

Abstract
For the past century, the total synthesis of natural products has served as the flagship of chemical synthesis and the principal driving force for discovering new chemical reactivity, evaluating physical organic theories, testing the power of existing synthetic methods, and enabling biology and medicine. This perspective article seeks to examine this time-honored and highly demanding art, distilling its essence in an effort to ascertain its power and future potential.
Essence (és'ns) n. the most significant part of a thing's nature; the sum of the intrinsic properties without which a thing would cease to be what it is, and which are not affected by accidental modifications. (The New Lexicon Webster's Dictionary)
Although the practice of total synthesis and the rationale behind its pursuit have changed throughout the course of its history, its most fundamental property has not. At its core, in its most essential form, natural product total synthesis is a vehicle for discovery, one that is perhaps unparalleled by any other endeavor in the realm of chemical synthesis (1–3). The reason follows: every natural product type isolated from the seemingly limitless chemical diversity in nature provides a unique set of research opportunities deriving from its distinctive three-dimensional architecture and biological properties. For instance, in the early part of the 20th century, efforts directed toward the synthesis of the antimalarial agent quinine (1) (Fig. 1) led to a sizeable body of knowledge regarding the construction of heteroaromatic systems and the unique physical properties of quinoline and piperidine rings. In more recent times, its structure has served as the basis for the design of several new classes of antimalarial drugs that have saved thousands of lives (4). Similarly, attempts to construct steroids such as progesterone (2) before World War II provided insights into how carbon–carbon bonds could both be forged and cleaved, with their partial or total synthesis ultimately rendering them available in quantities that are sufficient to propel them into useful drugs, such as the birth control pill, which is now used by millions of women around the world (5). In the 1950s, the highly sensitive β-lactam ring of penicillin (3) served as the impetus for John Sheehan (6) to develop carbodiimide-based reagents for the formation of peptide bonds. This discovery capped the first practical synthesis of this essential medicinal agent, enabled the synthesis of designed penicillins with activity profiles superior to the parent natural product, and revolutionized the entire peptide-synthesis field. In the 1960s and 1970s, members of the eicosanoid family of natural products, such as prostaglandin F2a (4), served as the artistic canvas on which E. J. Corey was inspired to create the first catalysts capable of orchestrating asymmetric Diels–Alder reactions. It also was the arena in which he and his group developed the now ubiquitous family of silyl-based protecting groups, one of the most general and powerful methods for the catalytic asymmetric reduction of ketones (Corey–Bakshi–Shibata reduction), and a series of prostaglandin analogs used for various purposes (7). During the same era, the unique structures possessed by molecules like vitamin B12 and cobyric acid (5) served as the testing ground for new reactions and fundamental physical organic principles, such as the Eschenmoser corrin synthesis (8) and the Woodward–Hoffman rules (9).
Fig. 1.

Structures of quinine (1), progesterone (2), penicillin (3), prostaglandin F2α (4), and cobyric acid (5), natural products whose total synthesis inspired and resulted in the development of manifold advances in chemistry, biology, and medicine.
This brief synopsis drawing from just five examples barely scratches the surface of the rich history of scientific breakthroughs in this field and the potential for making fundamental advances. Here, we offer several additional examples from our own experiences in the hope that they will illustrate the true wealth of discoveries that can emanate from more contemporary endeavors in total synthesis.
Selected Total Synthesis Endeavors
If the past few decades have taught us anything about the power of our synthetic tools, it is that we have yet to reach a level of efficiency and deftness commensurate to that possessed by nature. Yet, although we cannot exactly emulate the master chemical artisan, synthetic chemists can come close when they develop synthetic strategies toward natural products based on biogenetic considerations because what often results is a concise and elegant construction of molecular complexity. As an added benefit, such approaches also afford insights into how reactions can be combined productively into novel cascade events. Historical examples of such successes include W. S. Johnson's synthesis of progesterone by means of a series of cation-π cyclizations and our own construction of several members of the endiandric acid family of natural products by means of a cascade sequence that combined electrocyclizations and Diels–Alder reactions (10).
A more recent entry comes from our research program directed toward selected members of the bisorbicillinoid family of natural products (11). For instance, although the cage-like natural product trichodimerol (12) (Scheme 1) seemed hopelessly complex on initial inspection based on its dense array of chiral centers and unusual ring framework, closer analysis suggested that its entire architecture could arise in a single step from a far simpler, but highly reactive, intermediate: quinol 7 and its tautomer 8. The operations required to bring about this tantalizing proposal, however, were relatively complex because they involved a dimerization-based domino sequence comprised of two Michael reactions and two ketalizations. After careful experimentation, we were able to accomplish this ambitious series of events in the laboratory by carefully cleaving the acetate group within 6 under basic conditions to generate the reactive monomers and then quenching the reaction with NaH2PO4·H2O. A similar strategy protocol was employed by Barnes-Seeman and Corey in their successful synthesis of trichodimerol (12). Amazingly, if this protocol was altered just slightly, we could also convert 7 and 8 into two other members of the family, bisorbicillinol (13) and bisorbibutenolide (14), by coaxing them to participate in an intermolecular Diels–Alder union instead (11). Whether or not nature creates these natural products in the same way is unknown, but it is difficult to conceive that nature would employ a pathway that is any less expedient. In any case, these sequences point to one of the key directions for the future of chemical synthesis as pressure increases to generate molecular complexity rapidly through efficient and atom-economical processes that produce minimal chemical waste.
Scheme 1.

Insights into the synthetic efficiency of nature: total synthesis of trichodimerol (12) through a dimerization event based on a double Michael/ketalization sequence. A different pathway from 6 led to bisorbicillinol (13) and bisorbibutenolide (14).
Similar levels of beauty, challenge, and opportunity for discovery also exist in multistep total synthesis, especially when targeting a complex natural product with a unique conglomeration of structural motifs. Indeed, rare and challenging molecular features within secondary metabolites have long presented synthetic chemists with golden opportunities for invention, whether simply by inspiring creative strategies and tactics, or serving as a stringent testing ground that reveals weaknesses in the power of existing methodology to fashion such complexity effectively. The brevetoxins (15, 16) (Scheme 2), the toxic agents responsible for the “Red Tide” phenomena, certainly fit this bill with their exquisite array of trans-fused ether rings of various sizes (13). Of particular challenge were their medium-sized cyclic ethers because the technology that was available when we undertook their total synthesis in the 1980s did not possess the power to forge such strained structural domains in highly functionalized settings. By using this gap in methodology as an opportunity for discovery, we developed a series of synthetic reactions based on the power of heteroatoms (i.e., sulfur and phosphorous) to drive these entropically disfavored ring closures; four of these methods are shown in Scheme 2. Apart from finding widespread applicability to a range of other synthetic problems, these unique approaches were critical to the completion of both of these formidable targets, accomplishments that have more recently inspired a host of researchers to attempt the total synthesis of other cyclic polyethers, some of even greater size and complexity (14–20).
Scheme 2.

The brevetoxins: ornate architectures whose unique ring systems demanded the development of several novel synthetic technologies before yielding to total synthesis.
In addition to new science emanating from well planned endeavors, discoveries can also arise by serendipity when an attempt to construct a certain structural motif does not proceed as expected. Our adventures related to the total synthesis of the CP molecules (31 and 32) (Scheme 3) underscore this mode of discovery as forging its complex polycyclic architecture not only demanded the development of many planned sequences and new methods but also afforded several examples of unexpected chemical reactivity that led to the opportunistic development of new chemistry (21–24). For example, as shown in Scheme 3, treatment of intermediate 33 with Dess–Martin periodinane (DMP) at elevated temperatures was expected to provide 34. Instead, the sole product of this experiment was the polycyclic adduct 36, a compound of little use for the prosecution of our strategy to access the target molecules. Yet, its occurrence unlocked a new field of chemistry, as the determined pursuit of the mechanistic underpinnings of this reaction led to the rational design of several new reactions. Some of the most important of these processes include the synthesis of heterocycles from intermediate o-quinodimethanes generated from compounds such as 37, the formation of α,β-unsaturated compounds from saturated precursors (40→41), the deprotection of dithianes (42→43), the oxidation of heterocyclic systems (44→45), and the generation of p-quinones from anilides (46→47). These studies have also led to the design and synthesis of a number of new and highly selective classes of hypervalent iodine reagents (ref. 25 and references therein). Had the experiment seeking to convert 33 into 34 not been performed and had the rationale for its outcome never been explored, this field of study might have remained latent for many years to come, if ever uncovered at all.
Scheme 3.

The CP-molecules: a serendipitous discovery during their total synthesis, in what was expected to be a routine oxidation, served as the catalyst for the design and development of a series of new synthetic technologies.
As a final set of examples related to the theme of how challenging molecular connectivities of natural products can lead to new methodology, we mention our recent forays toward diazonamide A, a natural product whose originally proposed structure (48) has recently (26) been revised to 49 (Scheme 4). Both of these architectures offered unique chances to make discoveries of the designed and unexpected varieties. For instance, early efforts directed toward the generation of the quaternary center linking the two major macrocycles of 48 were based on effecting a 5-exo-tet cyclization on a precursor, such as 50, leading to 54 (Scheme 4). This transformation would, indeed, be realized eventually, opening a new entry for the formation of such benzofuranone adducts (27). At first, however, initial experiments led to the observation of an unintended cyclofragmentation cascade leading to the highly desirable 3-arylbenzofuran nucleus (53), a privileged structural motif found in numerous clinically used pharmaceuticals. This insight into chemical reactivity inspired us to extend this finding into the realm of split-and-pool combinatorial synthesis, ultimately providing technology for the facile and traceless preparation of diverse members of this important family of compounds (28).
Scheme 4.

The diazonamide A studies: confirming structural assignments and discovering new chemical reactivity.
The opportunity to design and develop another cascade sequence within the context of the diazonamide A program came with roadblocks confronted in forming a carbon–carbon bond to close the right macrocycle of the revised structure (49) (29) with enough functionality to assemble all its disparate heteroaromatic rings. Inspired by a series of seemingly unconnected literature precedents, we anticipated that we could accomplish this objective by combining three known reactions into a new single sequence. As shown in the conversion of 55 to 60, those operations were a heteropinacol reaction initiated by the power of SmI2 complexed to N,N-dimethylacetamide, in situ N–O bond cleavage to generate an amino alcohol, and subsequent peptide coupling. Not only did the successful realization of this cascade sequence represents the first macrocyclization using a heteropinacol reaction, it also fueled the completion of the revised structure of diazonamide A (49), an endeavor that finally proved its long-questioned structural disposition (30, 31). Reflecting on this program, as well as the other programs described thus far from a broader perspective, these endeavors illustrate that virtually all complex architectures are destined to lead to some form of discovery. Key to this state of affairs is the fact that total synthesis is uncompromising, i.e., no atom or functional group within the structure of a natural product can be altered or subtracted to make the task of synthesizing it any easier. Consequently, every failure along the way (and there are usually many failures in such research programs) forces the practitioner to be creative if progress is to be made toward the final destination.
It is also important to recognize that, no matter how unique they may be, natural products are not merely compounds meant to show off the synthetic skills of nature. Secondary metabolites almost always have a specific biochemical purpose, and more often than not, their presence has conferred some form of evolutionary advantage to the producing organism. This concept has important ramifications because if we can determine both how and why nature has evolved these molecules to achieve certain objectives, then we can likely unravel many of the mysteries of biology and gain the insights needed to impart function to molecules of our own imagining. Over the course of the past 15 years, one of the most important families of natural products that has helped to guide scientists in such directions is the enediyne antitumor antibiotics, the flagship of which is calicheamicin γ1I (61, Scheme 5) (32). These compounds exhibit their potency by cleaving single- and double-stranded DNA through a diradical species generated upon Bergman cycloaromatization of their enediyne motif. What is especially interesting, however, is the way that nature has loaded a uniquely engineered triggering system within each enediyne molecule to set this critical event into motion at just the right moment. For instance, in the case of calicheamicin γ1I, this process is initiated by the cleavage of its trisulfide moiety to reveal a thiol species followed by the attack of this newly unveiled motif onto its neighboring enone functionality through an intramolecular conjugate-addition reaction.
Scheme 5.

Calicheamicin γ1I and the enediynes: an opportunity to make fundamental discoveries in chemical synthesis, physical organic chemistry, molecular design, and chemical biology.
Such ingenious reactivity has inspired chemists to develop their own enediyne systems with equally sophisticated activation devices. As part of our program that culminated in the first total synthesis of calicheamicin γ1 in 1992 (33–35), we designed several such compounds by using information gathered from a series of physical organic studies that established the structural parameters required for cyclic enediynes to undergo Bergman cycloaromatization. As shown in Scheme 5, these molecules ranged from simple hydrocarbons (i.e., 62) activated by exposure to heat to far more ornate systems like 65 (which is a compound patterned after the natural product dynemicin A), whose reactivity was unleashed under basic conditions. The latter of these in vivo-activated designed enediynes exhibited cytotoxicity at picomolar concentrations (36). As such, this program illustrates how endeavors in total synthesis not only can lead to a better understanding of specific architectural domains but also how the reactive features of a natural product can be harnessed to deliver new classes of therapeutic agents with real-world applicability.
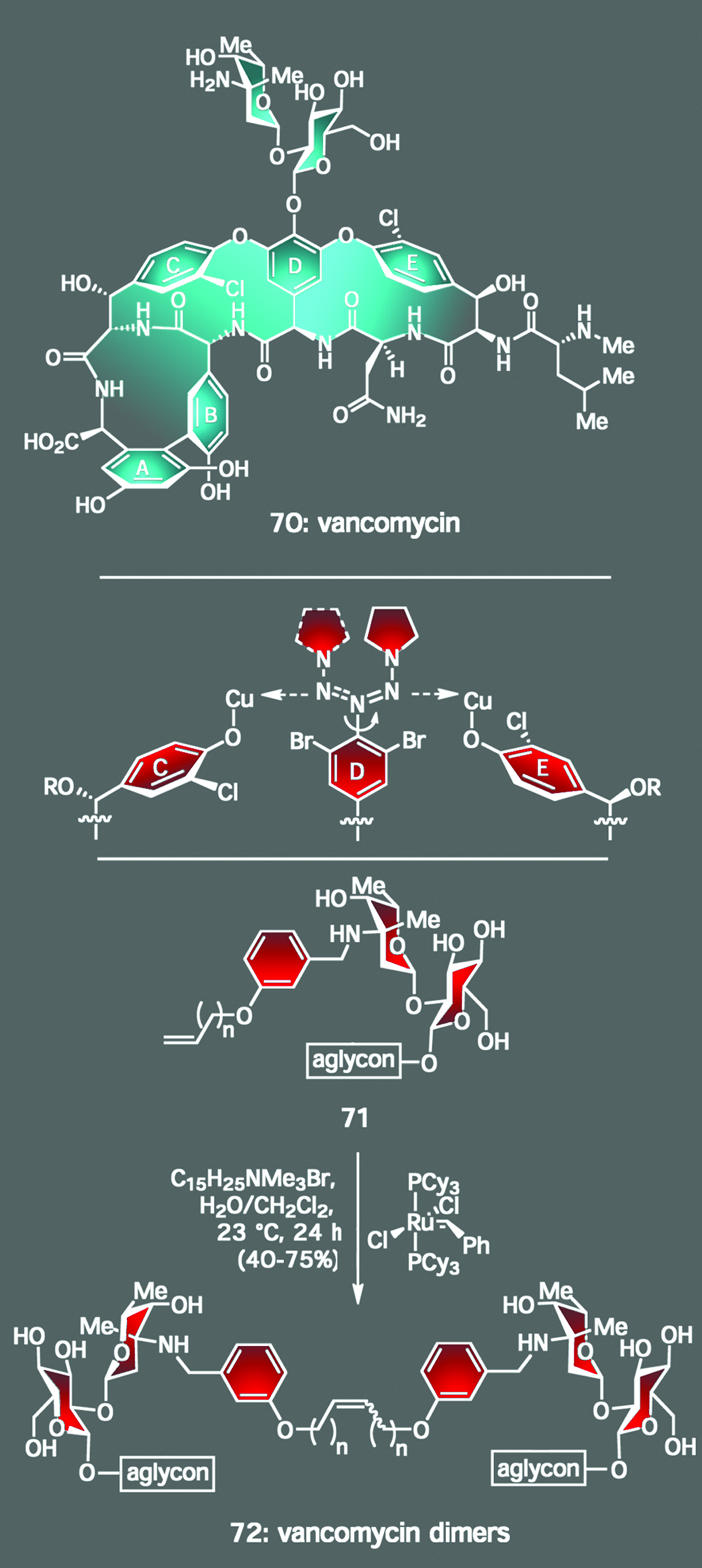
Similar themes drove our program toward the clinically used anticancer agent Taxol (69, Fig. 2) (37), in which the presence of two highly functionalized six-membered rings led to the design of unique Diels–Alder-based synthetic strategies (38) for their construction, and its novel activity inspired the design of several types of taxoid molecules (39–43). Such rationales also propelled our work with vancomycin (70, Scheme 6), the agent currently considered to be the antibiotic of last resort against deadly methicillin-resistant strains of Staphylococcus aureus (44). Here, our program sought to accomplish both the total synthesis of the molecule and, subsequently, the preparation of a series of vancomycin analogs to counter bacterial resistance to the parent natural product. Achieving the first of these objectives required the development of new synthetic methodology because the tests provided by the unique conglomeration of vancomycin's sensitive functional groups (including seven epimerizable arylglycines) and stereochemical complexity (including 18 chiral centers and three axes of atropisomerism) were rather rigorous. Most noteworthy among these discoveries was a triazene-driven aryl ether forming reaction catalyzed by copper salts to generate the two 16-membered macrocyclic rings of the target molecule, a transformation with broad synthetic scope (45–49). The second task, preparing vancomycin analogs with improved activity, was accomplished by means of an inventive use of a process known as dynamic combinatorial screening (also known as target-driven combinatorial synthesis). Drawing insight from the fact that two molecules of vancomycin can bind simultaneously, and reversibly, to its biological target (an l-Lys-d-Ala-d-Ala peptide subunit) through a number of hydrogen bonds, we hypothesized that we could create potent vancomycin-like antibiotics by creating a dimeric form of the natural product. Our method was to expose a collection of monomeric analogs to the biological target by using a reactive handle on these building blocks to trap the strongest binders as a new heterodimer. As indicated in Scheme 6, this goal was accomplished under ambient conditions through the power of olefin cross-metathesis by using catalytic amounts of a ruthenium catalyst (50) and a phase-transfer reagent (51). In each round of screening, the predominant product was also the most potent (based on synthesizing and testing all dimers separately), with several compounds possessing enhanced activity relative to vancomycin as well as high potency against several vancomycin-resistant bacterial strains. Again, total synthesis provided an outlet to develop not only new reactions and synthetic strategies but also tools that might prove key in combating bacterial resistance.
Fig. 2.
Taxol: total synthesis met the challenge through a convergent strategy using two Diels–Alder reactions.
Scheme 6.

Vancomycin: a synthetic target whose novel architecture afforded the opportunity to develop new chemistry, such as a triazene-driven method for bisaryl ether formation and a cross-metathesis reaction in water to form dimeric vancomycin analogs with activity greater than that of the natural product.
Our final entry for this article takes total synthesis one step further with another set of bioactive molecules. This group of compounds are the epothilones (73–76, Scheme 7), a family of natural products whose exploration has not only provided an opportunity to develop new synthetic technology but also is at the brink of providing real medical dividends for the treatment of disease (52). First isolated in the 1980s, these agents kill tumor cells with extremely high efficiency, possessing IC50 values in the low nanomolar range for most of the cell lines that constitute a typical first-round anticancer activity screen. Such potency rivals, and in some cases exceeds, that possessed by agents like Taxol and vinblastine, two compounds currently used in the clinic to manage cancer. Therefore, numerous researchers from academe as well as the pharmaceutical industry have been lured to pursue research programs directed toward their total synthesis in order to render them readily available and to create new, and potentially better, clinical candidates (53).
Scheme 7.

The epothilones: use of new solid-phase technology combined with an olefin metathesis-based cyclorelease strategy rapidly generated a number of structural analogs, ultimately culminating in a drug candidate (82) for cancer chemotherapy.
Some of our work toward these highly promising molecules is shown in Scheme 7. Unlike most synthetic plans in which the controlled installation of stereochemical elements is deemed to be of critical importance, we initially chose to develop a strategy toward the epothilones in which we could access as many structural isomers as possible with minimal effort. That goal was achieved by using the power of solid-phase synthesis along with reactions that could lead to stereochemical mixtures. Thus, after the synthesis of 77, a compound generated as a mixture of two diastereomers by a nonselective aldol reaction, we exploited the nonselective nature of ring-closing olefin metathesis to form both E- and Z-macrocycles. Accordingly, from one common resin-bound intermediate, we were able to access four structurally different epothilones (78–81), compounds that were separated by HPLC and independently elaborated to the final targets. This combinatorial approach, complemented by target-oriented synthesis of specifically designed analogs, led to several hundred distinct compounds whose biological screening established a clear set of structure–activity relationships for the epothilones (54). These studies ultimately paved the way for the intelligent design of novel epothilone-like structures possessing comparable or even more potent and selective anticancer properties than the parent natural products. One of these compounds (82) is now in clinical trials sponsored by Novartis, our collaborator for its development. Accordingly, this work further reinforces the value of natural product total synthesis in leading to drug candidates and eventually to useful medicines; in fact, approximately one-half of the top-selling pharmaceuticals on the market today either are natural products or are based on structures of natural products (55). As an aside, these investigations provided one of the first examples of a complex natural product being synthesized on solid-support. The use of the radiofrequency method of encoding combinatorial libraries in this study was also pioneering (56).
Conclusions
New reactions, cascade sequences and biomimetic strategies, clinical drug candidates, physical organic chemistry principles, structure elucidation, art, and excitement: the list of discoveries and concepts that consistently emanate from programs in total synthesis is rich in content and is increasing in length (57). We leave it for the rest of the insightful Perspectives and research articles in this Special Feature dedicated to natural product total synthesis to reinforce these categories and convey the richness of this art, and we close instead with some words penned in 2003 by E. J. Corey:
How many challenging and worthy synthetic targets remain to be discovered? How many truly powerful and general new synthetic strategies and synthetic reactions remain to be discovered? Is there a prospect for the development of entirely new ways of planning or executing synthesis? In my judgment the opportunities for new developments and discoveries are so vast that today's synthesis is best regarded as a youngster with a brilliant future (2).
Acknowledgments
We thank our collaborators whose names appear in the references and whose contributions made this work enjoyable and highly rewarding. This work was supported by the National Institutes of Health, the Skaggs Institute for Chemical Biology, American BioScience, Merck, Schering Plough, Hoffmann–La Roche, GlaxoWellcome, Amgen, Novartis, Bristol-Myers Squibb (fellowship to S.A.S.), Boehringer Ingelheim, Zeneca, CaPCURE, the George E. Hewitt Foundation, Pfizer (fellowship to S.A.S.), and the National Science Foundation (fellowship to S.A.S.).
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Nicolaou, K. C. & Sorensen, E. J. (1996) Classics in Total Synthesis: Targets, Strategies, Methods (VCH, Weinheim, Germany), p. 799.
- 2.Nicolaou, K. C. & Snyder, S. A. (2003) Classics in Total Synthesis II: More Targets, Strategies, Methods (Wiley–VCH, Weinheim, Germany), p. 639.
- 3.Nicolaou, K. C., Vourloumis, D., Winssinger, N. & Baran, P. S. (2000) Angew. Chem. Int. Ed. 39, 44-122. [PubMed] [Google Scholar]
- 4.Garfield, S. (2000) Mauve (Faber & Faber, London), p. 224.
- 5.Djerassi, C. (1990) Steroids Made It Possible (American Chemical Society, Washington. D.C.), p. 205.
- 6.Sheehan, J. C. (1984) The Enchanted Ring: The Untold Story of Penicillin (MIT Press, Cambridge), p. 224.
- 7.Corey, E. J. & Chen, X.-M. (1989) The Logic of Chemical Synthesis (Wiley, New York), p. 436.
- 8.Götschi, E., Hunkeler, W., Wild, H.-J., Schneider, P., Fuhrer, W., Gleason, J. & Eschenmosher, A. (1973) Angew. Chem. Int. Ed. 12, 910-912. [DOI] [PubMed] [Google Scholar]
- 9.Benfey, O. T. & Morris, P. J. T. (2001) Robert Burns Woodward: Artist and Architect in the World of Molecules (Chemical Heritage Foundation, Philadelphia), p. 470.
- 10.Nicolaou, K. C., Montagnon, T. & Snyder, S. A. (2003) Chem. Commun., 551-564. [DOI] [PubMed]
- 11.Nicolaou, K. C., Vassilikogiannakis, G., Simonsen, K. B., Baran, P. S., Zhong, Y.-L., Vidali, V. P., Pitsinos, E. N. & Couladouros, E. A. (2000) J. Am. Chem. Soc. 122, 3071-3079. [DOI] [PubMed] [Google Scholar]
- 12.Barnes-Seeman, D. & Corey, E. J. (1999) Org. Lett. 1, 1503-1504. [DOI] [PubMed] [Google Scholar]
- 13.Nicolaou, K. C. (1996) Angew. Chem. Int. Ed. 35, 589-607. [Google Scholar]
- 14.Nicolaou, K. C., Hwang, C.-K., Duggan, M. E., Nugiel, D. A., Abe, Y., Bal Reddy, K., DeFrees, S. A., Reddy, D. R., Awartani, R. A., Conley, S. R., et al. (1995) J. Am. Chem. Soc. 117, 10227-10238. [Google Scholar]
- 15.Nicolaou, K. C., Theodorakis, E. A., Rutjes, F. P. J. T., Sato, M., Tiebes, J., Xiao, X.-Y., Hwang, C.-K., Duggan, M. E., Yang, Z., Couladouros, E. A., et al. (1995) J. Am. Chem. Soc. 117, 10239-10251. [Google Scholar]
- 16.Nicolaou, K. C., Rutjes, F. P. J. T., Theodorakis, E. A., Tiebes, J., Sato, M. & Untersteller, E. (1995) J. Am. Chem. Soc. 117, 10252-10263. [Google Scholar]
- 17.Nicolaou, K. C., Bunnage, M. E., McGarry, D. G., Shi, S., Somers, P. K., Wallace, P. A., Chu, X.-J., Agrios, K. A., Gunzner, J. L. & Yang, Z. (1999) Chem. Eur. J. 5, 599-617. [Google Scholar]
- 18.Nicolaou, K. C., Wallace, P. A., Shi, S., Ouellette, M. A., Bunnage, M. E., Gunzner, J. L., Argrios, K. A., Shi, G.-Q. & Yang, Z. (1999) Chem. Eur. J. 5, 618-627. [Google Scholar]
- 19.Nicolaou, K. C., Shi, G.-Q., Gunzner, J. L., Gärtner, P., Wallace, P. A., Ouellette, M. A., Shi, S., Bunnage, M. E., Agrios, K. A., Veale, C. A., et al. (1999) Chem. Eur. J. 5, 628-645. [Google Scholar]
- 20.Nicolaou, K. C., Gunzner, J. L., Shi, G.-Q., Agrios, K. A., Gärtner, P. & Yang, Z. (1999) Chem. Eur. J. 5, 646-658. [Google Scholar]
- 21.Nicolaou, K. C. & Baran, P. S. (2002) Angew. Chem. Int. Ed. 41, 2678-2720. [DOI] [PubMed] [Google Scholar]
- 22.Nicolaou, K. C., Jung, J., Yoon, W. H., Fong, K. C., Choi, H.-S., He, Y., Zhong, Y.-L. & Baran, P. S. (2002) J. Am. Chem. Soc. 124, 2183-2189. [DOI] [PubMed] [Google Scholar]
- 23.Nicolaou, K. C., Baran, P. S., Zhong, Y.-L., Fong, K. C. & Choi, H.-S. (2002) J. Am. Chem. Soc. 124, 2190-2201. [DOI] [PubMed] [Google Scholar]
- 24.Nicolaou, K. C., Zhong, Y.-L., Baran, P. S., Jung, J., Choi, H.-S. & Yoon, W. H. (2002) J. Am. Chem. Soc. 124, 2202-2211. [DOI] [PubMed] [Google Scholar]
- 25.Nicolaou, K. C., Mathison, C. J. N. & Montagnon, T. (2004) J. Am. Chem. Soc. 126, 5192-5201. [DOI] [PubMed] [Google Scholar]
- 26.Li, J., Burgett, A. W. G., Esser, L., Amerzcua, C. & Harran, P. G. (2001) Angew. Chem. Int. Ed. 40, 4770-4773. [DOI] [PubMed] [Google Scholar]
- 27.Nicolaou, K. C., Snyder, S. A., Simonsen, K. B. & Koumbis, A. E. (2000) Angew. Chem. Int. Ed. 39, 3473-3478. [DOI] [PubMed] [Google Scholar]
- 28.Nicolaou, K. C., Snyder, S. A., Bigot, A. & Pfefferkorn, J. A. (2000) Angew. Chem. Int. Ed. 39, 1093-1096. [PubMed] [Google Scholar]
- 29.Li, J., Jeong, S., Esser, L. & Harran, P. G. (2001) Angew. Chem. Int. Ed. 40, 4765-4770. [DOI] [PubMed] [Google Scholar]
- 30.Nicolaou, K. C., Bella, M., Chen, D. Y.-K., Huang, X., Ling, T. & Snyder, S. A. (2002) Angew. Chem. Int. Ed. 41, 3495-3499. [DOI] [PubMed] [Google Scholar]
- 31.Nicolaou, K. C., Bheema Rao, P., Hao, J., Reddy, M. V., Rassias, G., Huang, X., Chen, D. Y.-K. & Snyder, S. A. (2003) Angew. Chem. Int. Ed. 42, 1753-1758. [DOI] [PubMed] [Google Scholar]
- 32.Nicolaou, K. C. (1993) Angew. Chem. Int. Ed. 32, 1377-1385. [Google Scholar]
- 33.Groneberg, R. D., Miyazaki, T., Stylianides, N. A., Schulze, T. J., Stahl, W., Schreiner, E. P., Suzuki, T., Iwabuchi, Y., Smith, A. L. & Nicolaou, K. C. (1993) J. Am. Chem. Soc. 115, 7593-7611. [Google Scholar]
- 34.Smith, A. L., Pitsinos, E., Hwang, C.-K., Mizuno, Y., Saimoto, H., Scarlato, G. R., Suzuki, T. & Nicolaou, K. C. (1993) J. Am. Chem. Soc. 115, 7612-7624. [Google Scholar]
- 35.Nicolaou, K. C., Hummel, C. W., Nakada, M., Shibayama, K., Pitsinos, E. N., Saimoto, H., Mizuno, Y., Baldenius, K.-U. & Smith, A. L. (1993) J. Am. Chem. Soc. 115, 7625-7635. [Google Scholar]
- 36.Nicolaou, K. C. & Smith, A. L. (1992) Acc. Chem. Res. 25, 497-503. [Google Scholar]
- 37.Nicolaou, K. C., Dai, W.-M. & Guy, R. K. (1994) Angew. Chem. Int. Ed. 33, 15-44. [Google Scholar]
- 38.Nicolaou, K. C., Snyder, S. A., Montagnon, T. & Vassilikogiannakis, G. E. (2002) Angew. Chem. Int. Ed. 41, 1668-1698. [DOI] [PubMed] [Google Scholar]
- 39.Nicolaou, K. C., Nantermet, P. G., Ueno, H., Guy, R. K., Couladouros, E. A. & Sorensen, E. J. (1995) J. Am. Chem. Soc. 117, 624-633. [Google Scholar]
- 40.Nicolaou, K. C., Liu, J.-J., Yang, Z., Ueno, H. Sorensen, E. J., Claiborne, C. F., Guy, R. K., Hwang, C.-K., Nakada, M. & Nantermet, P. G. (1995) J. Am. Chem. Soc. 117, 634-644. [Google Scholar]
- 41.Nicolaou, K. C., Yang, Z., Liu, J.-J., Nantermet, P. G., Claiborne, C. F., Renaud, J., Guy, R. K. & Shibayama, K. (1995) J. Am. Chem. Soc. 117, 645-652. [Google Scholar]
- 42.Nicolaou, K. C., Ueno, H., Liu, J.-J., Nantermet, P. G., Yang, Z., Renaud, J., Paulvannan, K. & Chadha, R. (1995) J. Am. Chem. Soc. 117, 653-659. [Google Scholar]
- 43.Nicolaou, K. C., Guy, R. K., Pitsinos, E. N. & Wrasidlo, W. (1994) Angew. Chem. Int. Ed. 33, 1583-1587. [Google Scholar]
- 44.Nicolaou, K. C., Boddy, C. N. C., Bräse, S. & Winssinger, N. (1999) Angew. Chem. Int. Ed. 38, 2096-2152. [DOI] [PubMed] [Google Scholar]
- 45.Nicolaou, K. C., Boddy, C. N. C., Natarajan, S., Yue, T.-Y., Li, H., Bräse, S. & Ramanjulu, J. M. (1997) J. Am. Chem. Soc. 119, 3421-3422. [Google Scholar]
- 46.Nicolaou, K. C., Li, H., Boddy, C. N. C., Ramanjulu, J. M., Yue, T.-Y., Natarajan, S., Chu, X.-J., Bräse, S. & Rübsam, F. (1999) Chem. Eur. J. 5, 2584-2601. [Google Scholar]
- 47.Nicolaou, K. C., Boddy, C. N. C., Li, H., Koumbis, A. E., Hughes, R., Natarajan, S., Jain, N. F., Ramanjulu, J. M. & Bräse, S. (1999) Chem. Eur. J. 5, 2602-2621. [Google Scholar]
- 48.Nicolaou, K. C., Koumbis, A. E., Takayanagi, M., Natarajan, S., Jain, N. F., Bando, T., Li, H. & Hughes, R. (1999) Chem. Eur. J. 5, 2622-2647. [Google Scholar]
- 49.Nicolaou, K. C., Mitchell, H. J., Jain, N. F., Hughes, R., Winssinger, N., Natarajan, S. & Koumbis, A. E. (1999) Chem. Eur. J. 5, 2648-2667. [Google Scholar]
- 50.Trnka, T. M. & Grubbs, R. H. (2001) Acc. Chem. Res. 34, 18-29. [DOI] [PubMed] [Google Scholar]
- 51.Nicolaou, K. C., Hughes, R., Cho, S. Y., Winssinger, N., Labischinski, H. & Endermann, R. (2001) Chem. Eur. J. 7, 3824-3843. [DOI] [PubMed] [Google Scholar]
- 52.Nicolaou, K. C., Roschanger, F. & Vourloumis, D. (1998) Angew. Chem. Int. Ed. 37, 2014-2045. [DOI] [PubMed] [Google Scholar]
- 53.Nicolaou, K. C., Ritzén, A. & Namoto, K. (2001) Chem. Commun. 1523-1535. [DOI] [PubMed]
- 54.Nicolaou, K. C., Winssinger, N., Pastor, J., Ninkovic, S., Sarabia, F., Vourloumis, D., Yang, Z., Li, T., Giannakakou, P. & Hamel, E. (1997) Nature 387, 268-272. [DOI] [PubMed] [Google Scholar]
- 55.Boger, D. L., Desharnais, J. & Capps, K. (2003) Angew. Chem. Int. Ed. 42, 4138-4176. [DOI] [PubMed] [Google Scholar]
- 56.Nicolaou, K. C., Xiao, X.-Y., Parandoosh, Z., Senyei, A. & Nova, M. P. (1995) Angew. Chem. Int. Ed. 34, 2289-2291. [Google Scholar]
- 57.Nicolaou, K. C. (2003) Tetrahedron 59, 6683-6738. [Google Scholar]