

Abstract
Signals generated by cryptochrome (CRY) blue-light photoreceptors are responsible for a variety of developmental and circadian responses in plants. The CRYs are also identified as circadian blue-light photoreceptors in Drosophila and components of the mammalian circadian clock. These flavoproteins all have an N-terminal domain that is similar to photolyase, and most have an additional C-terminal domain of variable length. We present here the crystal structure of the photolyase-like domain of CRY-1 from Arabidopsis thaliana. The structure reveals a fold that is very similar to photolyase, with a single molecule of FAD noncovalently bound to the protein. The surface features of the protein and the dissimilarity of a surface cavity to that of photolyase account for its lack of DNA-repair activity. Previous in vitro experiments established that the photolyase-like domain of CRY-1 can bind Mg·ATP, and we observe a single molecule of an ATP analog bound in the aforementioned surface cavity, near the bound FAD cofactor. The structure has implications for the signaling mechanism of CRY blue-light photoreceptors.
By necessity, plants are exquisitely sensitive to the presence of solar radiation. Plants use various nonchlorophyll photoreceptors to detect light (1). The signals initiated by these light receptors control the development and circadian rhythms of the plant. The three main photoreceptor proteins in the mouse-ear cress Arabidopsis thaliana are phytochromes, phototropins, and the cryptochromes (CRYs); the latter class of photoreceptors senses the presence of blue light. CRYs belong to a superfamily of flavoproteins that occurs in all kingdoms of life (2–4). In addition to the CRYs, this superfamily includes the folate and deazaflavin classes of cyclobutane pyrimidine dimer photolyases, the pyrimidine 6-4 pyrimidone photolyases and the so-called “cryptochromes DASH” (identified in Arabidopsis and Synechocystis, homology to CRYs from Homo and Drosophila). All members of this superfamily have an N-terminal photolyase homology (PHR) domain. The PHR domain binds a FAD cofactor and a second, light-harvesting chromophore (4–8). In cyclobutane pyrimidine dimer photolyase, energy transferred to the flavin cofactor after the absorption of a blue-light photon stimulates the transient transfer of an electron to a bound thymine dimer. The electron transfer initiates the repair of the lesion. At the C-terminal side of the PHR domain, the CRYs typically have another domain (called CCT in plants) that varies widely in its size and amino acid sequence (9).
In plants and insects, CRYs serve as blue-light photoreceptors (6, 10, 11). There is evidence that they also have this function in mammals, where CRYs are additionally known to be important components of the circadian-rhythm molecular machinery (12–14). Plants use the signals generated by blue-light photoreception by the CRYs to cue key developmental signals, such as inhibition of hypocotyl elongation and anthocyanin production (6, 10). In addition, plant CRYs are circadian photoreceptors (15). Arabidopsis has two blue-light photoreceptor cry genes, termed cry1 and cry2. The CRY1 protein detects blue light and initiates downstream signals that effect photomorphogenic changes in the plant. Photoreception by plant CRYs occurs in the PHR domain, and the CCT region contacts COP1, a protein that is downstream in the blue-light signaling pathway (16).
Although the biochemical mechanism of CRY signaling has remained obscure, recent experiments have revealed unexpected activities associated with the plant CRYs. Expression of a β-glucuronidase-CCT fusion protein in Arabidopsis induces a constitutive light response (17), indicating that this portion of the protein is vital for signaling and that the PHR domain must hold the CCT domain in a nonsignaling state in the dark. Also, two separate research groups have reported that Arabidopsis CRY1 undergoes an autophosphorylation reaction that is stimulated by blue light (18, 19). The blue-light enhancement of this phenomenon depends on the presence of the FAD cofactor, and the phosphorylated residue was identified as one or more serines. Further, it has been shown that CRY1 binds ATP in the presence of Mg2+ (18). The enzymatic and ATP-binding activities are found in the PHR region of CRY1. The function of the autophosphorylation in blue-light signaling is not yet established. Because photolyases do not possess this function, it appears that CRYs and photolyases use a similar protein fold to carry out very disparate types of catalysis.
Hindering the analysis of the functions of plant CRYs is the fact that no high-resolution model for the three-dimensional structure of a CRY blue-light photoreceptor is available. The crystal structure of CRY DASH from Synechocystis sp. PCC 6803 (CRY-DASH) has been solved (4), but the role of this protein as a photoreceptor is not clearly established. To shed light on the structure and function of CRY blue-light photoreceptors, we have determined the crystal structures at 2.6- and 2.45-Å resolution, respectively, of the native and ATP-bound PHR region of CRY1 from Arabidopsis (CRY1-PHR). The structures, which are similar to that of DNA photolyases, reveal that CRY1-PHR binds Mg·ATP at an unconventional site. The structure also reveals differences between CRY1-PHR and the photolyases that explain the lack of photolyase activity associated with CRY1 and has implications for the mechanism of blue-light signaling by CRYs.
Materials and Methods
Expression and Purification of CRY1-PHR. The expression plasmid for CRY1-PHR was assembled as described in ref. 5. This plasmid contains a gene for maltose-binding protein (MBP) fused to the N terminus of A. thaliana CRY1-PHR (residues 1–509). Escherichia coli cells containing this plasmid were grown in YTGK medium (10 g/liter Bacto-tryptone/16 g/liter yeast extract/10 ml/liter glycerol/5 g/liter NcCl/0.75 g/liter KCl) (20) at 30°C to an OD600 of 0.6. The temperature of the culture was then lowered to 25°C, and the expression of CRY1-PHR was induced by the addition of 0.1 mM isopropyl β-d-thiogalactoside to the medium. The induction proceeded overnight and was stopped by pelleting the cells and freezing them at -80°C. All purification procedures were carried out at 4°C. The cells were lysed by using lysozyme and sonication, and the cellular debris was removed by centrifugation. The supernatant was applied to an amylose affinity column (New England Biolabs), and MBP-CRY1-PHR was eluted according to the manufacturer's instructions. CRY1-PHR-containing fractions from this column were concentrated and applied to a Superdex 200 gel filtration column (Amersham Pharmacia Biosciences). After elution from this column, the MBP-CRY1-PHR protein was treated with Factor Xa (New England Biolabs) overnight to remove the MBP. The protein was separated from the protease and MBP by applying the mixture to a MonoQ (Amersham Pharmacia Biosciences) column and eluting with a gradient of NaCl. Fractions containing CRY1-PHR were pooled and concentrated.
Crystallization of CRY1-PHR. To a 0.8-μl drop of CRY1-PHR solution at ≈2.5 mg/ml, 0.2 μl of a 3 M solution of the nondetergent solubilizing agent sulfobetaine dimethylethylammonium propane sulfonate (SB-195, Hampton Research, Riverside, CA) was added. This solution was mixed with 1 μl of Drop Solution [100 mM Mes, pH 5.5/0.3 M 1,6-hexanediol (Hampton Research)/4% (vol/vol) glycerol/5 mM Na,K tartrate]. The drop was suspended over 1 ml of the Well Solution [Drop Solution with 12% (vol/vol) glycerol]. Drops were incubated at 26°C. Crystals of CRY1-PHR appeared after ≈1 week and took an additional 1–2 weeks to reach full size. Crystals were stabilized by transferring them to Stabilization Solution [100 mM Mes, pH 5.5/0.3 M 1,6-hexanediol/0.6 M SB-195/20 mM MgCl2/5 mM Na,K tartrate/15% (vol/vol) glycerol]. The crystals were incubated in this solution for 10 min at room temperature, then transferred to Cryostabilization Solution [Stabilization Solution with 35% (vol/vol) glycerol]. After 10 min in this solution, the crystals were flash-cooled in liquid propane and subsequently stored under liquid nitrogen. When included in the stabilization solutions, the concentration of the ATP-analog adenosine 5′-(β,γ-imido)triphosphate (AMP-PNP) was 10 mM.
Data Collection and Structure Determination. X-ray diffraction data from CRY1-PHR crystals were collected at 100 K at beamline 19-ID at the Structural Biology Center of the Advanced Photon Source at the Argonne National Laboratory (Argonne, IL). Table 1 contains the data-collection statistics. The data were indexed, integrated, and scaled by using the hkl2000 package (22). The structure was determined by using the molecular replacement protocols in cns (Version 1.1) (23). Atomic coordinates of a monomer of E. coli DNA photolyase, Protein Data Bank ID code 1DNP (24), stripped of waters, cofactors, and nonhomologous side chains, were used as the molecular replacement model. A single monomer of CRY1-PHR was located in the asymmetric unit. After density modification with cns, the resultant electron-density maps allowed the protein to be modeled from residues 13 to 498. The model was built and adjusted by using the program xtalview (25). The model was refined by using the simulated annealing, conjugate gradient minimization, and individual B-factor minimization procedures available in cns. See Table 1 for the final statistics on the structure.
Table 1. Data and refinement statistics.
| Structure | Native | AMP-PNP-bound |
|---|---|---|
| Space group | P6322 | P6322 |
| a, b, Å | 169.213 | 169.430 |
| c, Å | 104.521 | 104.245 |
| α, β, γ, ° | 90, 90, 120 | 90, 90, 90, 120 |
| Resolution, Å* | 50–2.6 | 50–2.45 |
| No. reflections | 319,411 | 388,897 |
| Unique reflections (F > 0) | 25,083 | 30,708 |
| Completeness, % | 95.9 (97.6) | 98.3 (91.4) |
| Avg. redundancy | 12.4 (9.9) | 11.4 (10.8) |
| Avg. I/σ | 26.5 (2.8) | 37.6 (3.0) |
| Rsym† | 0.097 (0.844) | 0.061 (0.604) |
| No. protein atoms | 3,891 | 3,897 |
| No. waters | 61 | 89 |
| No. Mg (II) ions | 3 | 3 |
| No. AMP-PNP | 0 | 1 |
| Rwork‡ | 0.205 | 0.204 |
| Rfree§ | 0.254 | 0.253 |
| rms deviation | ||
| Bond lengths, Å | 0.009 | 0.016 |
| Bond angles, ° | 1.6 | 1.9 |
| Ramachandran distribution¶ | ||
| Favored, % | 94.8 | 94.6 |
| Allowed, % | 100 | 100 |
Numbers in parentheses are for the highest-resolution shell of the data
Rsym = Σh Σi|Î(h) – Ii(h)|/Σh Σi Ii(h), where Ii(h) is the ith measurement of the intensity for Miller indices h, and Î(h) represents the mean intensity value of the symmetry (or Friedel) equivalent reflections of Miller indices h
Rwork = Σh‖Fo| – |Fc‖/Σh|Fo|
The formula for Rfree is the same as that for Rwork, except that it is calculated with a portion of the structure factors that had not been used for refinement
Using a modified Ramachandran plot (21)
AMP-PNP-Binding Experiments. The association constant for AMP-PNP binding to CRY1-PHR was measured at 4°C by using the gel-chromatographic method of Hummel and Dreyer (26). CRY1-PHR was purified as described above, then subjected to extensive dialysis against HD buffer [50 mM Hepes, pH 7.0/100 mM NaCl/20 mM MgCl2/20% (vol/vol) glycerol/0.5 mM DTT]. A Superose 12 30/10 column (Amersham Pharmacia Biosciences) was equilibrated with concentrations of AMP-PNP ([AMP-PNP]free) ranging from 8.8 to 217 μM. A 50-μl sample of solution containing 30 μM CRY1-PHR that had been equilibrated with AMP-PNP was injected onto the column, and the absorbance of the eluent was monitored at 254 nm. The flow rate of the column was 0.3 ml/min. Three such injections with varying concentrations of AMP-PNP were made for every [AMP-PNP]free. The area of the peak or trough that resulted at the elution volume of AMP-PNP (17.8 ml) was plotted versus the [AMP-PNP] added to the injected sample, and a linear fit of these data was calculated. The x-intercept of this line was taken as the equilibrium [AMP-PNP] ([AMP-PNP]eq). The concentration of bound AMP-PNP ([AMP-PNP]bound) was calculated by using the equation [AMP-PNP]bound = [AMP-PNP]eq - [AMP-PNP]free. From these data, a plot of ν (amount of AMP-PNP bound per mole of CRY1-PHR) versus [AMP-PNP]free was generated, and the program kaleidograph (Synergy Software, Reading, PA) was used to perform a least-squares fit of these data to the nonlinear equation:
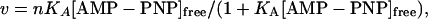 |
[1] |
where n is the number of binding sites on CRY1-PHR and KA is the association constant of CRY1-PHR and AMP-PNP (27).
Results and Discussion
The Crystal Structure of CRY1-PHR. We determined the crystal structure of CRY1-PHR by using E. coli DNA photolyase as a molecular-replacement search model (Fig. 1). We focused on the PHR region because initial attempts at expressing full-length CRY1 from A. thaliana in E. coli cells were unsuccessful (5). CRY1-PHR comprises two domains, the N-terminal α/β domain and the C-terminal α domain. The α/β domain (residues 13–139) adopts a dinucleotide-binding fold made up of a five-stranded parallel β-sheet surrounded by four α-helices and a 310-helix. The α domain (residues 217–495) encompasses 14 α-helices and two 310-helices; it is in this domain that the cofactor FAD is bound. Running between the two domains is a 77-aa segment that exhibits only limited regular secondary structure (three small α-helices and a 310-helix); we term this segment the connector region (residues 140–216). There is a disulfide bond between Cys-190 of the connector region and Cys-80 of the α/β domain (Fig. 1B). It is not known whether this bond exists in vivo. The disulfide bond does not seem to have a large effect on the tertiary structure of CRY1-PHR; it merely binds the connector region to the α/β domain. Therefore, the absence in vivo of the cystine bridge likely would affect only the local conformation of the connector region. Residues 1–12 and 498–509 are not visible in our electron-density maps and thus are not included in our CRY1-PHR model.
Fig. 1.

Structure of CRY1-PHR and its disulfide bond. (A) The structure of CRY1-PHR. Green, helices; purple, β-strands; dark blue, loop regions; orange, FAD cofactor; light blue, AMP-PNP, which is not bound in the native structure. (B) The disulfide bond in CRY1-PHR. The side chains of Cys-80 and Cys-190 are shown, with the carbons in dark blue and the sulfurs in yellow. Superimposed is a simulated-annealing omit map (Fo - Fc, contoured at 3 σ) (28). Figs. 1 and 3 were generated by using pymol.
Bound to CRY1-PHR is the cofactor FAD. The cofactor binds to the protein in a U-shaped conformation that is observed in all known photolyase structures, as well as in the structure of CRY-DASH. Although a folate-type chromophore [5,10-methenyltetrahydrofolate (MTHF)] was found to be associated with CRY1-PHR in previous experiments (5), our electron-density maps betray no evidence of a second cofactor. It is likely that this chromophore dissociated from CRY1-PHR during the numerous purification steps required to yield crystallization-quality protein. Because the pocket that binds MTHF in photolyase is largely filled with amino acid side chains in CRY1-PHR, the binding mode of MTHF to this protein is unclear. Attempts to soak CRY1-PHR crystals with MTHF significantly degraded the quality of the crystals.
On the surface of CRY1-PHR is a cavity that leads from the surface to the mostly buried FAD cofactor (Fig. 2A). One side of the cavity is lined predominantly with hydrophobic residues; the opposite side harbors mainly positively charged and polar amino acid side chains. The bottom of the pocket is formed primarily by the FAD cofactor. Between residues Leu-296 and Tyr-402 on the hydrophobic side of the pocket is electron density that we modeled as one molecule of 1,6-hexanediol, which was included in the crystallization medium (data not shown). A similar cavity on the surface of photolyase (Fig. 2B) is thought to harbor the UV-damaged pyrimidine dimer (24). Hereafter, we shall refer to this feature as the FAD-access cavity.
Fig. 2.

Surface features near to the FAD-access cavity. Shown are the surfaces of CRY1-PHR (A) and photolyase (B). The electrostatic potential is color-coded on the surface, with red and blue representing areas of negative and positive electrostatic potential, respectively. White line, boundary of the FAD-access cavities in both parts. Figs. 2 and 5C were generated by using grasp (29).
Comparison with Other Members of the CRY/Photolyase Superfamily. The structure of CRY1-PHR is very similar to that of DNA photolyase and CRY-DASH. Currently, the structures of DNA photolyase from three different bacterial species are known (24, 30, 31), as is the structure of CRY-DASH from Synechocystis sp. PCC 6803 (4). Table 2 shows the rms deviations that result from pairwise comparisons of the positions of the Cα atoms of CRY1-PHR to those of each of these proteins. CRY1-PHR differs the most from the Thermus thermophilus photolyase, but this difference (rms deviations of ≈1.5 Å over 382 comparable Cα positions) is still small. The largest differences occur in the connector region. The connector regions in all five proteins bridge equivalent secondary structural elements in the α/β and α domains, but they adopt widely divergent courses in doing so. A chain of tryptophan residues that has been proposed (9, 32) to transfer electrons between the FAD cofactor and Trp-324 (E. coli Trp-306) is intact in our CRY1-PHR model (data not shown). The aforementioned disulfide bond between the connector region and the α/β domain is unique to CRY1-PHR.
Table 2. Comparisons of other structures to CRY1-PHR.
| rms deviation*, Å | Equivalent Cαs | Identity†, % | |
|---|---|---|---|
| Photolyases | |||
| E. coli | 1.3 | 421 | 30 |
| Synechococcus | 1.3 | 432 | 27 |
| T. thermophilus | 1.5 | 382 | 29 |
| Cryptochrome | |||
| CRY-DASH | 1.3 | 423 | 26 |
rms deviation from CRY1-PHR
Amino-acid identity
Comparisons of the surface features of the members of the CRY/photolyase superfamily show a key difference between CRY1-PHR and the other proteins in the superfamily: The surface of CRY1-PHR is predominantly negatively charged, with a small concentration of positive charge near to the FAD-access cavity (Fig. 2 A). This contrasts with the photolyases and CRY-DASH, which have a positively charged groove on their surfaces near to the FAD-access cavity (Fig. 2B). In photolyases, this groove is posited to bind the phosphates of the double-stranded DNA surrounding a UV-induced lesion (24). Also, it was surmised that this region of the surface of CRY-DASH contributes to its ability to bind DNA (4). The lack of a positively charged groove on the surface of CRY1-PHR probably accounts for its lack of DNA-binding activity.
Several aspects of the FAD-access cavity differ from the cavities of the photolyases (Fig. 3) and CRY-DASH. There are two tryptophan residues that are important for specific thymine-dimer binding and DNA binding in E. coli photolyase: Trp-277 and Trp-384, respectively. These residues are changed, respectively, to Leu-296 and Tyr-402 in CRY1-PHR. CRY-DASH has a tryptophan at the former position (CRY-DASH Trp-292) and also has a tyrosine at the latter position (CRY-DASH Tyr-398). Some of the differences in this region result in a cavity that is more commodious than those of photolyase or CRY-DASH. Amino acids 363 and 417 of CRY1-PHR have smaller side chains than their counterparts in the other proteins. Also, the cavity of CRY1-PHR is rendered deeper by Leu-398 occupying the space that Tyr-281 or Phe-296 filled in photolyase and CRY-DASH, respectively (Fig. 3). Many of the amino acid differences result in a charge distribution in and around the cavity of CRY1-PHR that contrasts with those of the other proteins (Figs. 2 and 3). Overall, the FAD-access cavity of CRY1-PHR is larger and has a unique chemical environment when compared with the cavities of other members of the photolyase/CRY superfamily. These differences, when coupled with the negatively charged surface of CRY-PHR, effectively explicate the lack of thymine-dimer repair activity of this protein.
Fig. 3.

Comparison of the aligned FAD-access cavities of CRY1-PHR and E. coli photolyase. Residues that are different between the solvent-exposed linings of the FAD-access cavities of the two proteins are shown, except for CRY1-PHR residue Ser-293 (photolyase residue Glu-274). Green carbons and labels, residues from CRY1-PHR; brown carbons and labels, residues from photolyase; red, oxygen atoms; blue, nitrogen atoms; yellow, sulfur atom. The secondary structure and the FAD cofactors are shown faded for clarity.
ATP Binding to CRY1-PHR. To ascertain the location of the ATP-binding site of CRY1-PHR, we soaked our crystals in solutions containing nucleoside triphosphate and Mg2+, enabling us to determine at a 2.45-Å resolution the structure of CRY1-PHR with nucleotide bound (Fig. 1 and Table 1). Because ATP might be utilized by crystalline CRY1-PHR, creating a heterogeneous mixture of ATP and ADP, we used the nonhydrolyzable ATP analog AMP-PNP. In this analog, the bridging-oxygen atom between the β- and γ-phosphates is replaced by an imido (-NH-) group. We confirmed that AMP-PNP binds to CRY1-PHR in vitro by using a chromatographic methodology (Fig. 4). We found that AMP-PNP binds to CRY1-PHR with a Kd of 75 ± 27 μM and a stoichiometry of ≈1:1. Comparing the structures of native and nucleotide-bound CRY1-PHR, there appears to be no change in the conformation of the protein upon Mg·AMP-PNP binding. Electron-density maps generated from crystals soaked in this nucleotide clearly show that one molecule of Mg·AMP-PNP binds in the FAD-access cavity of CRY1-PHR (Fig. 5) (33). The adenine base moiety is located on the hydrophobic side of the pocket, sandwiched between residues Leu-296 and Tyr-402. This moiety has displaced the 1,6-hexanediol observed in the unliganded structure (see above). Hydrogen bonds to the nucleotide are made by several polar or charged residues (Fig. 5B). The FAD cofactor has a water-mediated contact to the bound nucleotide. No protein residues contact a Mg2+ cation associated with the triphosphate moiety of the nucleotide. Although the adenine and ribose moieties of the bound AMP-PNP penetrate deeply into the cavity, the phosphates are located near to the surface of the protein, where they are exposed to solvent (Fig. 5C). As mentioned above, CRY1-PHR apparently catalyzes the transfer of a phosphate from ATP to a serine residue (18). However, the nearest serine residue is ≈11 Å away from the γ-phosphate of bound AMP-PNP in our structure. The location of this ATP-binding site in CRY1-PHR is equivalent to the putative pyrimidine-dimer binding site in photolyases. Indeed, the position of the ribose moiety of AMP-PNP in our structure is very similar to that of a thymine base that was found to bind to the photolyase from T. thermophilus (30). The AMP-PNP probably could not assume this conformation in the cavity of photolyase or CRY-DASH because of steric clashes with residues at positions 300 and 363 (CRY1-PHR numbering) (data not shown). One implication of this ATP-binding site is that bound nucleotide sterically restricts access to the FAD cofactor; if ATP occupies this site in vivo, it seems unlikely that the FAD-access cavity could be a site for electron transfer to a small molecule substrate other than the nucleotide.
Fig. 4.

In vitro AMP-PNP binding to CRY1-PHR. Circles, individual data points; black line, a nonlinear least-squares fit to these data by using the equation in Materials and Methods. The KA for the binding is 13300 ± 4700 M-1, and n (the stoichiometry of binding) is 1.01 ± 0.15.
Fig. 5.

AMP-PNP binding to CRY1-PHR. (A) Stereo representation of the electron density of the AMP-PNP bound to CRY1-PHR. The final refined coordinates for AMP-PNP are shown, colored as in Fig. 3 with the following changes: yellow, carbon atoms; pink, phosphorus atoms. Superimposed on these coordinates is a simulated-annealing omit map (Fo - Fc, contoured at 3 σ) (28). This part of the figure was generated by using xtalview (25) and rendered with povray (www.povray.org). (B) The AMP-PNP-binding site. Shown is a ball-and-stick representation of the final refined coordinates of AMP-PNP and nearby protein residues. Dashed lines, hydrogen bonds. Atoms are colored as in A, with the following exceptions: green, carbons belonging to the protein; silver, carbons from the FAD. Distances are given in Ångströms. Both hydrogen bonds from Arg-360 to the β-phosphate of the AMP-PNP measure 3.4 Å. This part of the figure was generated in vmd (33) and rendered with povray. (C) AMP-PNP binding in the FAD-access cavity. The surface is the same as in Fig. 2. The AMP-PNP, shown as spheres, is colored as in A. A bound Mg2+ cation is not shown.
There are several exceptional features of this ATP-binding site. Among them are the paucity of protein-to-phosphate contacts and the lack of protein ligands to the Mg2+. Also noteworthy is the lack of a nearby serine that could attack the γ-phosphate. There are several possible explanations for these traits. The ATP-binding site may be incomplete; a second CRY1 could dock onto a CRY1 that has ATP bound, providing additional contacts to the cation and solvent-exposed phosphate moieties, which are presented at the entrance to the FAD-access cavity. The docking event also could supply a serine for attack on the γ-phosphate. This docking scenario would predict that the autophosphorylation reaction is intermolecular. The missing residues from the CCT could also contribute to ATP binding or to catalysis. Other factors that may contribute to the seemingly unproductive conformation of the nucleotide are the nonnative nature of AMP-PNP and the crystalline milieu. Finally, the observed ATP-binding site may simply be a crystallographic artifact, and the true site has not yet been observed.
Despite the elucidation of the crystal structure of CRY1-PHR, the mechanism of blue-light signaling by CRYs remains elusive. In two other classes of plant photoreceptors, the phytochromes and the phototropins, light reception triggers large structural changes in the chromophore that apparently lead to conformational changes in the protein (34–36). These conformational changes in turn could allow the photoreceptor to signal to downstream elements that a photon has been absorbed. There is no evidence, however, that the FAD bound to CRY1 undergoes such an alteration upon photon absorption or excitation-energy transfer from an antenna chromophore. Therefore, alternative proposals are necessary to explain the mechanism of blue-light signaling by CRY1. It is tempting to speculate that Cys-80 and Cys-190, which form a disulfide bond in our structure, could play a key role in such a signaling mechanism. The redox state of these cysteines could alter the structure of the connector region, which consequently could affect the ability of the protein to interact with a downstream signaling partner. There are some facts that argue against this model, however. The generality of such a mechanism is called into question by the fact that these two cysteines are not evolutionarily conserved in plant CRYs. Further, there is no clear pathway available to enable electrons to traverse the 30 Å from the FAD to the disulfide bond. A redox reaction involving the cysteines could proceed intermolecularly, with one CRY1 acting as the excited electron donor and another as the acceptor, but it is unknown whether CRY1 is capable of undergoing such a reaction. Additional biochemical and biophysical characterization is required to ascertain whether the observed cystine is relevant to the function of CRY1. There are several other possibilities for the blue-light signaling mechanism. Electron transfer to a small molecule or to the CCT, which is critical for downstream signaling, is one such possibility. Even with the FAD-access cavity occluded by ATP (Fig. 5C), electron transfer to or from the FAD could occur via a chain of conserved tryptophans that are known to traffic electrons in cyclobutane pyrimidine dimer photolyases (9, 32). The autophosphorylation reaction, which is enhanced in the presence of blue light (18, 19), also must be considered as a potential signal. Theoretical calculations suggest that the adenine moiety of FAD acts as an electron conduit between the isoalloxazine ring and the bound thymine dimer in photolyase (37). This hypothesis, coupled with the close proximity of atoms of the adenine moieties of the FAD and bound AMP-PNP (the closest atoms are 4.8 Å apart), suggests that electron transfer could occur from FAD to the adenine of bound ATP. The consequences of such a transfer are unknown.
Finally, the crystal structure at hand represents only 75% of full-length CRY1. The CCT, which is not present in our protein construct, is vital for interacting with components that are downstream of CRY1 in the blue-light signaling pathway (16, 38). When expressed as a β-glucuronidase fusion protein, the CCT acts constitutively to activate phenotypic responses associated with growth in blue light (17). Any complete structural analysis of CRYs must include this essential component. It should be very illuminating to view a structure of intact CRY1. Such a structure could show the interaction between the PHR and CCT domains and could further elucidate the mechanism of CRY signaling.
Acknowledgments
We thank Dr. A. Sancar for the CRY1-PHR plasmid and helpful discussions and the beamline personnel for their assistance. The x-ray diffraction data were collected at the Argonne National Laboratory's Structural Biology Center at the Advanced Photon Source, which is supported by the Office of Energy Research, U.S. Department of Energy, under Contract W-31-109-ENG-38. This research was supported in part by Welch Foundation Grant I-1185 (to J.D.) J.D. is an Investigator with the Howard Hughes Medical Institute.
Abbreviations: AMP-PNP, adenosine 5′-(β,γ-imido)triphosphate; CRY, cryptochrome; CRY1-PHR, PHR region of CRY1 of Arabidopsis thaliana; CCT, C-terminal region of CRY; DASH, identified in Arabidopsis and Synechocystis, homology to CRYs from Homo and Drosophila; CRY-DASH, Synechocystis sp. PCC 6803 CRY-DASH; MBP, maltose-binding protein; MTHF, 5,10-methenyltetrahdrofolate; PHR, photolyase homology region.
Data deposition: The coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org. [PDB ID codes 1U3C (CRY1-PHR) and 1U3D (CRY1-PHR with AMP-PNP bound)].
References
- 1.Gyula, P., Schafer, E. & Nagy, F. (2003) Curr. Opin. Plant Biol. 6, 446-452. [DOI] [PubMed] [Google Scholar]
- 2.Cashmore, A. R., Jarillo, J. A., Wu, Y.-J. & Liu, D. (1999) Science 284, 760-765. [DOI] [PubMed] [Google Scholar]
- 3.Hitomi, K., Okamoto, K., Daiyasu, H., Miyashita, H., Iwai, S., Toh, H., Ishiura, M. & Todo, T. (2000) Nucleic Acids Res. 28, 2353-2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brudler, R., Hitomi, K., Daiyasu, H., Toh, H., Kucho, K., Ishiura, M., Kanehisa, M., Roberts, V. A., Todo, T., Tainer, J. A. & Getzoff, E. D. (2003) Mol. Cell 11, 59-67. [DOI] [PubMed] [Google Scholar]
- 5.Malhotra, K., Kim, S.-T., Batschauer, A., Dawut, L. & Sancar, A. (1995) Biochemistry 34, 6892-6899. [DOI] [PubMed] [Google Scholar]
- 6.Ahmad, M. & Cashmore, A. R. (1993) Nature 366, 162-166. [DOI] [PubMed] [Google Scholar]
- 7.Hsu, D. S., Zhao, X., Zhao, S., Kazantsev, A., Wang, R.-P., Todo, T., Wei, Y.-F. & Sancar, A. (1996) Biochemistry 35, 13871-13877. [DOI] [PubMed] [Google Scholar]
- 8.Kleine, T., Lockhart, P. & Batschauer, A. (2003) Plant J. 35, 93-103. [DOI] [PubMed] [Google Scholar]
- 9.Sancar, A. (2003) Chem. Rev. 103, 2203-2237. [DOI] [PubMed] [Google Scholar]
- 10.Ahmad, M., Lin, C. & Cashmore, A. R. (1995) Plant J. 8, 653-658. [DOI] [PubMed] [Google Scholar]
- 11.Emery, P., So, W. V., Kaneko, M., Hall, J. C. & Rosbash, M. (1998) Cell 95, 669-679. [DOI] [PubMed] [Google Scholar]
- 12.Thompson, C. L., Selby, C. P., Partch, C. L., Plante, D. T., Thresher, R. J., Araujo, F. & Sancar, A. (2004) Mol. Brain Res. 122, 158-166. [DOI] [PubMed] [Google Scholar]
- 13.Thresher, R. J., Vitaterna, M. H., Miyamoto, Y., Kazantsev, A., Hsu, D. S., Petit, C., Selby, C. P., Dawut, L., Smithies, O., Takahashi, J. S. & Sancar, A. (1998) Science 282, 1490-1494. [DOI] [PubMed] [Google Scholar]
- 14.van der Horst, G. T. J., Muijtjens, M., Kobayashi, K., Takano, R., Kanno, S., Takao, M., de Wit, J., Verkerk, A., Eker, A. P. M., van Leenen, D., et al. (1999) Nature 398, 627-630. [DOI] [PubMed] [Google Scholar]
- 15.Somers, D. E., Devlin, P. F. & Kay, S. A. (1998) Science 282, 1488-1490. [DOI] [PubMed] [Google Scholar]
- 16.Yang, H.-Q., Tang, R.-H. & Cashmore, A. R. (2001) Plant Cell 13, 2573-2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang, H.-Q., Wu, Y.-J., Tang, R.-H., Liu, D., Liu, Y. & Cashmore, A. R. (2000) Cell 103, 815-827. [DOI] [PubMed] [Google Scholar]
- 18.Bouly, J.-P., Giovani, B., Djamei, A., Mueller, M., Zeugner, A., Dudkin, E. A., Batschauer, A. & Ahmad, M. (2003) Eur. J. Biochem. 270, 2921-2928. [DOI] [PubMed] [Google Scholar]
- 19.Shalitin, D., Yu, X., Maymon, M., Mockler, T. & Lin, C. (2003) Plant Cell 15, 2421-2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wynn, R. M., Davie, J. R., Chuang, J. L., Cote, C. D. & Chuang, D. T. (1998) J. Biol. Chem. 273, 13110-13118. [DOI] [PubMed] [Google Scholar]
- 21.Lovell, S. C., Davis, I. W., Arendall, W. B., III, de Bakker, P. I. W., Word, J. M., Prisant, M. G., Richardson, J. S. & Richardson, D. C. (2002) Proteins 50, 437-450. [DOI] [PubMed] [Google Scholar]
- 22.Otwinowski, Z. & Minor, W. (1997) Methods Enzymol. 276, 307-326. [DOI] [PubMed] [Google Scholar]
- 23.Brunger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N. S., et al. (1998) Acta Crystallogr. D 54, 905-921. [DOI] [PubMed] [Google Scholar]
- 24.Park, H.-W., Kim, S.-T., Sancar, A. & Deisenhofer, J. (1995) Science 268, 1866-1872. [DOI] [PubMed] [Google Scholar]
- 25.McRee, D. E. (1992) J. Mol. Graphics 10, 44-46. [Google Scholar]
- 26.Hummel, J. P. & Dreyer, W. J. (1962) Biochim. Biophys. Acta 63, 530-532. [DOI] [PubMed] [Google Scholar]
- 27.Klotz, I. M. (1946) Arch. Biochem. 9, 109-117. [PubMed] [Google Scholar]
- 28.Hodel, A., Kim, S.-H. & Brunger, A. T. (1992) Acta Crystallogr. A 48, 851-859. [Google Scholar]
- 29.Nicholls, A., Sharp, K. & Honig, B. (1991) Proteins 11, 281-296. [DOI] [PubMed] [Google Scholar]
- 30.Komori, H., Masui, R., Kauramitsu, S., Yokoyama, S., Shibata, T., Inoue, Y. & Miki, K. (2001) Proc. Natl. Acad. Sci. USA 98, 13560-13565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tamada, T., Kitadokoro, K., Higuchi, Y., Inaka, K., Yasui, A., de Ruiter, P. E., Eker, A. P. M. & Miki, K. (1997) Nat. Struct. Biol. 4, 887-891. [DOI] [PubMed] [Google Scholar]
- 32.Cheung, M. S., Daizadeh, I., Stuchebrukhov, A. A. & Heelis, P. F. (1999) Biophys. J. 76, 1241-1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Humphrey, W., Dalke, A. & Schulten, K. (1996) J. Mol. Graphics 14, 33-38. [DOI] [PubMed] [Google Scholar]
- 34.Crosson, S. & Moffat, K. (2002) Plant Cell 14, 1067-1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Harper, S. M., Neil, L. C. & Gardner, K. H. (2003) Science 301, 1541-1544. [DOI] [PubMed] [Google Scholar]
- 36.van der Horst, M. A. & Hellingwerf, K. J. (2004) Acc. Chem. Res. 37, 13-20. [DOI] [PubMed] [Google Scholar]
- 37.Antony, J., Medvedev, D. & Stuchebrukhov, A. A. (2000) J. Am. Chem. Soc. 122, 1057-1065. [Google Scholar]
- 38.Wang, H., Ma, L.-G., Li, J.-M., Zhao, H.-Y. & Deng, X. W. (2001) Science 294, 154-158. [DOI] [PubMed] [Google Scholar]