastan,I. and Gottesman,M.M. (1999) Biochemical, cellular, and pharmacological aspects of the multidrug transporter. Annu. Rev. Pharmacol. Toxicol., 39, 361–398. [DOI] [PubMed] [Google Scholar]
- 6.Thiebaut F., Tsuruo,T., Hamada,H., Gottesman,M.M., Pastan,I. and Willingham,M.C. (1987) Cellular localization of the multidrug-resistance gene product P-glycoprotein in normal human tissues. Proc. Natl Acad. Sci. USA, 84, 7735–7738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garraway L.A. and Chabner,B. (2002) MDR1 inhibition: less resistance or less relevance? Eur. J. Cancer, 38, 2337–2340. [DOI] [PubMed] [Google Scholar]
- 8.Leith C.P., Kopecky,K.J., Godwin,J., McConnell,T., Slovak,M.L., Chen,I.M., Head,D.R., Appelbaum,F.R. and Willman,C.L. (1997) Acute myeloid leukemia in the elderly: assessment of multidrug resistance (MDR1) and cytogenetics distinguishes biologic subgroups with remarkably distinct responses to standard chemotherapy. A Southwest Oncology Group study. Blood, 89, 3323–3329. [PubMed] [Google Scholar]
- 9.Kaye S.B. (1998) Multidrug resistance: clinical relevance in solid tumours and strategies for circumvention. Curr. Opin. Oncol., 10 (Suppl. 1), S15–S19. [PubMed] [Google Scholar]
- 10.Kankesan J., Vanama,R., Yusuf,A., Thiessen,J.J., Ling,V., Rao,P.M., Rajalakshmi,S. and Sarma,D.S. (2003) Effect of PSC 833, an inhibitor of P-glycoprotein on N-methyl-N-nitrosourea induced mammary carcinogenesis in rats. Carcinogenesis, 24, 1977–1984. [DOI] [PubMed] [Google Scholar]
- 11.Sadanand V., Kankesan,J., Yusuf,A., Stewart,C., Rutka,J.T., Thiessen,J.J., Ling,V., Rao,P.M., Rajalakshmi,S. and Sarma,D.S. (2003) Effect of PSC 833, a potent inhibitor of P-glycoprotein, on the growth of astrocytoma cells in vitro. Cancer Lett., 198, 21–27. [DOI] [PubMed] [Google Scholar]
- 12.Varma M.V., Ashokraj,Y., Dey,C.S. and Panchagnula,R. (2003) P-glycoprotein inhibitors and their screening: a perspective from bioavailability enhancement. Pharmacol. Res., 48, 347–359. [DOI] [PubMed] [Google Scholar]
- 13.Sparreboom A. and Nooter,K. (2000) Does P-glycoprotein play a role in anticancer drug pharmacokinetics? Drug Resist. Updat., 3, 357–363. [DOI] [PubMed] [Google Scholar]
- 14.Kurreck J. (2003) Antisense technologies. Improvement through novel chemical modifications. Eur. J. Biochem., 270, 1628–1644. [DOI] [PubMed] [Google Scholar]
- 15.Crooke S.T. (2004) Progress in antisense technology. Annu. Rev. Med., 55, 61–95. [DOI] [PubMed] [Google Scholar]
- 16.Agrawal S. and Kandimalla,E.R. (2001) Antisense and/or immunostimulatory oligonucleotide therapeutics. Curr. Cancer Drug Targets, 1, 197–209. [DOI] [PubMed] [Google Scholar]
- 17.Agrawal S. and Kandimalla,E.R. (2000) Antisense therapeutics: is it as simple as complementary base recognition? Mol. Med. Today, 6, 72–81. [DOI] [PubMed] [Google Scholar]
- 18.Dias N. and Stein,C.A. (2002) Antisense oligonucleotides: basic concepts and mechanisms. Mol. Cancer Ther., 1, 347–355. [PubMed] [Google Scholar]
- 19.Stein C.A. (2001) Antisense that comes naturally. Nat. Biotechnol., 19, 737–738. [DOI] [PubMed] [Google Scholar]
- 20.Dean N.M. and Bennett,C.F. (2003) Antisense oligonucleotide-based therapeutics for cancer. Oncogene, 22, 9087–9096. [DOI] [PubMed] [Google Scholar]
- 21.Pirollo K.F., Rait,A., Sleer,L.S. and Chang,E.H. (2003) Antisense therapeutics: from theory to clinical practice. Pharmacol. Ther., 99, 55–77. [DOI] [PubMed] [Google Scholar]
- 22.Herdewijn P. (2000) Heterocyclic modifications of oligonucleotides and antisense technology. Antisense Nucleic Acid Drug Dev., 10, 297–310. [DOI] [PubMed] [Google Scholar]
- 23.Koshkin A.A., Singh,S.K., Nielsen,P., Rajwanshi,V.K., Kumar,R., Meldgaard,M., Olsen,C.E. and Wengel,J. (1998) LNA (Locked Nucleic Acids): synthesis of the adenine, cytosine, guanine, 5-methylcytosine, thymine and uracil bicyclonucleoside monomers, oligomerisation, and unprecedented nucleic acid recognition. Tetrahedron, 54, 3607–3630. [Google Scholar]
- 24.Nielsen P.E., Egholm,M., Berg,R.H. and Buchardt,O. (1991) Sequence-selective recognition of DNA by strand displacement with a thymine-substituted polyamide. Science, 254, 1497–1500. [DOI] [PubMed] [Google Scholar]
- 25.Wang J., Verbeure,B., Luyten,I., Lescrinier,E., Froeyen,M., Hendrix,C., Rosemeyer,H., Seela,F., Van Aerschot,A. and Herdewijn,P. (2000) Cyclohexene nucleic acids (CeNA): serum stable oligonucleotides that activate RNase H and increase duplex stability with complementary RNA. J. Am. Chem. Soc., 122, 8595–8602. [Google Scholar]
- 26.Miller P.S. and Ts'o,P.O. (1987) A new approach to chemotherapy based on molecular biology and nucleic acid chemistry: matagen (masking tape for gene expression). Anticancer Drug Des., 2, 117–128. [PubMed] [Google Scholar]
- 27.Levin A.A. (1999) A review of the issues in the pharmacokinetics and toxicology of phosphorothioate antisense oligonucleotides. Biochim. Biophys. Acta, 1489, 69–84. [DOI] [PubMed] [Google Scholar]
- 28.Zamaratski E., Pradeepkumar,P.I. and Chattopadhyaya,J. (2001) A critical survey of the structure-function of the antisense oligo/RNA heteroduplex as substrate for RNase H. J. Biochem. Biophys. Methods, 48, 189–208. [DOI] [PubMed] [Google Scholar]
- 29.Larsen H.J., Bentin,T. and Nielsen,P.E. (1999) Antisense properties of peptide nucleic acid. Biochim. Biophys. Acta, 1489, 159–166. [DOI] [PubMed] [Google Scholar]
- 30.Agrawal S. and Zhao,Q. (1998) Mixed backbone oligonucleotides: improvement in oligonucleotide-induced toxicity in vivo. Antisense Nucleic Acid Drug Dev., 8, 135–139. [DOI] [PubMed] [Google Scholar]
- 31.Peyman A. and Uhlmann,E. (1996) Minimally modified oligonucleotides—combination of end-capping and pyrimidine-protection. Biol. Chem. Hoppe-Seyler, 377, 67–70. [DOI] [PubMed] [Google Scholar]
- 32.Rait A., Pirollo,K., Will,D.W., Peyman,A., Rait,V., Uhlmann,E. and Chang,E.H. (2000) 3′-End conjugates of minimally phosphorothioate-protected oligonucleotides with 1-O-hexadecylglycerol: synthesis and anti-ras activity in radiation-resistant cells. Bioconjug. Chem., 11, 153–160. [DOI] [PubMed] [Google Scholar]
- 33.Hamma T. and Miller,P.S. (1999) Syntheses of alternating oligo-2′-O-methylribonucleoside methylphosphonates and their interactions with HIV TAR RNA. Biochemistry, 38, 15333–15342. [DOI] [PubMed] [Google Scholar]
- 34.Alahari S.K., DeLong,R., Fisher,M.H., Dean,N.M., Viliet,P. and Juliano,R.L. (1998) Novel chemically modified oligonucleotides provide potent inhibition of P-glycoprotein expression. J. Pharm. Exp. Ther., 286, 419–428. [PubMed] [Google Scholar]
- 35.Alahari S.K., Dean,N.M., Fisher,M.H., Delong,R., Manoharan,M., Tivel,K.L. and Juliano,R.L. (1996) Inhibition of expression of the multidrug resistance-associated P-glycoprotein of by phosphorothioate and 5′ cholesterol-conjugated phosphorothioate antisense oligonucleotides. Mol. Pharmacol., 50, 808–819. [PubMed] [Google Scholar]
- 36.Puerta-Fernandez E., Romero-Lopez,C., Barroso-delJesus,A. and Berzal-Herranz,A. (2003) Ribozymes: recent advances in the development of RNA tools. FEMS Microbiol. Rev., 27, 75–97. [DOI] [PubMed] [Google Scholar]
- 37.Hannon G.J. (2002) RNA interference. Nature, 418, 244–251. [DOI] [PubMed] [Google Scholar]
- 38.Sioud M. (2004) Therapeutic siRNAs. Trends Pharmacol. Sci., 25, 22–28. [DOI] [PubMed] [Google Scholar]
- 39.Harborth J., Elbashir,S.M., Vandenburgh,K., Manninga,H., Scaringe,S.A., Weber,K. and Tuschl,T. (2003) Sequence, chemical, and structural variation of small interfering RNAs and short hairpin RNAs and the effect on mammalian gene silencing. Antisense Nucleic Acid Drug Dev., 13, 83–105. [DOI] [PubMed] [Google Scholar]
- 40.Fire A., Xu,S., Montgomery,M.K., Kostas,S.A., Driver,S.E. and Mello,C.C. (1998) Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature, 391, 806–811. [DOI] [PubMed] [Google Scholar]
- 41.Elbashir S.M., Harborth,J., Lendeckel,W., Yalcin,A., Weber,K. and Tuschl,T. (2001) Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature, 411, 494–498. [DOI] [PubMed] [Google Scholar]
- 42.Golub T.R., Slonim,D.K., Tamayo,P., Huard,C., Gaasenbeek,M., Mesirov,J.P., Coller,H., Loh,M.L., Downing,J.R., Caligiuri,M.A. et al. (1999) Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science, 286, 531–537. [DOI] [PubMed] [Google Scholar]
- 43.Semizarov D., Frost,L., Sarthy,A., Kroeger,P., Halbert,D.N. and Fesik,S.W. (2003) Specificity of short interfering RNA determined through gene expression signatures. Proc. Natl Acad. Sci. USA, 100, 6347–6352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Scacheri P.C., Rozenblatt-Rosen,O., Caplen,N.J., Wolfsberg,T.G., Umayam,L., Lee,J.C., Hughes,C.M., Shanmugam,K.S., Bhattacharjee,A., Meyerson,M. et al. (2004) Short interfering RNAs can induce unexpected and divergent changes in the levels of untargeted proteins in mammalian cells. Proc. Natl Acad. Sci. USA, 101, 1892–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sioud M. and Sorensen,D.R. (2003) Cationic liposome-mediated delivery of siRNAs in adult mice. Biochem. Biophys. Res. Commun., 312, 1220–1225. [DOI] [PubMed] [Google Scholar]
- 46.Ramachandran C. and Wellham,L.L. (2003) Effect of MDR1 phosphorothioate antisense oligodeoxynucleotides in multidrug-resistant human tumor cell lines and xenografts. Anticancer Res., 23, 2681–2690. [PubMed] [Google Scholar]
- 47.Pakunlu R.I., Cook,T.J. and Minko,T. (2003) Simultaneous modulation of multidrug resistance and antiapoptotic cellular defense by MDR1 and BCL-2 targeted antisense oligonucleotides enhances the anticancer efficacy of doxorubicin. Pharm. Res., 20, 351–359. [DOI] [PubMed] [Google Scholar]
- 48.Brigui I., Djavanbakht-Samani,T., Jolles,B., Pigaglio,S. and Laigle,A. (2003) Minimally modified phosphodiester antisense oligodeoxyribonucleotide directed against the multidrug resistance gene mdr1. Biochem. Pharmacol., 65, 747–754. [DOI] [PubMed] [Google Scholar]
- 49.Pan L., Tong,Y., Jin,Y., Zhou,S., Zhang,Y., Yang,X. and Mao,N. (2001) Reversing drug resistance in the ovarian carcinoma cell line SKOV3/mdr1 in vitro by antisense oligodeoxynucleotides. Chin. Med. J. (Engl), 114, 929–932. [PubMed] [Google Scholar]
- 50.Fisher A.A., Ye,D., Sergueev,D.S., Fisher,M.H., Shaw,B.R. and Juliano,R.L. (2002) Evaluating the specificity of antisense oligonucleotide conjugates. A DNA array analysis. J. Biol. Chem., 277, 22980–22984. [DOI] [PubMed] [Google Scholar]
- 51.Quattrone A., Papucci,L., Morganti,M., Coronnello,M., Mini,E., Mazzei,T., Colonna,F.P., Garbesi,A. and Capaccioli,S. (1994) Inhibition of MDR1 gene expression by antimessenger oligonucleotides lowers multiple drug resistance. Oncol. Res., 6, 311–320. [PubMed] [Google Scholar]
- 52.Kobayashi H., Takemura,Y. and Miyachi,H. (2001) Novel approaches to reversing anti-cancer drug resistance using gene-specific therapeutics. Hum. Cell, 14, 172–184. [PubMed] [Google Scholar]
- 53.Nagata J., Kijima,H., Hatanaka,H., Asai,S., Miyachi,H., Abe,Y., Yamazaki,H., Nakamura,M., Watanabe,N., Mine,T. et al. (2002) Reversal of drug resistance using hammerhead ribozymes against multidrug resistance-associated protein and multidrug resistance 1 gene. Int. J. Oncol., 21, 1021–1026. [PubMed] [Google Scholar]
- 54.Kuznetsova M., Fokina,A., Lukin,M., Repkova,M., Venyaminova,A. and Vlassov,V. (2003) Catalytic DNA and RNA for targeting MDR1 mRNA. Nucleosides Nucleotides Nucleic Acids, 22, 1521–1523. [DOI] [PubMed] [Google Scholar]
- 55.Kobayashi H., Dorai,T., Holland,J.F. and Ohnuma,T. (1993) Cleavage of human MDR1 mRNA by a hammerhead ribozyme. FEBS Lett., 319, 71–74. [DOI] [PubMed] [Google Scholar]
- 56.Holm P.S., Dietel,M. and Krupp,G. (1995) Similar cleavage efficiencies of an oligoribonucleotide substrate and an mdr1 mRNA segment by a hammerhead ribozyme. Gene, 167, 221–225. [DOI] [PubMed] [Google Scholar]
- 57.Wu H., Hait,W.N. and Yang,J.M. (2003) Small interfering RNA-induced suppression of MDR1 (P-glycoprotein) restores sensitivity to multidrug-resistant cancer cells. Cancer Res., 63, 1515–1519. [PubMed] [Google Scholar]
- 58.Nieth C., Priebsch,A., Stege,A. and Lage,H. (2003) Modulation of the classical multidrug resistance (MDR) phenotype by RNA interference (RNAi). FEBS Lett., 545, 144–150. [DOI] [PubMed] [Google Scholar]
- 59.Hendrix C., Rosemeyer,H., De Bouvere,B., Van Aerschot,A., Seela,F. and Herdewijn,P. (1997) 1′,5′-anhydrohexitol oligonucleotides: hybridisation and strand displacement with oligoribonucleotides, interaction with RNase H and HIV reverse transcriptase. Chemistry—A Eur. J., 3, 1513–1520. [Google Scholar]
- 60.Lescrinier E., Esnouf,R., Schraml,J., Busson,R., Heus,H.A., Hilbers,C.W. and Herdewijn,P. (2000) Solution structure of a HNA–RNA hybrid. Chem. Biol., 7, 719–731. [DOI] [PubMed] [Google Scholar]
- 61.Vandermeeren M., Preveral,S., Janssens,S., Geysen,J., Saison-Behmoaras,E., Van Aerschot,A. and Herdewijn,P. (2000) Biological activity of hexitol nucleic acids targeted at Ha-ras and intracellular adhesion molecule-1 mRNA. Biochem. Pharmacol., 59, 655–663. [DOI] [PubMed] [Google Scholar]
- 62.Flores M.V., Atkins,D., Stewart,T.S., Van Aerschot,A. and Herdewijn,P. (1999) Antimalarial antisense activity of hexitol nucleic acids. Parasitol. Res., 85, 864–866. [DOI] [PubMed] [Google Scholar]
- 63.De Bouvere B., Kerremans,L., Rozenski,J., Janssen,G., Van Aerschot,A., Claes,P., Busson,R. and Herdewijn,P. (1997) Improved synthesis of anhydrohexitol building blocks for oligonucleotide synthesis. Liebigs Ann.-Rec., 1453–1461. [Google Scholar]
- 64.DeWinter H., Lescrinier,E., Van Aerschot,A. and Herdewijn,P. (1998) Molecular dynamics simulation to investigate differences in minor groove hydration of HNA/RNA hybrids as compared to HNA/DNA complexes. J. Am. Chem. Soc., 120, 5381–5394. [Google Scholar]
- 65.Levesque L., Dean,N.M., Sasmor,H. and Crooke,S.T. (1997) Antisense oligonucleotides targeting human protein kinase C-alpha inhibit phorbol ester-induced reduction of bradykinin-evoked calcium mobilization in A549 cells. Mol. Pharmacol., 51, 209–216. [DOI] [PubMed] [Google Scholar]
- 66.Kane S.E., Reinhard,D.H., Fordis,C.M., Pastan,I. and Gottesman,M.M. (1989) A new vector using the human multidrug resistance gene as a selectable marker enables overexpression of foreign genes in eukaryotic cells. Gene, 84, 439–446. [DOI] [PubMed] [Google Scholar]