

Abstract
Humans in industrialized areas are continuously exposed to phthalate plasticizers, prompting concerns of their potential toxicities. Previous studies from our laboratory and others have shown that various phthalates activate several mammalian nuclear receptors, in particular the constitutive androstane receptor (CAR), the pregnane X receptor (PXR), and the peroxisomal proliferator-activated receptors (PPARs), although often at concentration levels of questionable relevance to human exposure. We discovered that di(2-ethylhexyl) phthalate (DEHP) and di-isononyl phthalate (DiNP), two of the highest volume production agents, were potent activators of human CAR2 (hCAR2), a unique human CAR splice variant and, to a lesser degree, human PXR (hPXR). These diphthalates undergo rapid metabolism in mammalian systems, initially to their major monophthalate derivatives MEHP and MiNP. Although MEHP and MiNP are reported activators of the rodent PPARs, with lower affinities for the corresponding human PPARs, it remains unclear whether these monophthalate metabolites activate hCAR2 or hPXR. In this investigation, we assessed the relative activation potential of selected monophthalates and other low molecular weight phthalates against hCAR, the most prominent hCAR splice variants, as well as hPXR and human PPAR. Using transactivation and mammalian two-hybrid protein interaction assays, we demonstrate that these substances indeed activate hCARs and hPXR but to varying degrees. MEHP and MiNP exhibit potent activation of hCAR2 and hPXR with higher affinities for these receptors than for the hPPARs. The rank order potency for MEHP and MiNP was hCAR2 > hPXR > hPPARs. Results from primary hepatocyte experiments also reflect the MEHP and MiNP upregulation of the respective human target genes. We conclude that both di- and monophthalates are potently selective hCAR2 activators and effective hPXR activators. These results implicate these targets as important mediators of selective phthalate effects in humans. The striking differential affinities for these compounds between human and rodent nuclear receptors further implies that biological results obtained from rodent models may be of only limited relevance for interpolating phthalate-mediated effects in humans.
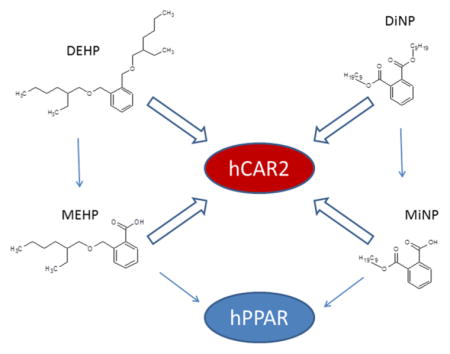
Graphical abstract
INTRODUCTION
Phthalates are ubiquitous environmental contaminants consisting of a diverse group of dialkyl or alkyl aryl derivatives of o-phthalic acid.1 Their physico- and chemico-properties are governed by the number of carbons in their alkyl side chains, and the higher molecular weight derivatives, such as dialkyl di(2-ethylhexyl) phthalate (DEHP) and di-isononyl phthalate (DiNP), are used in large volume production of PVC plastics, whereas smaller molecular weight derivatives are used in solvents, paints, cosmetics, and other household products. Biomonitoring studies have shown that virtually all human populations living in developed countries are exposed to these agents at some level.2–4
In male rodents, under varying treatment modalities, DEHP exposures are reported to decrease testosterone levels, reproductive organ weights, sperm counts, and sperm motility.5–7 Decreased estradiol levels and estrus cycle disruptions are two consistent findings in female rodents exposed to DEHP or its metabolite MEHP.8 Human data are limited, but phthalate levels in males have correlated with lowered testosterone levels, decreased sperm quality, and cryptorchidism.9–11 In females, phthalate exposure was associated with endometriosis,12,13 premature delivery,14,15 and pregnancy loss.16 More recently, phthalate exposure was suggested as a risk factor for Type II diabetes and obesity.17
The adverse effects of DEHP and other dialkyl phthalates are likely mediated by their metabolites, as in humans and most mammals the dialkyl phthalates are rapidly metabolized in the liver and other tissues to monophthalate derivatives, which can then be further oxidized.18,19 Human biomonitoring data show that following DEHP exposures, oxidized/conjugated MEHP derivatives are the primary metabolites in urine, whereas MEHP is the primary metabolite found in serum.20,21 These metabolites activate nuclear receptors, which result in the activation of target genes.
The nuclear receptors constitutive androstane receptor (CAR) and pregnane X receptor (PXR) coordinately regulate the expression of target genes involved in all phases of xenobiotic metabolism. In humans, the CAR gene undergoes alternative mRNA splicing to result in prominent variants, hCAR2 and hCAR3, with the reference or wild-type form termed hCAR1.22 hCAR2 and hCAR3 have 4 and 5 amino acid insertions, respectively; however, the modest alterations result in differences in the receptors’ ligand specificity and activation relative to those of hCAR1.23–25 It is noteworthy that these variants are not generated in rodents due to differences in the splice donor sequences flanking the respective exons within the rat and mouse genomes.25 Previously, our laboratory demonstrated that DEHP and DiNP are highly potent activators of the hCAR2 variant and, to a lesser extent, hPXR, whereas hCAR1 and hCAR3 exhibited distinct preferences for shorter chain diphthalates.25,26 The corresponding primary metabolites mono-(2-ethylhexyl)phthalate (MEHP) and monoisononyl phthalate (MiNP) are known activators of the rodent and human peroxisome proliferator activated receptors PPARα and PPARγ,27 which likely mediate some of the adverse effects of these chemicals. In addition, phthalate monoesters are reported to activate rodent and human PXR.28 Although described as activators, relatively high phthalate concentrations are typically necessary to generate the noted effects.
Interestingly, in vivo rodent studies indicate activation of rodent PPAR and CAR target genes following exposure to DEHP.29,30 Further, a recent study used yeast two-hybrid assays and molecular docking studies to demonstrate phthalate ester and monoester binding to wild-type human CAR1.31 However, the potential for activation of human CAR1 or its splice variants by monophthalates remains unclear. In this study, we assessed the activation of human CAR variants and PXR against various monophthalates and other lower molecular weight phthalate derivatives. Differences in the activation potential of these agents were clearly identified with hCAR2 and PXR exhibiting much higher affinities for MEHP and MiNP than those of hCAR1, hCAR3, or the human PPARs.
EXPERIMENTAL PROCEDURES
Chemicals and Reagents
General chemicals, di(2-ethylhexyl) phthalate (DEHP, CAS# 117-81-7), 5α-androstan-3α-ol (ANDRO, CAS# 7657-50-3), di-isononyl phthalate (DiNP, CAS# 68515-48-0), dimethyl sulfoxide (DMSO, CAS# 67-68-5), troglitazone (TG, acs-045, CAS# 97322-87-7), and WY14643 were purchased from Sigma-Aldrich. Benzyl butyl phthalate (BBP, CAS# 85-68-7), monobenzyl phthalate (MBzP, CAS# 2528-16-7), mono-(2-ethylhexyl)phthalate (MEHP, CAS# 4376-20-9), monoethyl phthalate (MEP, CAS# 2306-33-4), and monoiso-nonyl phthalate (MiNP, isometrically pure 9 carbon ester, CAS# EDF-014) were purchased from AccuStandard (New Haven, CT). Di(2-propyl heptyl) phthalate (DPHP, CAS# 53306-54-0) was purchased from Toronto Research Chemicals (Ontario, CA), and phthalic acid (PA, CAS# 88-99-3) was purchased from TCI America (Portland, OR). 6-(4-Chlorophenyl)imidazo[2,1-b][1,3]thiazole-5-carbaldehyde O-3,4-dichlorobenzyl) oxime (CITCO, CAS# 338404-52-7) was obtained from BIOMOL Research Laboratories (Plymouth Meeting, PA). TO901317 (TO, CAS# 293754-55-9) was obtained from Cayman Chemical Co. (Ann Arbor, MI). GW501516 (CAS# 50892-23-4) was purchased from Calbiochem. Primers were purchased from Integrated DNA Technologies (Coralville, IA).
Plasmid Constructs
The vectors, pTracer-CMV2-hCAR1 and -hCAR2, pcDNA3.1(+)-RXRα, pcDNA3.1(+)-RXRα ligand binding domain (LBD), 2B6-XREM-PBREM, 3A4-XREM, and PPRE luciferase reporters were described previously.22–24,32 The pTracer-CMV2-hCAR3, mammalian two-hybrid pmCAR-LBD, and VP16-SRC-1 vectors were also previously reported.23 For pTracer-CMV2-hPXR, hPXR was amplified from liver cDNA using the primers shown in Table 1. The pm-hPXR vector was a gift from Dr. Vanden Heuvel of Pennsylvania State University.33 Vectors containing PPAR isoforms were previously created in our laboratory. hPPARα was amplified from liver cDNA and cloned into the pcDNA3.1(−) vector. For the current studies, PPARα was amplified from the pcDNA3.1(−) clone using primers shown in Table 1 and subcloned into pTracer-CMV2. PPARβ/∂ and hPPARγ, amplified from liver cDNA or MGC clone 3447380 (GE Healthcare), respectively, were cloned into the pcDNA3.1(+) vector. For the current studies, PPARβ/∂ and -γ inserts were subcloned into pTracer-CMV2 using the KpnI and XbaI restriction sites. For mammalian two-hybrid assays, the PPARα and PPARγ-LBDs were subcloned from the pTracer-CMV2-PPAR constructs using the primers shown in Table 1 and inserted into the pm-GAL4-DNA binding domain vector (Clontech Matchmaker Mammalian Assay Kit 2, Mountain View, CA).
Table 1.
Primer Sequences Used for Creating Transactivation and Mammalian Two-Hybrid Assay Vectors
| construct | primer | sequence |
|---|---|---|
| CMV2-PXR | forward (EcoRI) | 5′-GATCGAATTCGACATGGAGGTGAGACCCAAAGAAAG-3′ |
| reverse (EcoRV) | 5′-GATCGATATCTCAGCTACCTGTGATGCCGAACAAC-3′ | |
| PPARα | forward (KpnI) | 5′-GATAGGTACCGCCACCATGGTGGACACGGAAAGC-3′ |
| reverse (NotI) | 5′-CATGCGGCCGCTCAGTACATGTCC-3′ | |
| Pm or VP16-PPARα_LBD | forward (BamHI) | 5′-GACGGATCCGAAGATTCTGAAACTGC-3′ |
| reverse (XbaI) | 5′-CTGCTCTAGATCAGTACATGTCCCTGTAG-3′ | |
| Pm or VP16-PPARγ_LBD | forward (BamHI) | 5′-TGAGGATCCGTCAGTACTGTCGGTTTCAG-3′ |
| reverse (XbaI) | 5′-CTGCAAGCTTCTAGTACAAGTCCTTGTAG-3′ |
Transactivation and Mammalian Two-Hybrid Assays
Culture conditions for maintenance of COS-1 cells (ATCC, Manassas, VA) were previously reported.25 COS-1 cells were used because they are devoid of endogenous CAR expression/activity as demonstrated in previous reports.23,24 For transfection and chemical treatments, the same medium was used except dextran/charcoal-treated FBS (HyClone, Logan, UT) replaced normal FBS. All transfections and chemical treatments for luciferase reporter and mammalian two-hybrid assays were performed in a 48-well format in triplicate or quadruplicate and repeated on separate occasions at least twice to verify concordance of the data between repeat experiments. Transfections for trans-activation and mammalian two-hybrid assays were performed as previously described.25 All test compounds were prepared in DMSO and levels never exceeded 0.2% (v/v). CITCO was used as a positive control for CAR activation.34 Because CAR1 is constitutively active, ANDRO (10 μM), a human CAR124 inverse-agonist, was included to decrease its activity, which can be restored in the presence of an agonist. WY14643 was used as a selective PPARα agonist,35 whereas GW501516 was used as a selective PPARβ/∂ agonist.36 TG was used as a selective PPARγ agonist.37 All chemical treatments were for 24 h, and luciferase assays were performed as previously reported.24 For dose–response studies, the data were analyzed using GraphPad Prism v6.04 software nonlinear regression analysis to determine EC50 and maximum activation values.
Culture and Treatment of Human Primary Hepatocytes
Primary human hepatocytes were obtained from anonymous donors through the Liver Tissue Cell Distribution System, University of Pittsburgh, Pittsburgh, PA, funded by NIH Contract #HHSN276201200017C, and approved for use by the respective institutional review boards. Donor information is shown in Table 2. The isolated hepatocytes were seeded at ~500,000 cells/well in 12 well collagen-coated plates in a sandwich format with matrigel overlay and cultured as previously described.38 Culture medium was replaced every 24 h. After 3 days, the medium was removed and replaced with treatment medium containing DMSO or 3 μM CITCO, 50 μM WY14643, 30 μM TG or varying concentrations of MEHP and MiNP. After 24 h, cells were washed in 1× phosphate buffered saline (pH 7.4), followed by the addition of 600 μL of Trizol reagent (Invitrogen). The Trizol solution was pipetted up and down to dislodge and lyse the cells and then transferred to 1.5 mL tubes and immediately stored at −80 °C. RNA was prepared according to the manufacturer’s directions, and concentrations and purity were determined using a Nanodrop 2000 (Thermo Scientific, Wilmington, DE). RNA integrity was assessed using a BioRad Experion (Hercules, CA), and RNA quality indicator values exceeded 7.5. RNA (2 μg) was used for synthesis of cDNA using the High Capacity Reverse Transcription cDNA kit (Applied Biosystems, Carlsbad, CA), and the remainder was stored at −80 °C. The cDNA reaction was performed on a BioRad C1000 Thermocycler using the following conditions: 25 °C for 10 m, 37 °C for 2 h, 85 °C for 5 m, and 4 °C forever. Once the reactions had cooled to 4 °C, cDNA was diluted 5-fold in nuclease-free water to a final concentration of 20 ng/μL and stored at −20 °C until use.
Table 2.
Human Hepatocyte Donor Information
| ID | age | sex | ethnicity | source | disease/treatment |
|---|---|---|---|---|---|
| HH1 | 71 | male | unknown | resection | neuroendocrine tumor metastasis, prior chemoembolization |
| HH2 | 62 | female | unknown | resection | hepatocellular carcinoma, no chemotherapy |
| HH3 | 59 | male | caucasian | resection | stage IV colon cancer, chemotherapy 6 months prior |
Real-Time PCR
Master mixes were prepared for each target. The volumes of components per duplicate reaction were 15 μL of SYBR Green mix, 0.6 μL of forward primer (100 nM final concentration), 0.6 μL of reverse primer (100 nM final concentration), 10.8 μL of nuclease-free water, and 3 μL of cDNA (20 ng/μL final concentration). SYBR green primer sets and amplification efficiencies are shown in Table 3. The duplicate aliquots from each master mix were transferred to a 96-well assay plate. The reactions were run on a BioRad CFX96 Real Time System equipped with a C1000 Thermocycler and CFX Manager Software v.2. The reaction conditions were as follows: 45 °C for 5 min, 95 °C for 3 min, 95 °C for 15 s, and 60 °C for 1 min (40 cycles total). Melt curves were run from 65 to 95 °C with an increment of 0.5 °C after each run. Standard curves using serial dilutions of human primary hepatocyte cDNA were run for all targets to determine reaction efficiencies.
Table 3.
SYBR Green Primers Used in Human Hepatocyte Gene Expression Analysis
| qPCR primer | sequence (5′-3′) | efficiency | |
|---|---|---|---|
| ACOX11 | FP | GAGCAGCAGGAGCGCTTCTT | 102 |
| RP | AGTTCCATGACCCATCTCTGTC | ||
| ANGPTL41 | FP | GTCCACCGACCTCCCGTTA | 89 |
| RP | CCTCATGGTCTAGGTGCTTGT | ||
| CPT21 | FP | CATACAAGCTACATTTCGGGACC | 106 |
| RP | AGCCCGGAGTGTCTTCAGAA | ||
| PDK41 | FP | GGAAGCATTGATCCTAACTGTGA | 87 |
| RP | GGTGAGAAGGAACATACACGATG | ||
| CYP2B62 | FP | GGTGTGCCCCACATTGTCA | 65 |
| RP | GGAGAGCAGTGCTCAGGATGA | ||
| CYP3A42 | FP | GGCCCACACCTCTGCCTT | 91 |
| RP | AAGCCCCACACTTTTCCATACTT | ||
| GAPDH2 | FP | CCCATCACCATCTTCCAGGAG | 96 |
| RP | GTTGTCATGGATGACCTTGGC |
Statistical Analyses
All statistical tests were performed using GraphPad Prism Software v. 6.04 (La Jolla, CA). For determining differences in activation of CAR by various treatments, two-way ANOVAs were performed, followed by a Dunnett’s multiple comparisons test for comparison to controls.
RESULTS
Activation of Human CAR Variants and Human PXR by Phthalates
On the basis of our previous identification of DEHP and DiNP as potent hCAR2 activators, we conducted transactivation assay screens for their respective monophthalate metabolites MEHP and MiNP, additional high molecular weight diphthalates, as well as several smaller molecular weight phthalate derivatives against hCAR1, hCAR2, hCAR3, and CAR-related hPXR (Figures 1 and 2). The positive control pan-CAR agonist CITCO restored androstanol repressed hCAR1 activity and activated hCAR2 and hCAR3 as expected. The PXR agonist TO903107 potently stimulated PXR activity, in agreement with our previous results.26 DEHP and DiNP activated hCAR2 and hPXR but exhibited only minimal activity against hCAR1 or hCAR3. MEHP and MiNP showed significant activation of hCAR2, moderate activation of hPXR, and slight activation of hCAR1 and hCAR3 at the highest doses. Interestingly, the higher molecular weight DPHP was also a strong activator of hCAR2 with little or no activation of hCAR1, hCAR3, or hPXR. The diphthalate BBP was a strong activator of hCAR3 with some activation of hCAR1 and hPXR but exhibited no activity with hCAR2. The lower molecular weight PA and MEP induced no or minimal activation of these receptors.
Figure 1.

Structures of phthalates used in this study.
Figure 2.

Activation of the 2B6-XREM-PBREM reporter by CAR1, CAR2, CAR3, or PXR after treatment with various phthalates. Results represent a single transfection experiment with all treatments in triplicate. COS-1 cells were transfected with the pcDNA3.1(+)-RXRα expression vector, 2B6-XREM-PBREM reporter, pRL-CMV vector for normalization of transfection efficiency, and one of CMV2-CAR1, CMV2-CAR2, CMV2-CAR3, or CMV2-PXR. All treatments were for 24 h, and all concentrations were micromolar (μM). The luciferase values were normalized for transfection efficiency and are expressed as fold-induction over DMSO control. Each data point represents the mean (± S.D.). #p < 0.05 compared with androstanol control; *p < 0.05 compared with DMSO control.
Activation of Human PPAR by Various Phthalates
Because the PPARs are classic phthalate targets, we also screened hPPARα, hPPARβ/∂, and hPPARγ with the various phthalates in transactivation assays (Figure 3). As expected, the positive control compounds for each receptor induced receptor activation of the PPRE reporter. Of the phthalates tested, only MBzP, MEHP, and MiNP stimulated receptor activation of the reporter, and these effects were only observed at the highest test dose (100 μM) of these compounds with the exception of MEHP, which activated hPPARγ at the 10 μM dose. It is also noteworthy that neither DEHP nor DiNP activated the hPPARs at doses that significantly activate hCAR and hPXR.
Figure 3.

Activation of the PPRE reporter by PPARs after treatment with various phthalates. Results represent a single transfection experiment with all treatments in quadruplicate. COS-1 cells were transfected with the pcDNA3.1(+)-RXRα expression vector, PPRE reporter, pRL-CMV vector for normalization of transfection efficiency, and one of CMV2-PPARα, CMV2-PPARβ/∂, or CMV2-PPARγ. All treatments were for 24 h, and concentrations were micromolar (μM). The data are represented as normalized luciferase values. The luciferase values were normalized for transfection efficiency and are expressed as fold-induction over DMSO control, and each data point represents the mean (± S.D.). *p < 0.05 compared with DMSO control.
Affinity Comparisons for MEHP, MiNP, and MBzP with hCAR2, hPXR, and hPPAR
On the basis of the finding that certain monophthalates activate hCAR and hPXR, we conducted more complete dose–response analyses using transactivation assays to determine the relative potencies of MEHP, MiNP, and MBzP for hCAR, hPXR, and the hPPARs. We first compared hCAR2 and hPPARα (Figure 4A) because both receptors are primarily expressed in the liver. The results showed that both MEHP and MiNP exhibit relatively high affinity for activation of hCAR2 at 2.2 and 2.8 μM, respectively, with very little activation of hPPARα. MBzP activated hCAR2 and PPARα with similar potency; however, EC50 values were not determined, as the dose response curves were incomplete, and higher doses were not tested, because it is unlikely that in vivo concentrations of human MBzP exposure would ever reach such high levels. In the next series of studies, we compared hCAR2 with hPXR, hPPARβ/∂, and hPPARγ (Figure 4B). The rank order potency for MEHP and MiNP was hCAR2 > hPXR > hPPARγ ≥ PPARβ/∂. There was only minor activation of all receptors by MBzP at the doses tested.
Figure 4.

Dose–response curves for MEHP, MiNP, and MBzP with CAR2 with PPARα (A) and with PXR, PPARβ/∂, and PPARγ (B). Results represent a single transfection experiment with all treatments in quadruplicate. COS-1 cells were transfected with the pcDNA3.1(+)-RXRα expression vector, PPRE reporter, pRL-CMV vector for normalization of transfection efficiency, and one of CMV2-CAR2, CMV2-PXR, CMV2-PPARα, CMV2-PPARβ/∂, or CMV2-PPARγ. All treatments were for 24 h, and all concentrations were micromolar (μM). The luciferase values were normalized for transfection efficiency and then corrected for background (DMSO) activity. All values represent the mean (± S.D.).
Mammalian Two Hybrid Analysis
We next used mammalian two hybrid assays to confirm the binding of monophthalates to hCAR2 and hPXR (Figure 5). Both hCAR and hPXR interacted with SRC1 in a dose-dependent manner in the presence of MEHP and MiNP, indicating direct binding of these substrates within the active site of these receptors. As in previous transactivation studies, MEHP and MiNP exhibited similar potency in activation of hCAR2, whereas no activation of hCAR2 or hPXR was observed at the doses of MBzP tested. We also evaluated these substrates with PPARα and PPARγ in mammalian two-hybrid assays but observed negligible activation at the doses tested (Figure 5).
Figure 5.

Mammalian two-hybrid analysis of MEHP, MiNP, and MBzP activation of CAR, PXR, PPARα, or PPARγ. COS-1 cells were transfected with the ligand binding domains of CAR, PXR, PPARα, or PPARγ in the pm (GAL4) vector, SRC1 in the VP16 vector, pcDNA3.1(+)-RXRα-LBD, pFR-luciferase reporter, and pRL-CMV vector for normalization. Chemical treatments were for 24 h. Postive controls were 3 μM CITCO for CAR2, 0.1 μM TO for PXR, 50 μM WY14643 for PPARα, and 30 μM TG for PPARγ. Data are represented as normalized luciferase values, and each data point represents the mean (±SD) of triplicate treatment wells from a representative transfection experiment.
Relative Affinities of Monophthalates and Diphthalates
We previously demonstrated that DEHP and DiNP are potent activators of hCAR2 and hPXR. Here, we investigated the relative potencies of MEHP vs DEHP and MiNP vs DiNP for hCAR2, hPXR, hPPARα, and hPPARγ. Even in these dose–response studies that used higher doses of DEHP and DiNP than the initial screening assays (shown in Figure 3), neither of the diphthalates activated the hPPARs (data not shown). Figure 6A shows that DEHP has higher affinities for hCAR2 and hPXR than those of MEHP. Maximum activation of hCAR2 by DEHP and MEHP is similar, whereas activation of hPXR by MEHP is approximately 75% that of DEHP maximum activation. Figure 6B shows that activation of hCAR2 by MiNP and DiNP is similar, whereas MiNP is a more potent activator of hPXR than DiNP. The derived EC50 values and maximum fold induction values are presented in Table 4.
Figure 6.

Dose–response curves for MEHP vs DEHP (A) or MiNP vs DiNP (B) with CAR2 and PXR. Results represent a single transfection experiment with all treatments in quadruplicate. COS-1 cells were transfected with the pcDNA3.1-RXRα expression vector, CYP2B6-PBREM-XREM (CAR2) or 3A4-XREM (PXR) reporter, the pRL-CMV vector for normalization of transfection efficiency, and either CMV2-CAR2 or CMV2-PXR. All treatments were for 24 h. The luciferase values were normalized for transfection efficiency and then corrected for background (DMSO) activity. All values represent the mean (± S.D.).
Table 4.
EC50 Values (μM) and Maximum Activation of hCAR2 and hPXR by DEHP, DiNP, and Their Metabolites MEHP and MiNP
| hCAR2 | hPXR | |||||||
|---|---|---|---|---|---|---|---|---|
|
|
|
|||||||
| EC50 | 95% CI | max fold activation | R2 | EC50 | 95% CI | max fold activation | R2 | |
| MEHP | 2.3 | 0.8–6.5 | 6.8 | 0.91 | 27.0 | 8.5–85.7 | 10.4 | 0.83 |
| DEHP | 0.1 | 0.05–0.18 | 7.1 | 0.94 | 3.5 | 1.2–10.1 | 13.7 | 0.89 |
| MiNP | 2.3 | 1.22–4.46 | 4.6 | 0.95 | 14.0 | 4.8–40.6 | 18.3 | 0.84 |
| DiNP | 0.7 | 0.35–1.22 | 4.2 | 0.95 | 29.5 | 5.0–172.8 | 7.8 | 0.68 |
Human Hepatocyte mRNA Expression after Phthalate Treatment
Primary human hepatocyte expression of hCAR and hPPAR target genes was assessed after exposure to varying concentrations of MEHP and MiNP (Figure 7). Only changes of greater than 2-fold are discussed here. CITCO, a pan-CAR agonist, induced expression of CAR target genes CYP2B6 in all three hepatocyte samples as well as CYP3A4 in HH1 and HH3. Samples HH1 and HH2 were treated with MEHP and MiNP at 0.1, 1, 3, and 10 μM. HH1 showed induction of CYP3A4 but not CYP2B6 at 10 μM MEHP. For HH3, we increased the doses of MEHP and MiNP to 1, 3, 10, and 30 μM. In this case, 30 μM MEHP increased expression of CYP2B6 but not CYP3A4. Although TG and WY14643 are considered PPAR agonists, they induced CYP2B6 and CYP3A4 expression in some hepatocyte samples. Interestingly, CITCO suppressed expression of PDK4 in all three samples.
Figure 7.

Effect of MEHP and MiNP on CAR/PXR and PPAR target gene expression in primary human hepatocytes. Cultured primary hepatocytes from human donors were treated with control compounds (DMSO, CITCO, WY14643, and TG) and various doses of MEHP and MiNP for 24 h. Total RNA was harvested and used for production of cDNA. RT-PCR was performed on cDNA samples isolated from the cells to quantify CYP2B6, CYP3A4, acyl CoA oxidase 1 (ACOX1), angiopoietin like 4 (ANGPTL4), carnitine palmitoyltransferase 2 (CPT2), pyruvate dehydrogenase kinase 4 (PDK4), and glyceraldehyde 3-phosphate dehydrogensase (GAPDH; as an internal reference). Values were expressed as fold induction relative to DMSO-treated cells and represent the average of two replicate samples.
WY14643 induced the PPAR target gene ACOX1 in HH3, but not in HH1 or HH2, and induced ANGPTL4 and CPT2 in HH1 and HH3 and PDK4 in all three donor samples. TG was a weak inducer of PPAR target genes in these studies, inducing only ANG and CPT in HH3 and PDK4 in HH1. Neither MEHP nor MiNP induced expression of any PPAR target genes in any of the hepatocyte samples. These results suggest that MEHP may activate hCAR and/or hPXR with little or no activity against the hPPARs in human hepatocytes.
DISCUSSION
Understanding the relative contributions of nuclear receptor activation in mediating the effects of phthalate exposure is critical for predicting human toxicity. CAR and PXR are key xenosensing members of the nuclear receptor family.39,40 The current studies identify a potential new role for CAR and PXR in mediating the effects of phthalates in humans.
Classically, monophthalates are thought of as PPAR ligands. However, species differences in activation of these receptors are well-known. Indeed, in vitro transactivation studies demonstrate differential monophthalate activation of mouse and human PPARα, β/∂, and γ.27,41,42 These previous studies show that monophthalates have higher affinity for and fold-activation of mouse PPARα compared with those of human PPARα.27,41 This is consistent with the observation that humans are resistant to the peroxisome-proliferative/hepatocarcinogenic effects of DEHP otherwise seen in rodent models.43 In addition, monophthalates have similar affinities for and activation of mouse and human PPARγ42 and activate mouse PPARβ/∂ but have minimal activity with human PPARβ/∂ at the doses studied.41 The findings presented in the current investigation, along with others, highlight important species differences in the hepatic response to phthalates.
Our previous studies demonstrated that the diphthalates DEHP and DiNP are highly potent activators of the specific hCAR2 splice variant, and also hPXR activators,26 with less activity with wild type hCAR1 and the variant hCAR3. However, as diphthalates are rapidly metabolized in vivo to their monophthalate derivatives, these enzymatic products have not been well characterized with respect to their human CAR activation potential. In the current investigation, we demonstrate that the principal monophthalate metabolites of DEHP and DiNP, MEHP and MiNP, respectively, are also potent activators of these receptor systems. MEHP and MiNP exhibit higher affinities for hCAR2 (EC50 = 2.3 and 2.3 μM, respectively) and hPXR (EC50 = 27 and 14 μM, respectively) than for the corresponding hPPARs (EC50 values could not be accurately determined due to low activation at the doses tested). A potential limitation of our study is that WY14643 only produced a 1.5-fold activation of hPPARα in the transactivation studies (Figure 3), suggesting this assay was not very sensitive. A possible explanation for the low fold activation is that we observed higher background activation of PPARα (DMSO-treated cells) in these assays. Nevertheless, our findings still clearly show that MEHP and MiNP are more potent activators of hCARs and hPXR than the hPPARs.
In addition to low activation of PPARs, we observed very little activation of hCAR1 and CAR3 by monophthalates. Recently, a yeast two-hybrid system and molecular docking studies were used to characterize the interaction of various phthalates with wild-type hCAR1.31 Although it is difficult to make direct comparisons due to differences in assay systems, their results agree with our previous and current studies that indicate little or no activation of hCAR1 by MEHP, DEHP, or DiNP. Another important consideration is that although both monophthalates and diphthalates activate hCAR2 and hPXR, only the monophthalates activate hPPARs.27 In these respects, although monophthalates are typically perceived as PPAR ligands, our current findings, along with others, highlight important species differences at the receptor level in the hepatic response to phthalates. Together, these results imply that hCAR2 and hPXR are likely the dominant mediators of phthalate effects in humans.
Results from primary human hepatocyte experiments presented here indicate that MEHP activates the hCAR and hPXR target genes CYP2B6 and CYP3A4 to a limited extent with no activation of PPAR target genes. We suspect that the limited induction of CAR/PXR target genes was due to further metabolism of MEHP and MiNP in the hepatocytes, such that the doses applied did not reach concentrations required for more robust responses. Further, low expression levels of hCAR2 in the donor hepatocytes might also contribute to the low response. Consistent with other studies, the PPAR ligands WY14643 and TG induced CYP2B6 and CYP3A4.44–46 In addition to being a PPARγ and α ligand, TG also activates hCAR variants and hPXR in transactivation assays.47 Further, the upstream region of the human CYP3A4 gene contains PPARα binding regions that are required for WY14643-mediated CYP3A4 induction.55 The upregulation of CAR target genes by PPAR selective agonists (and vice versa) in the human hepatocyte studies emphasizes the characteristic ligand overlap and regulatory cross-talk between nuclear receptors.
Physiologically, the PPARs are well characterized as metabolic sensors, critical for regulation of energy homeostasis. PPARα and PPARβ/∂ coordinate fatty acid oxidation in liver and muscle tissue, and PPARγ controls lipid storage in adipose tissue.48,49 Through a variety of complex mechanisms, PPARs also affect glucose metabolism and improve insulin sensitivity, and for these reasons, PPARγ agonists are useful in the treatment of diabetes.49 More recently, a role for CAR and PXR in the regulation of lipid and glucose metabolism has also emerged. In mouse models, hepatic lipid homeostasis is affected by activation of CAR, which through regulatory cross-talk mechanisms ultimately results in decreased lipid catabolism and decreased lipogenesis; in contrast, activation of PXR appears to promote lipogenesis50–52 CAR and PXR also decrease gluconeogenic enzyme expression through mechanisms that block transcription factor binding to promoter regions of these genes.52,53 Further, transgenic mouse models suggest a role for CAR and PXR in diabetes and obesity.54–56
Increasing concern has centered on a potential role for phthalates in the obesity and Type II diabetes (T2D) epidemic.57 For example, higher phthalate exposure, assessed through urinary monophthalate excretion, was associated with increased abdominal circumference and insulin resistance in adult males in the US.17 Further, US women with higher urinary monophthalate concentrations, including DEHP metabolites, were more likely to have diabetes.58 In these respects, the potential role of CAR, PXR, and the PPARs as mediators of the metabolic effects of phthalates is intriguing. Mechanistic studies have implicated PPARγ in MEHP-induced adipogenesis. Treatment of the preadipocyte cell line 3T3L1 with MEHP increased cellular lipid and triglyceride content, 41,42 whereas a PPARγ inhibitor or PPARγ siRNA significantly reduced MEHP-induced adipogenesis.42 Further, MEHP induced expression of PPARγ target genes involved in adipocyte differentiation in 3T3L1 cells.42 In vivo rodent studies demonstrated a role for PPARα in DEHP-mediated effects on hepatic energy metabolism. Wild-type mice treated with DEHP fed a control diet or high fat diet exhibited a lean phenotype, whereas PPARα knockout mice were not protected.59 Interestingly, these effects were not observed in PPARα-humanized mice, lending further evidence for the existence of receptor-mediated differences between rodents and humans.
It is clear that a complex system of regulatory cross-talk exists between CAR, PXR, and PPARs, and due to their underlying functional redundancy, it is challenging to quantify their respective roles as relative determinants of phthalate-related or other environmental chemical-induced toxicities in humans. However, the current findings suggest that, in humans, hCAR2 and PXR may be more important than the PPARs in mediating the hepatic effects of phthalate exposure. Further studies are required to elucidate the relative contributions of these receptor systems in potential adverse physiological effects of phthalates in humans. It is also noteworthy that due to differences in their primary gene structures, rodents are not capable of producing a splice variant equivalent to hCAR2.25 These and other known species differences in receptor activation suggest that results obtained from phthalate studies in rodents, including humanized mouse models, should be interpreted carefully.
Acknowledgments
Funding
This research was supported by a grant from the USPHS/NIH, General Medical Sciences Institute, GM066441 (CJO), and also by the National Institute of Food and Agriculture, U.S. Department of Agriculture, under award number 2014-06624.
ABBREVIATIONS
- ANDRO
5α-androstan-3α-ol
- BBP
benzyl butyl phthalate
- CAR
constitutive androstane receptor
- hCAR
human CAR
- hCAR2 or hCAR3
human CAR splice variants
- CITCO
6-(4-chlorophenyl)imidazo[2,1-b][1,3]thiazole-5-carbaldehyde O-3,4-dichlorobenzyl)oxime
- DEHP
di(2-ethylhexyl)phthalate
- DiNP
di-isononyl phthalate
- DPHP
di(2-propyl heptyl) phthalate
- MBzP
monobenzyl phthalate
- MEHP
mono-(2-ethylhexyl)phthalate
- MEP
monoethyl phthalate
- MiNP
monoiso-nonyl phthalate
- PA
phthalic acid
- PXR
pregnane X receptor
- PPAR
peroxisome proliferator-activated receptor
- hPPAR
human PPAR
- mPPAR
mouse PPAR
- TG
troglitazone
- TO
TO901317
Footnotes
The authors declare no competing financial interest.
References
- 1.Erickson BE. Pressure on Plastics. Chem Eng News. 2015;93(10–15):10–15. [Google Scholar]
- 2.Wittassek M, Koch HM, Angerer J, Bruning T. Assessing exposure to phthalates - the human biomonitoring approach. Mol Nutr Food Res. 2011;55:7–31. doi: 10.1002/mnfr.201000121. [DOI] [PubMed] [Google Scholar]
- 3.Saravanabhavan G, Murray J. Human biological monitoring of diisononyl phthalate and diisodecyl phthalate: a review. J Environ Public Health. 2012;2012:810501. doi: 10.1155/2012/810501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Qian H, Chen M, Kransler KM, Zaleski RT. Assessment of chemical coexposure patterns based upon phthalate biomonitoring data within the 2007/2008 National Health and Nutrition Examination Survey. J Exposure Sci Environ Epidemiol. 2015;25:249–255. doi: 10.1038/jes.2014.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Park JD, Habeebu SS, Klaassen CD. Testicular toxicity of di-(2-ethylhexyl)phthalate in young Sprague-Dawley rats. Toxicology. 2002;171:105–115. doi: 10.1016/s0300-483x(01)00567-4. [DOI] [PubMed] [Google Scholar]
- 6.Shirota M, Saito Y, Imai K, Horiuchi S, Yoshimura S, Sato M, Nagao T, Ono H, Katoh M. Influence of di-(2-ethylhexyl)phthalate on fetal testicular development by oral administration to pregnant rats. J Toxicol Sci. 2005;30:175–194. doi: 10.2131/jts.30.175. [DOI] [PubMed] [Google Scholar]
- 7.Culty M, Thuillier R, Li W, Wang Y, Martinez-Arguelles DB, Benjamin CG, Triantafilou KM, Zirkin BR, Papadopoulos V. In utero exposure to di-(2-ethylhexyl) phthalate exerts both short-term and long-lasting suppressive effects on testosterone production in the rat. Biol Reprod. 2008;78:1018–1028. doi: 10.1095/biolreprod.107.065649. [DOI] [PubMed] [Google Scholar]
- 8.Laskey JW, Berman E. Steroidogenic assessment using ovary culture in cycling rats: effects of bis(2-diethylhexyl)-phthalate on ovarian steroid production. Reprod Toxicol. 1993;7:25–33. doi: 10.1016/0890-6238(93)90006-s. [DOI] [PubMed] [Google Scholar]
- 9.Swan SH, Main KM, Liu F, Stewart SL, Kruse RL, Calafat AM, Mao CS, Redmon JB, Ternand CL, Sullivan S, Teague JL. Decrease in anogenital distance among male infants with prenatal phthalate exposure. Environ Health Perspect. 2005;113:1056–1061. doi: 10.1289/ehp.8100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pan G, Hanaoka T, Yoshimura M, Zhang S, Wang P, Tsukino H, Inoue K, Nakazawa H, Tsugane S, Takahashi K. Decreased serum free testosterone in workers exposed to high levels of di-n-butyl phthalate (DBP) and di-2-ethylhexyl phthalate (DEHP): a cross-sectional study in China. Environ Health Perspect. 2006;114:1643–1648. doi: 10.1289/ehp.9016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pak VM, McCauley LA, Pinto-Martin J. Phthalate exposures and human health concerns: A review and implications for practice. AAOHN Journal. 2011;59:228–233. doi: 10.3928/08910162-20110426-01. [DOI] [PubMed] [Google Scholar]
- 12.Cobellis L, Latini G, De FC, Razzi S, Paris I, Ruggieri F, Mazzeo P, Petraglia F. High plasma concentrations of di-(2-ethylhexyl)-phthalate in women with endometriosis. Hum Reprod. 2003;18:1512–1515. doi: 10.1093/humrep/deg254. [DOI] [PubMed] [Google Scholar]
- 13.Reddy BS, Rozati R, Reddy BV, Raman NV. Association of phthalate esters with endometriosis in Indian women. BJOG. 2006;113:515–520. doi: 10.1111/j.1471-0528.2006.00925.x. [DOI] [PubMed] [Google Scholar]
- 14.Latini G, De Felice C, Presta G, Del Vecchio A, Paris I, Ruggieri F, Mazzeo P. In utero exposure to di-(2-ethylhexyl)phthalate and duration of human pregnancy. Environ Health Perspect. 2003;111:1783–1785. doi: 10.1289/ehp.6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Whyatt RM, Adibi JJ, Calafat AM, Camann DE, Rauh V, Bhat HK, Perera FP, Andrews H, Just AC, Hoepner L, Tang D, Hauser R. Prenatal di(2-ethylhexyl)phthalate exposure and length of gestation among an inner-city cohort. Pediatrics. 2009;124:e1213–e1220. doi: 10.1542/peds.2009-0325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mu D, Gao F, Fan Z, Shen H, Peng H, Hu J. Correction to Levels of Phthalate Metabolites in Urine of Pregnant Women and Risk of Clinical Pregnancy Loss. Environ Sci Technol. 2015;49:13081. doi: 10.1021/acs.est.5b04255. [DOI] [PubMed] [Google Scholar]
- 17.Stahlhut RW, van Wijngaarden E, Dye TD, Cook S, Swan SH. Concentrations of urinary phthalate metabolites are associated with increased waist circumference and insulin resistance in adult U.S. males. Environ Health Perspect. 2007;115:876–882. doi: 10.1289/ehp.9882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kluwe WM. Overview of phthalate ester pharmacokinetics in mammalian species. Environ Health Perspect. 1982;45:3–9. doi: 10.1289/ehp.82453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koch HM, Preuss R, Angerer J. Di(2-ethylhexyl)phthalate (DEHP): human metabolism and internal exposure– an update and latest results. Int J Androl. 2006;29:155–165. doi: 10.1111/j.1365-2605.2005.00607.x. [DOI] [PubMed] [Google Scholar]
- 20.Koch HM, Bolt HM, Angerer J. Di(2-ethylhexyl)phthalate (DEHP) metabolites in human urine and serum after a single oral dose of deuterium-labelled DEHP. Arch Toxicol. 2004;78:123–130. doi: 10.1007/s00204-003-0522-3. [DOI] [PubMed] [Google Scholar]
- 21.Hogberg J, Hanberg A, Berglund M, Skerfving S, Remberger M, Calafat AM, Filipsson AF, Jansson B, Johansson N, Appelgren M, Hakansson H. Phthalate diesters and their metabolites in human breast milk, blood or serum, and urine as biomarkers of exposure in vulnerable populations. Environ Health Perspect. 2008;116:334–339. doi: 10.1289/ehp.10788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Auerbach SS, Ramsden R, Stoner MA, Verlinde C, Hassett C, Omiecinski CJ. Alternatively spliced isoforms of the human constitutive androstane receptor. Nucleic Acids Res. 2003;31:3194–3207. doi: 10.1093/nar/gkg419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Auerbach SS, Stoner MA, Su S, Omiecinski CJ. Retinoid X receptor-alpha-dependent transactivation by a naturally occurring structural variant of human constitutive androstane receptor (NR1I3) Mol Pharmacol. 2005;68:1239–1253. doi: 10.1124/mol.105.013417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Auerbach SS, Dekeyser JG, Stoner MA, Omiecinski CJ. CAR2 displays unique ligand binding and RXRalpha heterodimerization characteristics. Drug Metab Dispos. 2007;35:428–439. doi: 10.1124/dmd.106.012641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dekeyser JG, Stagliano MC, Auerbach SS, Prabhu KS, Jones AD, Omiecinski CJ. Di(2-ethylhexyl) phthalate is a highly potent agonist for the human constitutive androstane receptor splice variant CAR2. Mol Pharmacol. 2009;75:1005–1013. doi: 10.1124/mol.108.053702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dekeyser JG, Laurenzana EM, Peterson EC, Chen T, Omiecinski CJ. Selective phthalate activation of naturally occurring human constitutive androstane receptor splice variants and the pregnane X receptor. Toxicol Sci. 2011;120:381–391. doi: 10.1093/toxsci/kfq394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hurst CH, Waxman DJ. Activation of PPARalpha and PPARgamma by environmental phthalate monoesters. Toxicol Sci. 2003;74:297–308. doi: 10.1093/toxsci/kfg145. [DOI] [PubMed] [Google Scholar]
- 28.Hurst CH, Waxman DJ. Environmental phthalate monoesters activate pregnane X receptor-mediated transcription. Toxicol Appl Pharmacol. 2004;199:266–274. doi: 10.1016/j.taap.2003.11.028. [DOI] [PubMed] [Google Scholar]
- 29.Eveillard A, Lasserre F, de Tayrac M, Polizzi A, Claus S, Canlet C, Mselli-Lakhal L, Gotardi G, Paris A, Guillou H, Martin PG, Pineau T. Identification of potential mechanisms of toxicity after di-(2-ethylhexyl)-phthalate (DEHP) adult exposure in the liver using a systems biology approach. Toxicol Appl Pharmacol. 2009;236:282–292. doi: 10.1016/j.taap.2009.02.008. [DOI] [PubMed] [Google Scholar]
- 30.Ren H, Aleksunes LM, Wood C, Vallanat B, George MH, Klaassen CD, Corton JC. Characterization of peroxisome proliferator-activated receptor alpha–independent effects of PPARalpha activators in the rodent liver: di-(2-ethylhexyl) phthalate also activates the constitutive-activated receptor. Toxicol Sci. 2010;113:45–59. doi: 10.1093/toxsci/kfp251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang H, Zhang Z, Nakanishi T, Wan Y, Hiromori Y, Nagase H, Hu J. Structure-dependent activity of phthalate esters and phthalate monoesters binding to human constitutive androstane receptor. Chem Res Toxicol. 2015;28:1196–1204. doi: 10.1021/acs.chemrestox.5b00028. [DOI] [PubMed] [Google Scholar]
- 32.Stoner MA, Auerbach SS, Zamule SM, Strom SC, Omiecinski CJ. Transactivation of a DR-1 PPRE by a human constitutive androstane receptor variant expressed from internal protein translation start sites. Nucleic Acids Res. 2007;35:2177–2190. doi: 10.1093/nar/gkm090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vanden Heuvel JP, Thompson JT, Frame SR, Gillies PJ. Differential activation of nuclear receptors by perfluorinated fatty acid analogs and natural fatty acids: a comparison of human, mouse, and rat peroxisome proliferator-activated receptoralpha, -beta, and -gamma, liver X receptor-beta, and retinoid X receptor-alpha. Toxicol Sci. 2006;92:476–489. doi: 10.1093/toxsci/kfl014. [DOI] [PubMed] [Google Scholar]
- 34.Maglich JM, Parks DJ, Moore LB, Collins JL, Goodwin B, Billin AN, Stoltz CA, Kliewer SA, Lambert MH, Willson TM, Moore JT. Identification of a novel human constitutive androstane receptor (CAR) agonist and its use in the identification of CAR target genes. J Biol Chem. 2003;278:17277–17283. doi: 10.1074/jbc.M300138200. [DOI] [PubMed] [Google Scholar]
- 35.Kliewer SA, Forman BM, Blumberg B, Ong ES, Borgmeyer U, Mangelsdorf DJ, Umesono K, Evans RM. Differential expression and activation of a family of murine peroxisome proliferator-activated receptors. Proc Natl Acad Sci U S A. 1994;91:7355–7359. doi: 10.1073/pnas.91.15.7355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oliver WR, Jr, Shenk JL, Snaith MR, Russell CS, Plunket KD, Bodkin NL, Lewis MC, Winegar DA, Sznaidman ML, Lambert MH, Xu HE, Sternbach DD, Kliewer SA, Hansen BC, Willson TM. A selective peroxisome proliferator-activated receptor delta agonist promotes reverse cholesterol transport. Proc Natl Acad Sci U S A. 2001;98:5306–5311. doi: 10.1073/pnas.091021198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Berger J, Bailey P, Biswas C, Cullinan CA, Doebber TW, Hayes NS, Saperstein R, Smith RG, Leibowitz MD. Thiazolidinediones produce a conformational change in peroxisomal proliferator-activated receptor-gamma: binding and activation correlate with antidiabetic actions in db/db mice. Endocrinology. 1996;137:4189–4195. doi: 10.1210/endo.137.10.8828476. [DOI] [PubMed] [Google Scholar]
- 38.Goyak KM, Johnson MC, Strom SC, Omiecinski CJ. Expression profiling of interindividual variability following xenobiotic exposures in primary human hepatocyte cultures. Toxicol Appl Pharmacol. 2008;231:216–224. doi: 10.1016/j.taap.2008.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baes M, Gulick T, Choi HS, Martinoli MG, Simha D, Moore DD. A new orphan member of the nuclear hormone receptor superfamily that interacts with a subset of retinoic acid response elements. Mol Cell Biol. 1994;14:1544–1552. doi: 10.1128/mcb.14.3.1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kliewer SA, Moore JT, Wade L, Staudinger JL, Watson MA, Jones SA, McKee DD, Oliver BB, Willson TM, Zetterstrom RH, Perlmann T, Lehmann JM. An orphan nuclear receptor activated by pregnanes defines a novel steroid signaling pathway. Cell. 1998;92:73–82. doi: 10.1016/s0092-8674(00)80900-9. [DOI] [PubMed] [Google Scholar]
- 41.Bility MT, Thompson JT, McKee RH, David RM, Butala JH, Vanden Heuvel JP, Peters JM. Activation of mouse and human peroxisome proliferator-activated receptors (PPARs) by phthalate monoesters. Toxicol Sci. 2004;82:170–182. doi: 10.1093/toxsci/kfh253. [DOI] [PubMed] [Google Scholar]
- 42.Feige JN, Gelman L, Rossi D, Zoete V, Metivier R, Tudor C, Anghel SI, Grosdidier A, Lathion C, Engelborghs Y, Michielin O, Wahli W, Desvergne B. The endocrine disruptor monoethyl-hexyl-phthalate is a selective peroxisome proliferator-activated receptor gamma modulator that promotes adipogenesis. J Biol Chem. 2007;282:19152–19166. doi: 10.1074/jbc.M702724200. [DOI] [PubMed] [Google Scholar]
- 43.Huber WW, Grasl-Kraupp B, Schulte-Hermann R. Hepatocarcinogenic potential of di(2-ethylhexyl)phthalate in rodents and its implications on human risk. Crit Rev Toxicol. 1996;26:365–481. doi: 10.3109/10408449609048302. [DOI] [PubMed] [Google Scholar]
- 44.Rakhshandehroo M, Hooiveld G, Muller M, Kersten S. Comparative analysis of gene regulation by the transcription factor PPARalpha between mouse and human. PLoS One. 2009;4:e6796. doi: 10.1371/journal.pone.0006796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rogue A, Lambert C, Josse R, Antherieu S, Spire C, Claude N, Guillouzo A. Comparative gene expression profiles induced by PPARgamma and PPARalpha/gamma agonists in human hepatocytes. PLoS One. 2011;6:e18816. doi: 10.1371/journal.pone.0018816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thomas M, Burk O, Klumpp B, Kandel BA, Damm G, Weiss TS, Klein K, Schwab M, Zanger UM. Direct transcriptional regulation of human hepatic cytochrome P450 3A4 (CYP3A4) by peroxisome proliferator-activated receptor alpha (PPARalpha) Mol Pharmacol. 2013;83:709–718. doi: 10.1124/mol.112.082503. [DOI] [PubMed] [Google Scholar]
- 47.Faucette SR, Zhang TC, Moore R, Sueyoshi T, Omiecinski CJ, LeCluyse EL, Negishi M, Wang H. Relative activation of human pregnane X receptor versus constitutive androstane receptor defines distinct classes of CYP2B6 and CYP3A4 inducers. J Pharmacol Exp Ther. 2007;320:72–80. doi: 10.1124/jpet.106.112136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Desvergne B, Wahli W. Peroxisome proliferator-activated receptors: nuclear control of metabolism. Endocr Rev. 1999;20:649–688. doi: 10.1210/edrv.20.5.0380. [DOI] [PubMed] [Google Scholar]
- 49.Monsalve FA, Pyarasani RD, Delgado-Lopez F, Moore-Carrasco R. Peroxisome proliferator-activated receptor targets for the treatment of metabolic diseases. Mediators Inflammation. 2013;2013:549627. doi: 10.1155/2013/549627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wada T, Gao J, Xie W. PXR and CAR in energy metabolism. Trends Endocrinol Metab. 2009;20:273–279. doi: 10.1016/j.tem.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 51.Gao J, Xie W. Pregnane X receptor and constitutive androstane receptor at the crossroads of drug metabolism and energy metabolism. Drug Metab Dispos. 2010;38:2091–2095. doi: 10.1124/dmd.110.035568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moreau A, Vilarem MJ, Maurel P, Pascussi JM. Xenoreceptors CAR and PXR activation and consequences on lipid metabolism, glucose homeostasis, and inflammatory response. Mol Pharmaceutics. 2008;5:35–41. doi: 10.1021/mp700103m. [DOI] [PubMed] [Google Scholar]
- 53.Gao J, Xie W. Targeting xenobiotic receptors PXR and CAR for metabolic diseases. Trends Pharmacol Sci. 2012;33:552–558. doi: 10.1016/j.tips.2012.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gao J, He J, Zhai Y, Wada T, Xie W. The constitutive androstane receptor is an anti-obesity nuclear receptor that improves insulin sensitivity. J Biol Chem. 2009;284:25984–25992. doi: 10.1074/jbc.M109.016808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dong B, Saha PK, Huang W, Chen W, Abu-Elheiga LA, Wakil SJ, Stevens RD, Ilkayeva O, Newgard CB, Chan L, Moore DD. Activation of nuclear receptor CAR ameliorates diabetes and fatty liver disease. Proc Natl Acad Sci U S A. 2009;106:18831–18836. doi: 10.1073/pnas.0909731106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.He J, Gao J, Xu M, Ren S, Stefanovic-Racic M, O’Doherty RM, Xie W. PXR ablation alleviates diet-induced and genetic obesity and insulin resistance in mice. Diabetes. 2013;62:1876–1887. doi: 10.2337/db12-1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Thayer KA, Heindel JJ, Bucher JR, Gallo MA. Role of environmental chemicals in diabetes and obesity: a National Toxicology Program workshop review. Environ Health Perspect. 2012;120:779–789. doi: 10.1289/ehp.1104597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.James-Todd T, Stahlhut R, Meeker JD, Powell SG, Hauser R, Huang T, Rich-Edwards J. Urinary phthalate metabolite concentrations and diabetes among women in the National Health and Nutrition Examination Survey (NHANES) 2001–2008. Environ Health Perspect. 2012;120:1307–1313. doi: 10.1289/ehp.1104717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Feige JN, Gerber A, Casals-Casas C, Yang Q, Winkler C, Bedu E, Bueno M, Gelman L, Auwerx J, Gonzalez FJ, Desvergne B. The pollutant diethylhexyl phthalate regulates hepatic energy metabolism via species-specific PPARalpha-dependent mechanisms. Environ Health Perspect. 2010;118:234–241. doi: 10.1289/ehp.0901217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Spandidos A, Wang X, Wang H, Seed B. PrimerBank: a resource of human and mouse PCR primer pairs for gene expression detection and quantification. Nucleic Acids Res. 2010;38:D792–D799. doi: 10.1093/nar/gkp1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Omiecinski CJ, Coslo DM, Chen T, Laurenzana EM, Peffer RC. Multi-species Analyses of Direct Activators of the Constitutive Androstane Receptor. Toxicol Sci. 2011;123(2):550. doi: 10.1093/toxsci/kfr191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chen T, Laurenzana EM, Coslo DM, Chen F, Omiecinski CJ. Proteasomal interaction as a critical activity modulator of the human constitutive androstane receptor. Biochem J. 2014;458(1):95–107. doi: 10.1042/BJ20130685. [DOI] [PMC free article] [PubMed] [Google Scholar]