


Abstract
We have developed a new primer design method based on the QuickChange™ site-directed mutagenesis protocol, which significantly improves the PCR amplification efficiency. This design method minimizes primer dimerization and ensures the priority of primer-template annealing over primer self-pairing during the PCR. Several different multiple mutations (up to 7 bases) were successfully performed with this partial overlapping primer design in a variety of vectors ranging from 4 to 12 kb in length. In comparison, all attempts failed when using complete-overlapping primer pairs as recommended in the standard QuickChange™ protocol. Our protocol was further extended to site-saturation mutagenesis by introducing randomized codons. Our data indicated no specific sequence selection during library construction, with the randomized positions resulting in average occurrence of each base in each position. This method should be useful to facilitate the preparation of high-quality site saturation libraries.
INTRODUCTION
Site-directed mutagenesis (SDM) is an invaluable tool to modify DNA sequences in molecular biological studies and genetic engineering. It is also widely used for studying protein structure–function relationships. Numerous mutagenesis methods have been developed based upon the PCR (1–4). The simplest and most broadly applicable protocol is currently the QuickChange™ Site-Directed Mutagenesis System (QCM) developed by Stratagene (La Jolla, CA). With this approach, the mutation is introduced in a single PCR with one pair of complementary primer containing the mutation of interest. However, this method is restricted to primer pairs of 25–45 bases in length with melting temperature (Tm) ≥ 78°C. Otherwise, primer dimer formation will become more favorable compared to the primer-template annealing, in particular for primer pairs with multiple mismatches, and the protocol has to be modified case by case, or more steps have to be carried out, e.g. by using an alternative two-stage PCR protocol (5). We encountered difficulties with the QuickChange™ protocol in the course mutational studies with catalytic antibodies, where we were completely unable to introduce any mutation following the recommended protocol. These difficulties led us to explore primer-design modifications. Here, we report a simple modification of the primer design, which not only overcomes the limitation of the melting temperature of primer design, but also makes multiple mutations available. This method enables DNA modifications, which were simply not possible with the standard QCM protocol.
Directed protein evolution is usually accomplished by generating random or targeted mutations in the protein coding sequence and screening the mutant library for functional improvements. With saturation mutagenesis, it is possible to create a library of mutants containing all possible mutations at one or more pre-determined target positions in a gene sequence. This method is used in directed evolution experiments to expand the number of amino acid substitutions accessible by random mutagenesis. In combination with high-throughput screening methods, the researchers have successfully used saturation mutagenesis to improve such enzymatic properties as thermostability (6,7) and enantioselectivity (8). However, most of them have been accomplished by standard cassette insertion method that needs additional cloning step after generating the saturation library. Recently, it has been reported that site-directed saturation mutagenesis could be carried out with QuickChange™ Multi Site-Directed Mutagenesis kit, but this method need phosphorylation of all oligos (9). Here, we show that our primer design can readily be extended to carry out saturation mutagenesis with standard primers in a single PCR.
MATERIALS AND METHODS
Plasmid pBAD-14D9hu (6 kb) (10), pBAD-10F11hu (6 kb) (11), pBAD-16E7hu (6 kb), pQE-14D9ScFv (4 kb) and pET-14D9-NusA (8 kb) (12), which cloned antibody Fv 14D9, 10F11, 16E7 gene fragments were used as mutagenesis template. Plasmid pCMVΔR8.91 (12 kb) containing HIV gene fragment was used to test the feasibility of larger template mutagenesis. All primers designed to introduce the site-directed mutation were synthesized and purified by MWG-biotech AG (Ebersberg, Germany). The melting temperature (Tm) was calculated with the formula given by Stratagene (http://www.stratagene.com/manuals/200519.pdf).
The PCR amplifications were carried out with Expand™ High Fidelity PCR system (Roche, Switzerland). The 50 μl PCR reaction was carried out with 50–100 ng templates, 0.4–2 μM primer pair, 200 μM dNTPs and 2 U of DNA polymerase. The extension reaction was initiated by pre-heating the reaction mixture to 94°C for 3 min; 16 cycles of 94°C for 1 min, 52°C for 1 min and 68°C for 8, 12, 16 or 24 min according to the length of template; followed by incubation at 68°C for 1 h. The PCR-amplification products were evaluated by agarose gel electrophoresis. The PCRs were purified by QIAquick™ PCR purification kit (Qiagen, Germany) and further treated with restriction enzyme DpnI (Fermentas). An aliquot of 1 μl above PCR product was transformed into XL1-Blue chemo-competent cells and inoculated on Luria–Bertani (LB) plate containing 100 μg/ml ampicillin. A total of 10 colonies were selected and their plasmids were isolated by mini-prep. The positive mutants were selected by respective restriction enzymatic digestion.
Site-saturation mutagenesis experiments were carried out with the same PCR procedure as described above. Out of 50 μl, 4 μl of purified PCR product was directly transformed into 200 μl TOP10 competent cells. The transformant was incubated in 5 ml LB medium containing 100 μg/ml ampicillin overnight. The randomized plasmid library was isolated by mini-prep and used for DNA sequencing. All mutants including randomized library were sequenced by Microsynth GmbH (Balgach, Switzerland).
For all primers, mutagenized positions are denoted in lowercase; randomized positions are in boldface; and the restriction sites are underlined. Backward primers designed with completely complementary role are marked with asterisk.
DH101A_f 5′-GTAACTTCTTTGcgTACTGGGGCCAAGGtACCACTCTCAC-3′ (KpnI)
DH101A_r* 5′-GTGAGAGTGGTaCCTTGGCCCCAGTAcgCAAAGAAGTTAC-3′ (KpnI)
DH101A_r 5′-CCAGTAcgCAAAGAAGTTACCATAGAAGATGGCAC-3′
AL46T_f 5′-CCTAAAaccCTGATTTACTCGACtagtTACCGGTACAGTG-3′ (SpeI)
AL46T_r 5′-CGAGTAAATCAGggtTTTAGGAGGTTGCCCTGG-3′
FH98A_f 5′-GTGCCATCgcaTATGGTAACTTCTTTGAC-3′ (NdeI)
FH98A_r 5′-GTTACCATAtgcGATGGCACAGTAATAGACTGC-3′ (NdeI)
WH104Y_f 5′-GATTCATTTCTAGTCTatTTTACGTTCTGGGGCC-3′
WH104Y_r 5′-CGTAAAatAGACTAGAAATGAATCTCCgCggATACAGTG-3′ (SacII)
WH104Y_f* 5′-CACTGTATccGcGGAGATTCATTTCTAGTCTatTTTACG -3′ (SacII)
SH100A_f 5′-GTATccGcGGAGATgCATTTCTAGTCTGG-3′ (SacII)
SH100A_r 5′-GAAATGcATCTCCgCggATACAGTGATACATG-3′ (SacII)
LL101F_f 5′-CATTTTCCGtTCGCGTTCGGTGCTGGGACC-3′
LL101F_r 5′-CGCGAaCGGAAAATGgGAtCCTTGAAAGCAG-3′ (BamHI)
DH50A_f 5′-GGAGcTATTTACCCTGGAtccGGGAATACTTACTAC-3′ (BamHI)
DH50A_r 5′-CCCggaTCCAGGGTAAATAgCTCCAATCCACTCAAGTCC-3′ (BamHI)
EL39A_f 5′-CCTATTTAGcATGGTAtCTGCAGAAACCAGGCC-3′ (KpnI deletion)
EL39A_r 5′-GCAGaTACCATgCTAAATAGGTGTTTCCATTAC-3′
SH99A_f 5′-GGTACTACGGTgcTGGCGCTGTCTCCTGGGGC-3′
SH99A_r 5′-CAGCGCCAgcACCGTAGTACCCgCggGCACAGAAATAGAC-3′ (SacII)
CMV_f 5′-GCTCGAcGCCGCCgCGGTGACCTTCAGAC-3′ (XhoI deletion)
CMV_r 5′-CGcGGCGGCgTCGAGCTTATAGCAAAATCC-3′ (XhoI deletion)
CMV_r* 5′-GTCTGAAGGTCACCGcGGCGGCgTCGAGC-3′ (XhoI deletion)
SSM1_f 5′-CTGTGCCATCNNKTATGGTAACTTCTTTGACTACTGG-3′
SSM1_r 5′-GAAGTTACCATAMNNGATGGCACAGTAATAGACTGC-3′
SSM2_f 5′-CTGTGCCATCNNKNNKGGTAACTTCTTTGACTACTGGGGCC-3′
SSM2_r 5′-GAAGTTACCMNNMNNGATGGCACAGTAATAGACTGCAGAG-3′
RESULTS
The primer design schemes used in this experiment are shown in Figure 1. The primers in the pair must complement each other at the 5′-terminus instead of the 3′-terminus to avoid primer self-extension. To determine the efficiency of this primer design, nine different positions from three different plasmids pBAD-14D9hu, pBAD-10F11hu and pBAD-16E7hu were selected for mutagenesis. The properties of designed primers are shown in Table 1. Tm values were varied from 73.6 to 86.6°C. The Tm differences between primer-to-template annealing and primer-pair self-annealing was also varied from 1.2 to ∼20°C. We introduced mutations ranging from one single base (LL101F) up to seven bases (AL46T). All nine PCR reactions were carried out using the same procedure and generated satisfactory quantities of amplification products (Figure 2, lane 2–4 and 6–11). In comparison, completely overlapping primers were also tested in two positions (DH101 and WH104). Both reactions failed to produced any amplification product (Figure 2, lanes 1 and 5), even though these primers were designed according to all rules of the standard QCM protocol. To examine the amplification efficiency with templates of different sizes, pQE-14D9ScFv and pET-14D9ScFv-NusA, which contain the same gene fragment as pBAD-14D9hu, were also used for mutagenesis with the FH98A primer pair. Similar quantities of amplification product were obtained, indicating that the size of the template is not a limiting factor. When using a plasmid larger than 12 kb, amplification succeeded when using a partially complementary primer pair (Figure 2, lane 14), but not with a completely overlapping primers. Ten colonies of each mutation were analyzed by restriction digestion using the corresponding enzyme, leading to estimate mutational efficiencies ranging from 40 to 100%.
Figure 1.
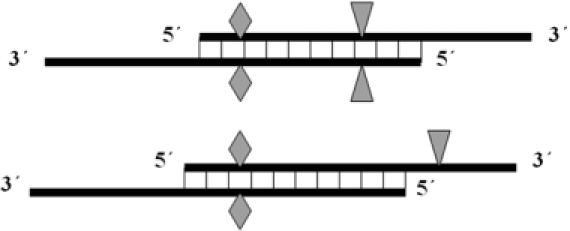
Schematic diagrams of the partial overlapping primer design. Diamonds indicate the targeted mutation; additional silent mutations used for positive mutant screening by restriction endonuclease digestion are shown as triangles.
Table 1. The Tm comparison between primer-template and primer pair.
| Primer | Length/mutation (bases) | Tm (°C) | Complementarity of primer pair (bases) | Self-annealing Tm (°C) |
|---|---|---|---|---|
| DH101A_f | 40/3 | 83.1 | 20 | 66.2 |
| DH101A_r | 35/2 | 78.9 | ||
| DH101A_r* | 40/3 | 83.1 | 40 | 83.1 |
| AL46T_f | 40/7 | 76.1 | 21 | 67 |
| AL46T_r | 33/3 | 79.1 | ||
| FH98A_f | 29/3 | 73.6 | 20 | 68.3 |
| FH98A_r | 33/3 | 76.7 | ||
| WH104Y_f | 34/2 | 76.5 | 24 | 65.4 |
| WH104Y_r | 39/5 | 76 | ||
| WH104Y_f* | 39/5 | 76 | 39 | 76 |
| SH100A_f | 29/4 | 75.4 | 21 | 70.9 |
| SH100A_r | 32/4 | 75.6 | ||
| LL101F_f | 30/1 | 82.6 | 15 | 58.4 |
| LL101F_r | 31/3 | 77.9 | ||
| DH50A_f | 36/4 | 78.2 | 24 | 77 |
| DH50A_r | 39/4 | 82.3 | ||
| EL39A_f | 33/2 | 78.9 | 21 | 67 |
| EL39A_r | 33/2 | 75.2 | ||
| SH99A_f | 32/2 | 86.6 | 21 | 76.7 |
| SH99A_r | 40/5 | 85.2 | ||
| CMV_f | 29/2 | 85.7 | 16 | 75.1 |
| CMV_r | 30/2 | 81.6 | ||
| CMV_r* | 29/2 | 85.7 | 29 | 85.7 |
All values were calculated according to the Tm calculation formula from Stratagene (http://www.stratagene.com/manuals/200519.pdf).
Figure 2.
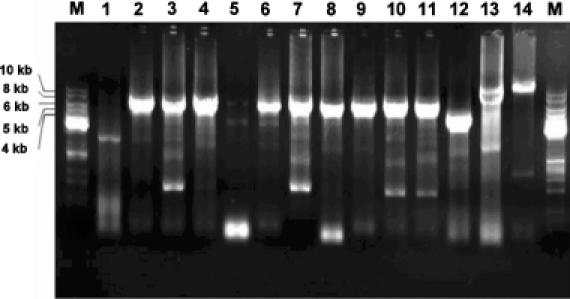
Agarose gel electrophoresis of mutagenesis reactions. Lane M: 1 kb DNA ladder standard (Fermentas). Lanes 1–4 use template pBAD-14D9: lane 1, DH101A*; lane 2, DH101A; lane 3, AL46T; and lane 4, FH98A. Lanes 5–8 use template pBAD-10F11: lane 5, WH104Y*; lane 6, WH104Y; lane 7, SH100A; and lane 8, LL101F. Lanes 9–11 use template pBAD-16E7: lane 9, DH50A; lane 10, EL39A; and lane 11, SH99A; lane 12, FH98A with template pQE-14D9ScFv; lane 13, FH98A with template pET-14D9ScFv-NusA; and lane 14, CMV.
Encouraged by the experiments above, we next tried to extend our protocol to site-saturation mutagenesis. The FH98 position in pBAD-14D9hu was randomly chosen as saturation mutagenesis candidate. To ensure the diversity of PCR amplification, 10 bases were placed before the randomized codon (NNK). PCR amplification was not influenced significantly by the presence of a randomized codon. Even two amino acids could be randomized with different annealing temperature (Figure 3). The sequence diversity of the resulting PCR amplification libraries was estimated by sequencing (Figure 4, left). Occurrence frequency was varied from 10 to 37.1% at position N1, from 19.5 to 35.4% at position N2, from 46.1 to 53.7% at position M (Table 2). The PCR product was transformed into Escherichia coli competent cell, and the diversity of saturation library was again monitored by sequencing. One additional G peak was observed after transformation into E.coli at position M, and two bigger ‘A’ peaks at positions N1 and N2 compared to the PCR product (Figure 4). These may be attributed to traces of intact plasmid left over from an incomplete DpnI digestion of the methylated template. Indeed, intact plasmids generally transform much more efficiently than any other forms, such that trace amounts of the plasmid remaining after DpnI digestion might have been selected over plasmids containing the randomized library during transformation. The randomized frequencies at positions N1 and N2 were calculated by subtracting the ‘G’ signal at position M and showed average ranges at three positions (Table 2), indicating that the circularization of linear randomized PCR product could be carried out in vivo without any significant selection.
Figure 3.
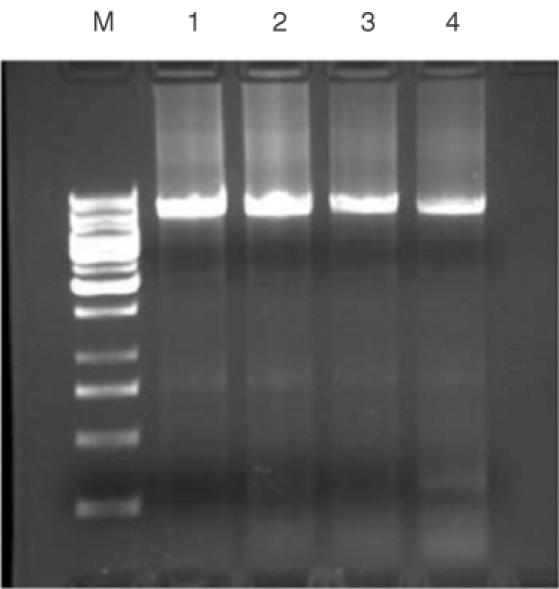
Agarose gel electrophoresis of site-directed saturation mutagenesis reactions. Lane M: 1 kb DNA ladder standard (Fermentas); lane 1, randomized PCR SSM1 with annealing temperature of 52°C; lane 2, randomized PCR SSM1 with annealing temperature of 57°C; lane 3, randomized PCR SSM2 with annealing temperature of 52°C; and randomized PCR SSM2 with annealing temperature of 57°C.
Figure 4.
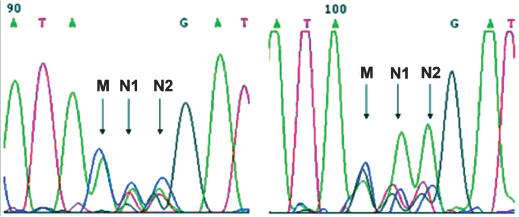
Sequencing results of site-directed saturation mutagenesis. Left, the PCR product (Figure 3, lane 1); Right, site-directed saturation library. Sequencing chromatography is represented using the software Chromas version 1.41. (http://trishul.sci.gu.edu.au/~conor/chromas.html/)
Table 2. Randomization frequency of each nucleotide at H98 position.
| PCR product (%) | Randomized library (%) | |||||||
|---|---|---|---|---|---|---|---|---|
| G | C | A | T | G | C | A | T | |
| Position M | — | 53.7 | 46.3 | — | 34.3 | 40 | 25.7 | — |
| Position N1 | 10 | 37.1 | 28.6 | 24.3 | 12 | 15.7 | 54.2 | 18.1 |
| Position N2 | 20.7 | 35.4 | 24.4 | 19.5 | 11.7 | 18.1 | 52.1 | 18.1 |
Occurrence frequencies were calculated according to signal amplitude at each position from sequencing chromatography.
DISCUSSION
The simple and efficient modification for QCM reported here overcomes and sometimes completely eliminates the problems associated with primer pair self-annealing. With this modification, the primer can be designed as for routine PCR experiments since the Tm value of the primer does not appear to be a strict determining factor anymore. Partial overlapping automatically enlarges the Tm difference between primer-to-template annealing and primer-pair self-annealing, which may be as low as 1.2°C. Furthermore, our experiments show that amplification does not really improve with larger Tm gap primer pairs, indicating that a small Tm-change is already enough to ensure the priority of template-primer annealing over primer pair self-annealing.
A few rules can be deduced from our experiment: (i) at least eight non-overlapping bases should be introduced at the 3′-terminus of primer; (ii) the targeted mutation(s) should be included into both primers, and a silent mutation should be present in at least one of them; and (iii) at least one G or C should be placed at the end of each terminus.
The advantages of the protocol presented here for SDM compared to standard QCM protocol can be summarized as follows: (i) a much broader variety of sequences can be used as mutation target since no Tm limitation has to be considered; (ii) larger sequences can be spanned without increasing primer length, which enables to chose optimal silent mutations; (iii) the mutation can be placed as close as four bases away from the 5′-terminus and at least 6–8 bases from the 3′-terminus, instead of 10–15 bases at both sides in the standard QCM protocol; and (iv) more mutations can be introduced in one primer, with a mutation content up to 17.5% (AL46T, 7 out of 40 bases), this becomes more important if some silent mutations have to be employed to facilitate downstream selection.
The experiments above also demonstrate an efficient site-saturation mutagenesis protocol requiring only a single PCR amplification. The application of our protocol for site-saturation mutagenesis generates a PCR product containing randomized codons at both 5′ and 3′ terminus. No repeated sequence was observed in the final library (confirmed by sequencing), suggesting that the occurrence of a relatively long (>10 bp) complementary sequence at each terminus made possible by the shifted primer design helped to ensure that circularization occurred without selection of particular sequences. Improvements in the efficiency of library construction are probably possible by the use of a more efficient template elimination procedure to reduce background and the use of electroporation in place of the classical chemical transformation method used here.
Acknowledgments
ACKNOWLEDGEMENTS
This work was financially supported by the University of Berne and The Swiss National Science Foundation.
REFERENCES
- 1.Cormack B. (1994) Introduction of a point mutation by sequential PCR steps. Curr. Protoc. Mol. Biol., 2, 8.5.7–8.5.9. [Google Scholar]
- 2.Aiyar A., Xiang,Y. and Leis,J. (1996) Site-directed mutagenesis using overlap extension PCR. Methods Mol. Biol., 57, 177–191. [DOI] [PubMed] [Google Scholar]
- 3.Ishii T.M., Zerr,P., Xia,X.M., Bond,C.T., Maylie,J. and Adelman,J.P. (1998) Site-directed mutagenesis. Methods Enzymol., 293, 53–71. [DOI] [PubMed] [Google Scholar]
- 4.Ling M.M. and Robinson,B.H. (1997) Approaches to DNA mutagenesis: an overview. Anal. Biochem., 254, 157–178. [DOI] [PubMed] [Google Scholar]
- 5.Wang W. and Marcolm,B.A. (1999) Two-stage PCR protocol allowing introduction of multiple mutations, deletions and insertions using QuickChange™ site-directed mutagenesis. BioTechniques, 26, 680–682. [DOI] [PubMed] [Google Scholar]
- 6.Miyazaki K., Wintrode,P.L., Grayling,R.A., Rubingh,D.N. and Arnold,F.H. (2000) Directed evolution study of temperature adaptation in a psychrophilic enzyme. J. Mol. Biol., 297, 1015–1026. [DOI] [PubMed] [Google Scholar]
- 7.Miyazaki K. and Arnold,F.H. (1999) Exploring nonnatural evolutionary pathways by saturation mutagenesis: rapid improvement of protein function. J. Mol. Evol., 49, 716–720. [DOI] [PubMed] [Google Scholar]
- 8.Wada M., Hsu,C.C., Franke,D., Mitchell,M., Heine,A., Wilson,I. and Wong,C.H. (2003) Directed evolution of N-acetylneuraminic acid aldolase to catalyze enantiomeric aldol reactions. Bioorg. Med. Chem., 11, 2091–2098. [DOI] [PubMed] [Google Scholar]
- 9.Hogrefe H.H., Cline,J., Youngblood,G.L. and Allen,R.M. (2002) Creating randomized amino acid libraries with the QuickChange Multi Site-Directed Mutagenesis kit. BioTechniques, 33, 1151–1165. [DOI] [PubMed] [Google Scholar]
- 10.Zheng L., Baumann,U. and Reymond,J.-L. (2004) Molecular mechanism of enantioselective proton transfer to carbon in catalytic antibody 14D9. Proc. Natl Acad. Sci. USA, 101, 3387–3392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zheng L., Goddard,J.-P., Baumann,U. and Reymond,J.-L. (2004) Expression improvement and mechanistic study of the retro-Diels-Alderase catalytic antibody 10F11 by site-directed mutagenesis. J. Mol. Biol., in press. [DOI] [PubMed] [Google Scholar]
- 12.Zheng L., Baumann,U. and Reymond,J.-L. (2003) Production of a functional catalytic antibody ScFv-NusA fusion protein in bacterial cytoplasm. J. Biochem., 133, 577–581. [DOI] [PubMed] [Google Scholar]