

Abstract
A concise and efficient total synthesis of the spermidine alkaloid (-)-isooncinotine (1) incorporating a 22-membered lactam ring is outlined. The approach is largely catalysis-based, involving a selective iron-catalyzed alkyl–aryl cross-coupling reaction of a difunctionalized pyridine substrate, a heterogeneous asymmetric hydrogenation step to set the chiral center of the target, and a highly integrated ring-closing metathesis/hydrogenation sequence to forge the saturated macrocyclic edifice in a single operation.
Despite the prevalence of the biogenetic bases spermidine and spermine in nature and their widespread use as tools in biochemical research (1), little is known about the physiological properties of the small but structurally rather unique class of alkaloids incorporating these polyamines into their macrocyclic skeletons (2–4). This may be due, in part, to the difficulties in obtaining pure samples of these compounds from the natural sources† as well as to the fact that most syntheses described thus far provided only racemic material. Prototype spermidine alkaloids are (-)-isooncinotine (1), (-)-oncinotine (2), (-)-neooncinotine (3), and oncinodin-12-one (4), which were isolated from the stem bark of Oncinotis nitida and Oncinotis tenuiloba (Apocynaceae) (5–7) and have been repeatedly targeted in the past (8–12) (Fig. 1). Only recently has one member of this series (i.e., compound 2) been made available in an optically active form through total synthesis by using a diastereoselective 1,3-dipolar cycloaddition for the formation of the stereogenic center at the junction of the piperidine and the macrocyclic ring (13).
Fig. 1.

Structures of macrocyclic spermidine alkaloids.
In pursuit of previous work in the field of polyamine alkaloids (14, 15), we were prompted to develop a complementary, practical, and potentially scaleable entry into this family of natural products, choosing the 22-membered macrolactam (-)-isooncinotine 1 as our initial target. The envisaged route, however, should not only provide material for further evaluation but also meet a set of stringent chemical criteria. Specifically, it is felt that contemporary organic chemistry must (i) prioritize catalysis-based bond formations, (ii) try to control absolute stereochemistry by asymmetric catalysis, and (iii) aim at high convergence combined with an overall “economy of steps” (16). Furthermore, the total synthesis of 1 should provide us with the possibility to scrutinize methodology developed in our laboratories. Outlined below is the successful reduction of this plan to practice.
Materials and Methods
General. All reactions were carried out under Ar in flame-dried glassware. The solvents used were purified by distillation over the drying agents indicated and were transferred under Ar: tetrahydrofuran (THF) (Na), CH2Cl2 (P4O10), MeCN, Et3N, pyridine, NMP, hexamethylphosphoramide (HMPA), tetramethylethylenediamine (CaH2), dimethylformamide (DMF) (dibutyltin dilaurate/Desmodur), MeOH, EtOH (Mg), and toluene (Na/K). For flash chromatography, Merck silica gel 60 (230–400 mesh) was used. For NMR, spectra were recorded on a DPX 300 or AV 400 spectrometer (Bruker) in the solvents indicated; chemical shifts (δ) are given in parts per million relative to tetramethylsilane, and coupling constants (J) are given in hertz. For IR, a Nicolet FT-7199 spectrometer or Perkin–Elmer Fourier transform-IR Diamant Spectrum One (ATR) was used; wavenumbers (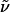 ) are given in cm-1. For MS [electron ionization (EI)], a Finnigan MAT 8200 (70 eV) was used, and for high-resolution MS (HRMS), a Finnigan MAT 95 was used. Elemental analyses were performed by H. Kolbe (Mülheim, Germany). All commercially available compounds were used as received.
) are given in cm-1. For MS [electron ionization (EI)], a Finnigan MAT 8200 (70 eV) was used, and for high-resolution MS (HRMS), a Finnigan MAT 95 was used. Elemental analyses were performed by H. Kolbe (Mülheim, Germany). All commercially available compounds were used as received.
Hex-5-enoic Acid (3-Aminopropyl)amide (16). Methyl 5-hexenoate (1.66 g, 13 mmol) was dissolved in 1,3-diaminopropane 15 (100 ml), and the mixture was stirred at 60°C for 3 days. The excess of the 1,3-diaminopropane was removed in vacuo to yield the title compound as a pale-yellow syrup (2.10 g, 95%). 1H-NMR (400 MHz, CDCl3): δ 6.47 (s, 1H), 5.79 (ddt, J = 6.7, 10.3, 17.1 Hz, 1H), 5.00 (m, 2H), 3.36 (m, 2H), 2.79 (t, J = 6.4 Hz, 2H), 2.17 (m, 2H), 2.09 (m, 2H), 1.74 (m, 2H), 1.63 (m, 2H), 1.38 (s, 2H); 13C-NMR (100 MHz, CDCl3): δ 172.8, 137.9, 115.1, 40.1, 37.8, 36.0, 33.1, 32.3, 24.8; IR (film): 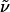 3,292, 3,075, 2,931, 2,865, 1,642, 1,554, 1,438, 1,361, 1,309, 1,259, 993, 912 cm-1; MS (EI): m/z (relative intensity) 153 (2), 152 (2), 141 (14), 140 (9), 116 (17), 100 (7), 99 (18), 98 (55), 97 (10), 87 (43), 73 (30), 72 (15), 69 (14), 57 (40), 56 (32), 44 (100), 41 (48), 39 (14), 30 (93). HRMS (EI) calcd. for C9H18N2O: 170.141912; found: 170.141877.
3,292, 3,075, 2,931, 2,865, 1,642, 1,554, 1,438, 1,361, 1,309, 1,259, 993, 912 cm-1; MS (EI): m/z (relative intensity) 153 (2), 152 (2), 141 (14), 140 (9), 116 (17), 100 (7), 99 (18), 98 (55), 97 (10), 87 (43), 73 (30), 72 (15), 69 (14), 57 (40), 56 (32), 44 (100), 41 (48), 39 (14), 30 (93). HRMS (EI) calcd. for C9H18N2O: 170.141912; found: 170.141877.
Hex-5-enoic Acid [3-(2-Nitrobenzenesulfonylamino)propyl]amide (17). A solution of compound 16 (1.58 g, 9.28 mmol) and 2-nitrobenzenesulfonyl chloride (2.06 g, 9.28 mmol) in pyridine (3 ml, 37.12 mmol) and CH2Cl2 was stirred for 1 h at ambient temperature. The solution was diluted with CH2Cl2 before it was washed with HCl (2 M, 10 ml) and saturated aqueous NaHCO3 (2 × 20 ml) and dried over Na2SO4. After evaporation of the solvent, the residue was purified by flash chromatography (CH2Cl2/MeOH, 30:1) to give compound 17 as a yellow syrup (2.41 g, 73%). 1H-NMR (400 MHz, CDCl3): δ 8.11 (m, 1H), 7.83 (m, 1H), 7.73 (m, 2H), 6.15 (t, J = 6.4 Hz, 1H), 6.05 (m, 1H), 5.76 (ddt, J = 6.7, 10.3, 17.0 Hz, 1H), 4.99 (m, 2H), 3.34 (dd, J = 6.3, 12.5 Hz, 2H), 3.14 (dd, J = 6.4, 12.7 Hz, 2H), 2.18 (m, 2H), 2.06 (m, 2H), 1.71 (m, 4H); 13C-NMR (100 MHz, CDCl3): δ 173.8, 148.0, 137.8, 133.9, 133.5, 132.7, 130.6, 125.1, 115.2, 40.7, 36.0, 35.8, 33.1, 30.0, 24.7; IR (film): 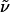 3,394, 3,300, 3,094, 2,935, 2,870, 1,641, 1,593, 1,542, 1,440, 1,417, 1,365, 1,340, 1,165, 1,025, 995, 915, 783 cm-1; MS (EI): m/z (relative intensity) 355 (4 [M+]), 260 (9), 186 (29), 169 (30), 140 (10), 115 (12), 114 (15), 107 (7), 87 (16), 69 (22), 56 (100), 44 (26), 41 (30), 30 (20). HRMS (EI) calcd. for C15H21N3O5S: 356.128019 (M + H); found: 356.128338 (M + H). Anal. calcd. for C15H21N3O5S: C, 50.69, H, 5.96, N, 11.82; found: C, 50.57, H, 5.98, N, 11.75.
3,394, 3,300, 3,094, 2,935, 2,870, 1,641, 1,593, 1,542, 1,440, 1,417, 1,365, 1,340, 1,165, 1,025, 995, 915, 783 cm-1; MS (EI): m/z (relative intensity) 355 (4 [M+]), 260 (9), 186 (29), 169 (30), 140 (10), 115 (12), 114 (15), 107 (7), 87 (16), 69 (22), 56 (100), 44 (26), 41 (30), 30 (20). HRMS (EI) calcd. for C15H21N3O5S: 356.128019 (M + H); found: 356.128338 (M + H). Anal. calcd. for C15H21N3O5S: C, 50.69, H, 5.96, N, 11.82; found: C, 50.57, H, 5.98, N, 11.75.
Hex-5-enoic Acid {3-[(4-Bromobutyl)-(2-nitrobenzenesulfonyl)amino]-propyl}amide (18). A solution of compound 17 (2.5 g, 7.0 mmol) in DMF (10 ml) was added to a suspension of K2CO3 (9.34 g, 67.6 mmol) and 1,4-dibromobutane (5.04 ml, 42.2 mmol) in DMF (50 ml). After stirring for 45 min at 60°C, the DMF was distilled off, and the residue was dissolved in CH2Cl2 (30 ml) and washed with brine (15 ml). The organic layer was dried over Na2SO4, the solvent was evaporated, and the residue was purified by flash chromatography (EtOAc) to provide 18 as a yellow syrup (3.04 g, 88%). 1H-NMR (400 MHz, CD2Cl2): δ 7.92 (m, 1H), 7.72 (m, 2H), 7.64 (m, 1H), 6.21 (m, 1H), 5.79 (ddt, J = 6.7, 10.3, 17.1 Hz, 1H), 4.98 (m, 2H), 3.35 (m, 6H), 3.23 (dd, J = 6.3, 12.7 Hz, 2H), 2.14 (m, 2H), 2.05 (m, 2H), 1.73 (m, 8H); 13C-NMR (100 MHz, CD2Cl2): δ 173.0, 148.3, 138.4, 134.1, 133.1, 132.3, 130.4, 124.5, 115.1, 47.3, 45.7, 36.4, 36.0, 33.5, 33.4, 29.8, 28.6, 27.0, 25.1; IR (film): 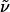 3,427, 3,300, 3,075, 2,934, 2,874, 1,643, 1,544, 1,459, 1,439, 1,373, 1,345, 1,300, 1,256, 1,161, 993, 915, 852, 777, 744, 584 cm-1; MS (EI): m/z (relative intensity) 356 (3), 305 (43), 303 (44), 251 (7), 249 (7), 192 (98), 190 (100), 186 (32), 170 (26), 154 (28), 137 (12), 135 (13), 100 (10), 84 (10), 56 (13), 55 (24), 44 (17), 30 (16). HRMS (EI) calcd. for C19H28BrN3O5S: 490.101144 (M + H); found: 490.101587 (M + H). Anal. calcd. for C19H28BrN3O5S: C, 46.53, H, 5.75, N, 8.57; found: C, 46.62, H, 5.68, N, 8.51.
3,427, 3,300, 3,075, 2,934, 2,874, 1,643, 1,544, 1,459, 1,439, 1,373, 1,345, 1,300, 1,256, 1,161, 993, 915, 852, 777, 744, 584 cm-1; MS (EI): m/z (relative intensity) 356 (3), 305 (43), 303 (44), 251 (7), 249 (7), 192 (98), 190 (100), 186 (32), 170 (26), 154 (28), 137 (12), 135 (13), 100 (10), 84 (10), 56 (13), 55 (24), 44 (17), 30 (16). HRMS (EI) calcd. for C19H28BrN3O5S: 490.101144 (M + H); found: 490.101587 (M + H). Anal. calcd. for C19H28BrN3O5S: C, 46.53, H, 5.75, N, 8.57; found: C, 46.62, H, 5.68, N, 8.51.
2-(6-Benzyloxyhexyl)-6-chloropyridine (7). (6-Bromohexyloxymethyl)-benzene (3.0 g, 11.1 mmol) was added to a suspension of magnesium turnings (323 mg, 13.3 mmol) activated with a crystal of iodine in THF (40 ml), and the mixture was refluxed for 2 h before it was allowed to reach ambient temperature. Unreacted magnesium was filtered off under Ar. An aliquot of the resulting solution of 6-benzyloxyhexylmagnesium bromide 6 (6.5 mmol) was added dropwise over a period of 30 min to a solution of 2,6-dichloropyridine 5 (876 mg, 5.92 mmol), Fe(acac)3 (105 mg, 0.3 mmol, 5 mol %), and NMP (5.1 ml, 53.28 mmol) in THF (30 ml) at 0°C. After complete addition, the reaction was stirred for 15 min at ambient temperature before it was quenched with saturated aqueous NH4Cl (10 ml). A standard extractive work-up (CH2Cl2) followed by flash chromatography (hexanes/EtOAc, 30:1) of the crude product afforded compound 7 as a colorless oil (1.482 g, 83%). 1H-NMR (300 MHz, CDCl3): δ 7.53 (dd, J = 7.6, 7.8 Hz, 1H), 7.25–7.35 (m, 5H), 7.13 (d, J = 7.8 Hz, 1H), 7.04 (d, J = 7.6 Hz, 1H), 4.49 (s, 2H), 3.46 (t, J = 6.5 Hz, 2H), 2.75 (m, 2H), 1.67 (m, 4H), 1.39 (m, 4H); 13C-NMR (75 MHz, CDCl3): δ 163.6, 150.7, 138.8, 138.7, 128.3, 127.6, 127.4, 121.3, 121.0, 72.8, 70.3, 37.9, 29.6, 29.6, 29.1, 26.0; IR (film): 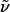 3,086, 3,063, 3,030, 2,932, 2,856, 2,792, 1,585, 1,559, 1,495, 1,453, 1,438, 1,408, 1,362, 1,167, 1,137, 1,099, 1,028, 987, 790, 736, 697 cm-1; MS (EI): m/z (relative intensity) 268 (10), 216 (2), 214 (18), 212 (55), 199 (18), 197 (54), 196 (8), 142 (27), 140 (82), 129 (21), 127 (65), 92 (15), 91 (100), 65 (13). HRMS (EI) calcd. for C18H22ClNO: 304.146817 (M + H); found: 304.146581 (M + H).
3,086, 3,063, 3,030, 2,932, 2,856, 2,792, 1,585, 1,559, 1,495, 1,453, 1,438, 1,408, 1,362, 1,167, 1,137, 1,099, 1,028, 987, 790, 736, 697 cm-1; MS (EI): m/z (relative intensity) 268 (10), 216 (2), 214 (18), 212 (55), 199 (18), 197 (54), 196 (8), 142 (27), 140 (82), 129 (21), 127 (65), 92 (15), 91 (100), 65 (13). HRMS (EI) calcd. for C18H22ClNO: 304.146817 (M + H); found: 304.146581 (M + H).
(R)-3-[6-(6-Benzyloxyhexyl)-pyridin-2-yl]-4-isopropyl-oxazolidin-2-one (9). A sealed tube was charged with (R)-4-isopropyloxazolidinone 8 (0.65 g, 5.0 mmol), K2CO3 (0.99 g, 9.6 mmol), CuI (137 mg, 0.72 mmol), toluene (5.5 ml), N,N′-dimethylethylenediamine (152 μl, 1.43 mmol), and 2-chloropyridine 7 (1.45 g, 4.8 mmol), and the resulting mixture was stirred at 140°C for 72 h. After reaching ambient temperature, the mixture was filtered, and the residue was washed with EtOAc (100 ml). The combined filtrates were evaporated, and the residue was purified by flash chromatography (hexane/EtOAc, 5:1) to give product 9 as a colorless solid (1.71 g, 90%). 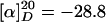 (c 1.3, CHCl3). 1H-NMR (400 MHz, CDCl3): δ 7.94 (d, J = 8.0 Hz, 1H), 7.59 (dd, J = 8.3, 7.5 Hz, 1H), 7.36–7.25 (m, 5H), 6.85 (d, J = 7.2 Hz, 1H), 4.91 (dt, J = 3.7, 8.8 Hz, 1H), 4.50 (s, 2H), 4.36 (t, J = 8.9 Hz, 1H), 4.27 (dd, J = 3.8, 8.9 Hz, 1H), 3.46 (t, J = 6.5 Hz, 2H), 2.74–2.65 (m, 2H), 2.53–2.45 (m, 1H), 1.75–1.58 (m, 4H), 1.45–1.32 (m, 4H), 0.93 (d, J = 7.1 Hz, 3H), 0.84 (d, J = 7.0 Hz, 3H); 13C-NMR (100 MHz, CDCl3): δ 160.3, 155.5, 149.8, 138.6, 138.1, 128.3, 127.6, 127.5, 118.0, 111.2, 72.8, 70.4, 62.9, 58.9, 37.6, 29.7, 29.0, 29.0, 27.8, 26.1, 17.9, 14.4; IR (film):
(c 1.3, CHCl3). 1H-NMR (400 MHz, CDCl3): δ 7.94 (d, J = 8.0 Hz, 1H), 7.59 (dd, J = 8.3, 7.5 Hz, 1H), 7.36–7.25 (m, 5H), 6.85 (d, J = 7.2 Hz, 1H), 4.91 (dt, J = 3.7, 8.8 Hz, 1H), 4.50 (s, 2H), 4.36 (t, J = 8.9 Hz, 1H), 4.27 (dd, J = 3.8, 8.9 Hz, 1H), 3.46 (t, J = 6.5 Hz, 2H), 2.74–2.65 (m, 2H), 2.53–2.45 (m, 1H), 1.75–1.58 (m, 4H), 1.45–1.32 (m, 4H), 0.93 (d, J = 7.1 Hz, 3H), 0.84 (d, J = 7.0 Hz, 3H); 13C-NMR (100 MHz, CDCl3): δ 160.3, 155.5, 149.8, 138.6, 138.1, 128.3, 127.6, 127.5, 118.0, 111.2, 72.8, 70.4, 62.9, 58.9, 37.6, 29.7, 29.0, 29.0, 27.8, 26.1, 17.9, 14.4; IR (film): 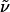 3,087, 3,063, 3,029, 2,933, 2,856, 1,759, 1,590, 1,579, 1,454, 1,397, 1,372, 1,318, 1,209, 1,116, 1,060, 978, 796, 738, 698 cm-1; MS (EI): m/z (relative intensity) 396 (M+, 19), 353 (5), 305 (25), 290 (63), 283 (10), 261 (7), 247 (30), 233 (79), 220 (37), 193 (6), 178 (12), 165 (12), 152 (8), 147 (5), 132 (7), 121 (10), 108 (18), 91 (100). HRMS (EI) calcd. for C24H32N2O3: 396.241292; found: 396.241003.
3,087, 3,063, 3,029, 2,933, 2,856, 1,759, 1,590, 1,579, 1,454, 1,397, 1,372, 1,318, 1,209, 1,116, 1,060, 978, 796, 738, 698 cm-1; MS (EI): m/z (relative intensity) 396 (M+, 19), 353 (5), 305 (25), 290 (63), 283 (10), 261 (7), 247 (30), 233 (79), 220 (37), 193 (6), 178 (12), 165 (12), 152 (8), 147 (5), 132 (7), 121 (10), 108 (18), 91 (100). HRMS (EI) calcd. for C24H32N2O3: 396.241292; found: 396.241003.
(R)-6-Piperidin-2-yl-hexan-1-ol Hydrochloride (10·HCl). A mixture of wet Pd(OH)2/C [20% (wt/wt), 400 mg], pyridine 9 (1.61 g, 4.03 mmol), MeOH (15 ml), and acetic acid (15 ml) was stirred in an autoclave under a hydrogen atmosphere [120 bars (1 bar = 100 kPa)] at 35°C for 24 h. The mixture was filtered through a short pad of Celite, which was subsequently washed with MeOH (30 ml). Hydrochloric acid (12 M, 666 μl, 8.0 mmol) was added, and the solvent was evaporated until no acetic acid was left. Oxazolidinone 8 (475 mg, 91%) was recovered by repeated washing with tert-butyl methyl ether/hexanes to yield a colorless residue (880 mg). This solid was dissolved in MeOH (3.1 ml) and HCl (12 M, 3.1 ml), and the resulting solution was stirred for 13 h. Evaporation to dryness gave 10·HCl (697 mg, 78%) as a white solid. The enantiomeric excess (ee) of 10 was determined to be 94% by GC analysis of the corresponding N-trifluoroacetamide derivative. 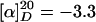 (c 1.02, CH3OH). 1H-NMR (400 MHz, CD3OD): δ 3.47 (t, J = 6.5 Hz, 2H), 3.29–3.22 (m, 1H), 3.02–2.87 (m, 2H), 1.95–1.91 (m, 1H), 1.83–1.77 (m, 2H), 1.66–1.32 (m, 13H); 13C-NMR (100 MHz, CD3OD): δ 63.1, 58.5, 46.3, 35.1, 33.7, 30.5, 30.0, 26.9, 26.3, 23.8, 23.4; IR (film):
(c 1.02, CH3OH). 1H-NMR (400 MHz, CD3OD): δ 3.47 (t, J = 6.5 Hz, 2H), 3.29–3.22 (m, 1H), 3.02–2.87 (m, 2H), 1.95–1.91 (m, 1H), 1.83–1.77 (m, 2H), 1.66–1.32 (m, 13H); 13C-NMR (100 MHz, CD3OD): δ 63.1, 58.5, 46.3, 35.1, 33.7, 30.5, 30.0, 26.9, 26.3, 23.8, 23.4; IR (film): 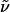 3,443, 3,187, 2,939, 2,851, 2,816, 2,569, 2,523, 2,408, 1,589, 1,461, 1,311, 1,074, 1,056, 921, 731 cm-1; MS (EI): m/z (relative intensity) 184 (1), 84 (100), 56 (6). HRMS (EI) calcd. for C11H24ClNO: 186.185789 ([M - Cl]+); found: 186.185904 ([M - Cl]+). GC of the corresponding N-trifluoroacetamide derivative: IVADEX 1, 25 m, 140°C iso, 0.6 bar H2, tR = 21.3 min (S), tR = 22.1 min (R): 94% ee (R).
3,443, 3,187, 2,939, 2,851, 2,816, 2,569, 2,523, 2,408, 1,589, 1,461, 1,311, 1,074, 1,056, 921, 731 cm-1; MS (EI): m/z (relative intensity) 184 (1), 84 (100), 56 (6). HRMS (EI) calcd. for C11H24ClNO: 186.185789 ([M - Cl]+); found: 186.185904 ([M - Cl]+). GC of the corresponding N-trifluoroacetamide derivative: IVADEX 1, 25 m, 140°C iso, 0.6 bar H2, tR = 21.3 min (S), tR = 22.1 min (R): 94% ee (R).
(R)-Hex-5-enoic Acid {3-[{4-[2-(6-Hydroxyhexyl)piperidinyl]butyl}-(2-nitrobenzene-sulfonyl)amino]propyl}amide (11). A solution of 10·HCl (300 mg, 1.35 mmol), bromide 18 (663 mg, 1.35 mmol), sodium iodide (202 mg, 6.75 mmol), and potassium carbonate (373 mg, 2.70 mmol) in EtOH (20 ml) was refluxed for 3 days. Additional bromide 18 (132 mg, 0.27 mmol) was added, and reflux was continued for 24 h. The solvent was evaporated, the residue was dissolved in CH2Cl2, and the solution was filtered through Celite and dried over Na2SO4. After removal of the solvent, the residue was purified by flash chromatography (CH2Cl2/MeOH/NH3, 30:1:0.5) to provide compound 11 as a yellow oil (584 mg, 73%). 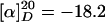 (c 1.11, CH2Cl2). 1H-NMR (400 MHz, CDCl3): δ 7.96 (m, 1H), 7.70 (m, 2H), 7.63 (m, 1H), 6.16 (m, 1H), 5.80 (ddt, J = 6.6, 10.3, 17.1 Hz, 1H), 5.01 (m, 2H), 3.63 (t, J = 6.5 Hz, 2H), 3.33 (m, 6H), 2.75 (m, 1H), 2.58 (m, 1H), 2.31 (m, 1H), 2.13 (m, 6H), 1.95 (s, 1H), 1.76 (m, 4H), 1.21–1.65 (m, 20H); 13C-NMR (100 MHz, CDCl3): δ 173.2, 148.1, 137.9, 133.6, 133.1, 131.7, 130.4, 124.1, 115.2, 62.7, 60.1, 52.5, 51.6, 47.9, 45.1, 35.9, 35.8, 33.1, 32.7, 30.8, 30.0, 29.8, 27.9, 26.4, 25.7, 25.7, 25.2, 24.7, 23.4, 22.9; IR (film):
(c 1.11, CH2Cl2). 1H-NMR (400 MHz, CDCl3): δ 7.96 (m, 1H), 7.70 (m, 2H), 7.63 (m, 1H), 6.16 (m, 1H), 5.80 (ddt, J = 6.6, 10.3, 17.1 Hz, 1H), 5.01 (m, 2H), 3.63 (t, J = 6.5 Hz, 2H), 3.33 (m, 6H), 2.75 (m, 1H), 2.58 (m, 1H), 2.31 (m, 1H), 2.13 (m, 6H), 1.95 (s, 1H), 1.76 (m, 4H), 1.21–1.65 (m, 20H); 13C-NMR (100 MHz, CDCl3): δ 173.2, 148.1, 137.9, 133.6, 133.1, 131.7, 130.4, 124.1, 115.2, 62.7, 60.1, 52.5, 51.6, 47.9, 45.1, 35.9, 35.8, 33.1, 32.7, 30.8, 30.0, 29.8, 27.9, 26.4, 25.7, 25.7, 25.2, 24.7, 23.4, 22.9; IR (film): 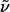 3,415, 3,294, 3,075, 2,932, 2,859, 1,647, 1,545, 1,460, 1,440, 1,373, 1,347, 1,261, 1,162, 1,125, 1,059, 913, 852, 777, 745, 584 cm-1; MS (EI): m/z (relative intensity) 495 (10), 493 (100), 463 (4), 408 (5), 397 (2), 306 (2), 238 (2), 223 (3), 198 (11), 110 (12), 84 (20). HRMS (EI) calcd. for C30H50N4O6S: 595.352932 (M + H); found: 595.352331 (M + H). Anal. calcd. for C30H50N4O6S: C, 60.58, H, 8.47; found: C, 60.68, H, 8.56.
3,415, 3,294, 3,075, 2,932, 2,859, 1,647, 1,545, 1,460, 1,440, 1,373, 1,347, 1,261, 1,162, 1,125, 1,059, 913, 852, 777, 745, 584 cm-1; MS (EI): m/z (relative intensity) 495 (10), 493 (100), 463 (4), 408 (5), 397 (2), 306 (2), 238 (2), 223 (3), 198 (11), 110 (12), 84 (20). HRMS (EI) calcd. for C30H50N4O6S: 595.352932 (M + H); found: 595.352331 (M + H). Anal. calcd. for C30H50N4O6S: C, 60.58, H, 8.47; found: C, 60.68, H, 8.56.
(R)-Hex-5-enoic Acid {3-[(2-Nitrobenzenesulfonyl)-{4-[2-(6-oxohexyl)-piperidinyl]butyl}-amino]propyl}amide. To a solution of oxalyl chloride (0.09 ml, 0.99 mmol) in CH2Cl2 (8 ml) at -60°C were added DMSO (0.13 ml, 1.77 mmol) and a solution of alcohol 11 (390 mg, 0.66 mmol) in CH2Cl2 (4 ml). Et3N (0.5 ml, 3.56 mmol) was introduced after 15 min, and the solution was stirred at -60°C for 40 min and at ambient temperature for 1 h before it was quenched with saturated aqueous NaHCO3 (5 ml). The aqueous layer was extracted with CH2Cl2 (3 × 10 ml), and the combined organic phases were successively washed with H2O (10 ml) and brine (10 ml) before being dried over Na2SO4. The residue was purified by flash chromatography (CH2Cl2/MeOH, 30:1 → CH2Cl2/MeOH + NEt3, 30:1 + 0.1%) to provide the title compound as a yellow oil (316 mg, 81%). 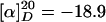 (c 1.03, CH2Cl2). 1H-NMR (300 MHz, CD2Cl2): δ 9.73 (t, J = 1.8 Hz, 1H), 7.93 (m, 1H), 7.68 (m, 3H), 6.00 (m, 1H), 5.81 (ddt, J = 6.6, 10.3, 17.1 Hz, 1H), 5.00 (m, 2H), 3.29 (m, 6H), 2.74 (m, 1H), 2.58 (m, 1H), 2.40 (dt, J = 1.8, 7.3 Hz, 1H), 2.17 (m, 7H), 1.19–1.80 (m, 23H); 13C-NMR (75 MHz, CD2Cl2): δ 203.1, 172.9, 148.5, 138.6, 134.0, 133.5, 132.2, 130.5, 124.5, 115.1, 60.5, 52.8, 51.6, 48.2, 45.6, 44.2, 36.3, 36.2, 33.5, 30.7, 30.2, 29.9, 28.5, 26.6, 25.8, 25.6, 25.1, 23.6, 23.6, 22.4; IR (film):
(c 1.03, CH2Cl2). 1H-NMR (300 MHz, CD2Cl2): δ 9.73 (t, J = 1.8 Hz, 1H), 7.93 (m, 1H), 7.68 (m, 3H), 6.00 (m, 1H), 5.81 (ddt, J = 6.6, 10.3, 17.1 Hz, 1H), 5.00 (m, 2H), 3.29 (m, 6H), 2.74 (m, 1H), 2.58 (m, 1H), 2.40 (dt, J = 1.8, 7.3 Hz, 1H), 2.17 (m, 7H), 1.19–1.80 (m, 23H); 13C-NMR (75 MHz, CD2Cl2): δ 203.1, 172.9, 148.5, 138.6, 134.0, 133.5, 132.2, 130.5, 124.5, 115.1, 60.5, 52.8, 51.6, 48.2, 45.6, 44.2, 36.3, 36.2, 33.5, 30.7, 30.2, 29.9, 28.5, 26.6, 25.8, 25.6, 25.1, 23.6, 23.6, 22.4; IR (film): 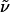 2,931, 2,860, 1,721, 1,646, 1,542, 1,439, 1,371, 1,345, 1,254, 1,161, 1,124, 995, 911, 851, 776, 774 cm-1; MS (EI): m/z (relative intensity) 495 (10), 493 (100), 463 (5), 406 (6), 308 (3), 248 (2), 223 (4), 196 (8), 186 (2), 154 (7), 138 (2), 126 (2), 110 (7), 100 (2), 98 (10), 84 (12). HRMS (EI) calcd. for C30H48N4O6S: 593.337282 (M + H); found: 593.336726 (M + H). Anal. calcd. for C30H48N4O6S: C, 60.78, H, 8.16; found: C, 60.52, H, 8.07.
2,931, 2,860, 1,721, 1,646, 1,542, 1,439, 1,371, 1,345, 1,254, 1,161, 1,124, 995, 911, 851, 776, 774 cm-1; MS (EI): m/z (relative intensity) 495 (10), 493 (100), 463 (5), 406 (6), 308 (3), 248 (2), 223 (4), 196 (8), 186 (2), 154 (7), 138 (2), 126 (2), 110 (7), 100 (2), 98 (10), 84 (12). HRMS (EI) calcd. for C30H48N4O6S: 593.337282 (M + H); found: 593.336726 (M + H). Anal. calcd. for C30H48N4O6S: C, 60.78, H, 8.16; found: C, 60.52, H, 8.07.
(R)-Hex-5-enoic Acid (3-{[4-(2-hept-6-Enylpiperidinyl)butyl]-(2-nitrobenzenesulfonyl)amino}propyl)amide (12). A suspension of methyltriphenylphosphonium bromide (629 mg, 1.76 mmol) in HMPA (0.61 ml, 3.52 mmol) and THF (5 ml) was treated with a solution of potassium bis(trimethylsilyl)amide (0.5 M in toluene, 3.5 ml, 1.76 mmol) at 0°C. The resulting bright-yellow solution was added to a solution of the aldehyde described above (260 mg, 0.44 mmol) in THF (5 ml)at -78°C. The mixture was stirred at that temperature for 5 min and then for 30 min at ambient temperature before it was quenched with H2O. A standard extractive work-up (CH2Cl2) followed by flash chromatography (CH2Cl2/MeOH, 30:1 → CH2Cl2/MeOH:aqueous NH4OH (25%), 30:1:0.5) afforded compound 12 as an orange oil (212 mg, 82%). 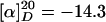 (c 1.04, CH2Cl2). 1H-NMR (300 MHz, CD2Cl2): δ 7.94 (m, 1H), 7.67 (m, 3H), 6.08 (m, 1H), 5.81 (m, 2H), 4.98 (m, 4H), 3.30 (m, 6H), 2.80 (m, 1H), 2.64 (m, 1H), 2.19 (m, 8H), 1.26–1.80 (m, 23H); 13C-NMR (75 MHz, CD2Cl2): δ 173.0, 148.5, 139.6, 138.6, 134.0, 133.4, 132.2, 130.6, 124.5, 115.1, 114.3, 60.9, 52.5, 51.5, 45.7, 36.3, 36.2, 34.1, 33.6, 30.6, 29.8, 29.6, 29.3, 28.5, 26.6, 26.0, 25.2, 23.3, 23.2; IR (film):
(c 1.04, CH2Cl2). 1H-NMR (300 MHz, CD2Cl2): δ 7.94 (m, 1H), 7.67 (m, 3H), 6.08 (m, 1H), 5.81 (m, 2H), 4.98 (m, 4H), 3.30 (m, 6H), 2.80 (m, 1H), 2.64 (m, 1H), 2.19 (m, 8H), 1.26–1.80 (m, 23H); 13C-NMR (75 MHz, CD2Cl2): δ 173.0, 148.5, 139.6, 138.6, 134.0, 133.4, 132.2, 130.6, 124.5, 115.1, 114.3, 60.9, 52.5, 51.5, 45.7, 36.3, 36.2, 34.1, 33.6, 30.6, 29.8, 29.6, 29.3, 28.5, 26.6, 26.0, 25.2, 23.3, 23.2; IR (film): 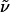 3,279, 3,073, 2,929, 2,858, 1,641, 1,542, 1,438, 1,371, 1,344, 1,255, 1,160, 1,123, 1,059, 993, 909, 851, 774, 742 cm-1; MS (EI): m/z (relative intensity) 495 (10), 493 (100), 463 (4), 404 (6), 223 (2), 194 (6), 154 (4), 138 (2). HRMS (EI) calcd. for C31H50N4O5S: 591.358017 (M + H); found: 591.357182 (M + H). Anal. calcd. for C31H50N4O5S: C, 63.02, H, 8.53; found: C, 62.82, H, 8.40.
3,279, 3,073, 2,929, 2,858, 1,641, 1,542, 1,438, 1,371, 1,344, 1,255, 1,160, 1,123, 1,059, 993, 909, 851, 774, 742 cm-1; MS (EI): m/z (relative intensity) 495 (10), 493 (100), 463 (4), 404 (6), 223 (2), 194 (6), 154 (4), 138 (2). HRMS (EI) calcd. for C31H50N4O5S: 591.358017 (M + H); found: 591.357182 (M + H). Anal. calcd. for C31H50N4O5S: C, 63.02, H, 8.53; found: C, 62.82, H, 8.40.
(R)-Hex-5-enoic Acid {3-[4-(2-hept-6-Enylpiperidinyl)butylamino]-propyl}amide (13). Mercaptoacetic acid (0.08 ml, 1.10 mmol) and LiOH (99 mg, 4.11 mmol) were added to a solution of compound 12 (162 mg, 0.27 mmol) in DMF (6 ml). The mixture was stirred for 1 h before it was diluted with H2O (1:1) and extracted with CH2Cl2 (3 × 5 ml). The combined organic phases were successively washed with saturated aqueous NaHCO3 (2 × 10 ml), H2O (10 ml), and brine (10 ml) before being dried over Na2SO4. Evaporation of the solvent followed by flash chromatography of the residue (CH2Cl2/MeOH:NH3, 30:1:0.5 → 5:1:0.5) furnished compound 13 as an orange oil (93 mg, 84%). 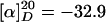 (c 1.05, CH2Cl2). 1H-NMR (300 MHz, CDCl3): δ 6.80 (m, 1H), 5.80 (m, 2H), 4.98 (m, 4H), 3.35 (m, 2H), 2.83 (m, 1H), 2.66 (m, 5H), 2.01–2.39 (m, 9H), 1.20–1.78 (m, 23H); 13C-NMR (75 MHz, CDCl3): δ 172.7, 139.1, 138.0, 115.2, 114.2, 60.2, 53.3, 51.8, 49.9, 48.4, 38.9, 36.1, 33.7, 33.2, 31.0, 30.2, 29.6, 28.9, 28.7, 28.3, 25.7, 25.5, 24.8, 23.8, 23.5; IR (film):
(c 1.05, CH2Cl2). 1H-NMR (300 MHz, CDCl3): δ 6.80 (m, 1H), 5.80 (m, 2H), 4.98 (m, 4H), 3.35 (m, 2H), 2.83 (m, 1H), 2.66 (m, 5H), 2.01–2.39 (m, 9H), 1.20–1.78 (m, 23H); 13C-NMR (75 MHz, CDCl3): δ 172.7, 139.1, 138.0, 115.2, 114.2, 60.2, 53.3, 51.8, 49.9, 48.4, 38.9, 36.1, 33.7, 33.2, 31.0, 30.2, 29.6, 28.9, 28.7, 28.3, 25.7, 25.5, 24.8, 23.8, 23.5; IR (film): 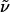 3,276, 3,075, 2,930, 2,856, 2,796, 1,642, 1,549, 1,442, 1,370, 1,260, 1,125, 1,049, 992, 909 cm-1; MS (EI): m/z (relative intensity) 405 (4, [M+]), 308 (100), 226 (9), 225 (42), 223 (11), 194 (20), 183 (13), 180 (17), 154 (26), 138 (9), 84 (26). HRMS (EI) calcd. for C25H47N3O: 406.379736 (M + H); found: 406.379190 (M + H).
3,276, 3,075, 2,930, 2,856, 2,796, 1,642, 1,549, 1,442, 1,370, 1,260, 1,125, 1,049, 992, 909 cm-1; MS (EI): m/z (relative intensity) 405 (4, [M+]), 308 (100), 226 (9), 225 (42), 223 (11), 194 (20), 183 (13), 180 (17), 154 (26), 138 (9), 84 (26). HRMS (EI) calcd. for C25H47N3O: 406.379736 (M + H); found: 406.379190 (M + H).
(R)-Docosahydro-4a,9,13-triazabenzocyclodocosen-14-one [(–)-(R)-isooncinotine] (1). HCl (3 ml, 2 M in ether) was added to a solution of diene 13 (58 mg, 0.14 mmol) in CH2Cl2 (3 ml), and the resulting mixture was stirred for 15 min at ambient temperature. The solvent was evaporated, and the residue was dried in vacuo for 30 min. The resulting hydrochloride was dissolved in CH2Cl2 (4 ml) and added simultaneously with a solution of the indenylidene complex 14 (6 mg) in CH2Cl2 (4 ml) to a refluxing solution of complex 14 (8 mg) in CH2Cl2 (80 ml) over a period of 2 h. Reflux was continued for 14 h before the mixture was concentrated to approximately half of the original volume and transferred to an autoclave. Stirring for 12 h at 70°C under an atmosphere of H2 (50 bars) followed by extraction with saturated NaHCO3 (10 ml) and flash chromatography of the crude material (CH2Cl2/MeOH, 10:1 → CH2Cl2/MeOH:NH3, 10:1:0.5 → 4:1:0.5) provided pure (-)-1 (41 mg, 76%). 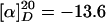 (c 1.00, MeOH),
(c 1.00, MeOH), 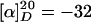 (c 0.067, MeOH) [ref. 6:
(c 0.067, MeOH) [ref. 6: 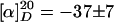 (c 0.067, MeOH)]. 1H-NMR (300 MHz, CDCl3): δ 7.60 (m, 1H), 3.38 (m, 2H), 2.60–2.89 (m, 5H), 2.37 (m, 1H), 2.16 (m, 4H), 1.26–1.64 (m, 32H); 13C-NMR (75 MHz, CDCl3): δ 173.0, 59.9, 53.4, 52.4, 50.3, 49.4, 39.8, 36.9, 30.8, 30.5, 29.7, 28.9, 28.7, 28.5, 28.4, 28.0, 27.6, 25.7, 24.0, 23.7, 23.3; IR (film):
(c 0.067, MeOH)]. 1H-NMR (300 MHz, CDCl3): δ 7.60 (m, 1H), 3.38 (m, 2H), 2.60–2.89 (m, 5H), 2.37 (m, 1H), 2.16 (m, 4H), 1.26–1.64 (m, 32H); 13C-NMR (75 MHz, CDCl3): δ 173.0, 59.9, 53.4, 52.4, 50.3, 49.4, 39.8, 36.9, 30.8, 30.5, 29.7, 28.9, 28.7, 28.5, 28.4, 28.0, 27.6, 25.7, 24.0, 23.7, 23.3; IR (film): 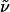 3,274, 3,067, 2,927, 2,854, 2,802, 1,645, 1,548, 1,461, 1,442, 1,370, 1,263, 1,123, 1,049, 924, 833, 752, 732 cm-1; MS (EI): m/z (relative intensity) 379 (48 [M+]), 378 (11), 377 (2), 338 (6), 336 (17), 323 (7), 322 (13), 307 (7), 297 (9), 293 (6), 283 (8), 281 (16), 280 (7), 268 (5), 266 (7), 255 (100), 253 (10), 227 (7), 226 (6), 225 (4), 208 (4), 183 (11), 181 (9), 171 (6), 168 (5), 155 (7), 142 (4), 140 (11), 137 (8), 136 (4), 129 (6), 125 (46), 124 (56), 109 (4), 98 (96), 97 (26), 96 (22), 67 (7), 56 (20), 55 (31), 44 (24), 43 (14), 41 (19), 30 (21). HRMS (EI) calcd. for C23H45N3O: 380.364086 (M + H); found: 380.363169 (M + H). Anal. calcd. for C23H45N3O: C, 72.77, H, 11.95; found: C, 72.83, H, 12.06.
3,274, 3,067, 2,927, 2,854, 2,802, 1,645, 1,548, 1,461, 1,442, 1,370, 1,263, 1,123, 1,049, 924, 833, 752, 732 cm-1; MS (EI): m/z (relative intensity) 379 (48 [M+]), 378 (11), 377 (2), 338 (6), 336 (17), 323 (7), 322 (13), 307 (7), 297 (9), 293 (6), 283 (8), 281 (16), 280 (7), 268 (5), 266 (7), 255 (100), 253 (10), 227 (7), 226 (6), 225 (4), 208 (4), 183 (11), 181 (9), 171 (6), 168 (5), 155 (7), 142 (4), 140 (11), 137 (8), 136 (4), 129 (6), 125 (46), 124 (56), 109 (4), 98 (96), 97 (26), 96 (22), 67 (7), 56 (20), 55 (31), 44 (24), 43 (14), 41 (19), 30 (21). HRMS (EI) calcd. for C23H45N3O: 380.364086 (M + H); found: 380.363169 (M + H). Anal. calcd. for C23H45N3O: C, 72.77, H, 11.95; found: C, 72.83, H, 12.06.
Results and Discussion
Strategic Considerations. Although (-)-isooncinotine 1 certainly lends itself to retrosynthetic analysis following established guidelines, it was perceived that our goal of achieving high convergence through catalysis might materialize more effectively by following nonconventional logic. To this end, we have deliberately chosen a remote C—C bond within the aliphatic tether of the target rather than the lactam as the site of macrocyclization, because this allows one to implement ring-closing metathesis (RCM) as a key transformation (Scheme 1). Although RCM was shown recently to be among the most efficient entries into carbo- and heterocyclic rings of virtually any size (17–19), the efficiency of RCM-based macrocyclizations is known to be strongly affected by heteroelements in proximity to the reacting alkenes, which can attenuate the reactivity of the emerging metal carbenes by formation of (stable) chelate complexes (20, 21). To avoid such complications, the C.15—C.16 bond (isooncinotine numbering) seemed best suited for this maneuver, because the carbonyl group of the amide is then sufficiently remote. Protonation of the amine groups in the cyclization precursor A would prevent them from interfering and, at the same time, avoid a more cumbersome protecting-group management.
Scheme 1.

Retrosynthetic analysis of (-)-isooncinotine 1.
The diene substrate A required for RCM derives from a suitable 2-substituted piperidine derivative B carrying the only chiral center of the target. Although such a compound could be formed by one of the established methods for the preparation of enantiopure piperidines described in the literature (22), the asymmetric hydrogenation of a suitably substituted pyridine derivative C would constitute an inherently more attractive solution. Although methods for the asymmetric hydrogenation of heteroarenes were largely missing in the registry of organic synthesis, a very recent development promises to fill the gap (23). The essence of this method is sketched in Scheme 2. Thus, protonation of a substituted pyridine derivative D bearing an chiral oxazolidinone at C.2 activates the heterocycle for hydrogenation and avoids catalyst poisoning. More importantly, however, the ensuing hydrogen bond favors conformation E, in which the auxiliary is coplanar with the pyridine ring; this in turn results in an effective shielding of one of the π-faces of the aromatic system by the isopropyl substituent and hence allows hydrogen to be delivered from the surface of a heterogeneous catalyst only to the opposite side of the substrate. Traceless cleavage of the resulting aminal F releases the auxiliary in situ and leads to the formation of the optically active piperidine H after hydrogenation of the resulting iminium (and/or enaminium) salt G (23). It was envisaged that the required pyridine substrate D can be secured by a method allowing for the selective cross coupling of inexpensive chloroarenes by using cheap, nontoxic, and benign iron salts as the precatalysts in lieu of the conventional palladium- or nickel-based procedures (24, 25).
Scheme 2.

Formation of optically active piperidine derivatives by asymmetric hydrogenation of pyridines with concomitant release of the chiral auxiliary (23).
Total Synthesis. Modern cross-coupling reactions are a domain of palladium and nickel catalysts, which are distinguished by an impressively wide scope and an excellent compatibility with many functional groups (26). Their favorable application profile usually overcompensates for the disadvantages resulting from the high costs of the palladium precursors, the concerns about the toxicity of nickel salts, the need for ancillary ligands, and the extended reaction times that are necessary in many cases. Encouraged by early reports of Kochi (27), our group recently showed that cheap, stable, nontoxic, and environmentally benign iron salts such as Fe(acac)3 constitute a promising alternative in certain cases (24, 25, 28, 29). They are able to catalyze, for example, the cross coupling of aromatic and heteroaromatic chlorides or sulfonates with various Grignard reagents with exceptional ease and unprecedentedly high reaction rates. When applied to substrates bearing more than one leaving group, this method allows one to perform selective, serial, or exhaustive alkylation reactions (24, 30).
This favorable characteristic of the iron-catalyzed protocol was exploited en route to 1 as shown in Scheme 3. Thus, the required pyridine derivative 7 was formed in excellent yield after addition of the functionalized Grignard reagent 6 derived from 6-benzyloxy-1-bromohexane to a solution of 2,6-dichloropyridine 5 in THF/NMP in the presence of Fe(acac)3 (5 mol %) at 0°C. Under these conditions, no product derived from the conceivable double alkylation of the substrate can be detected.‡ Chloride 7 was then subjected to a Cu(I)-catalyzed amidation (31), which allows for the convenient introduction of the chiral oxazolidin-2-one 8 serving as the stereochemical control element in the subsequent asymmetric hydrogenation reaction (23). It is gratifying that this key transformation worked exquisitely well in the presence of Pd(OH)2 on charcoal as precatalyst in MeOH/HOAc at 35°C using an initial H2 pressure of 120 bars, providing the desired piperidine derivative 10 in 78% yield and excellent optical purity (ee = 94%). Not only was the chiral auxiliary 8 cleaved off under the chosen reaction conditions and recovered in 91% yield, but the benzyl ether protecting group for the terminal alcohol in 9 was also concomitantly removed.
Scheme 3.

[a] Fe(acac)3 catalytic, THF/NMP, 0°C, 83%. [b] Oxazolidin-2-one 8, CuI catalytic, N,N′-dimethylethylenediamine, K2CO3, toluene, 140°C, 90%. [c] Pd(OH)2/C catalytic, MeOH, HOAc, H2 [120 atm (1 atm = 101.3 kPa)], 35°C, 78%, ee = 94%. [d] Bromide 18, K2CO3, NaI, EtOH, reflux, 73%. [e] Oxalyl chloride, DMSO, CH2Cl2, NEt3, -60°C, 81%. [f] {Ph3P-CH3}+ Br-, potassium hexamethyldisilazide, THF, HMPA, -78°C → room temperature, 82%. [g] HSCH2COOH, LiOH, DMF, 84%. [h] (i) HCl/Et2O, CH2Cl2; (ii) complex 14 catalytic, CH2Cl2, reflux; (iii) H2 (50 bars), 70°C, 76%.
Compound 18 as a suitable surrogate for the spermidine part of the target was readily prepared from 1,3-diaminopropane 15, which was monoacylated with methyl 5-hexenoate followed by conversion of the remaining amine in 16 into the corresponding o-nitrobenzenesulfonyl (nosyl, Ns) derivative 17 (32, 33) (Scheme 4). Taking advantage of the higher acidity of the sulfonamide (34), a selective N-alkylation of 17 with 1,4-dibromobutane was achieved to give product 18 on a multigram scale. Reaction of this compound with piperidine 10 in the presence of NaI provided alcohol 11, which was converted into alkene 12 via Swern oxidation and subsequent Wittig reaction; therefore, it was essential to use potassium hexamethyldisilazide as the base in THF/HMPA to ensure high yields. Cleavage of the nosyl group on treatment of 12 with mercaptoacetic acid and LiOH in DMF (35) furnished diene 13 as the substrate required for the envisaged macrocyclization reaction.
Scheme 4.

[a] 5-Hexenoic acid methyl ester, 60°C, 95%. [b] 2-Nitrobenzenesulfonyl chloride, pyridine, CH2Cl2, 73%. [c] 1,4-Dibromobutane, K2CO3, DMF, 60°C, 88%.
In line with our expectations, this key transformation proceeded smoothly in the presence of the ruthenium indenylidene catalyst 14 (36–38), previously developed in our laboratory as a cheap, stable, and practical alternative to the classical Grubbs catalyst Cl2(PCy3)2Ru CHPh (39). Protonation of the substrate is mandatory to avoid coordination of the amines to the metal center, which would result in loss of the catalytic activity of the ruthenium complex (40). It is also important to note that the 22-membered cycloalkene formed by RCM does not have to be isolated but can be converted into isooncinotine by stirring the crude reaction mixture in an autoclave under H2 (50 bars, 70°C) for 12 h. Under these conditions, the alkylidene complex is converted into a ruthenium hydride, which serves as homogeneous catalyst saturating the double bond (30, 41, 42). The analytical and spectroscopic data of (-)-isooncinotine 1 thus obtained from diene 13 in 76% over three steps in “one pot” are in excellent agreement with those reported in the literature (6). Moreover, this total synthesis firmly establishes the (R)-configuration of the alkaloid, which was only deduced previously by comparison of the chiroptical properties of degradation products with those of (-)-N-methyl-coniine (6).
CHPh (39). Protonation of the substrate is mandatory to avoid coordination of the amines to the metal center, which would result in loss of the catalytic activity of the ruthenium complex (40). It is also important to note that the 22-membered cycloalkene formed by RCM does not have to be isolated but can be converted into isooncinotine by stirring the crude reaction mixture in an autoclave under H2 (50 bars, 70°C) for 12 h. Under these conditions, the alkylidene complex is converted into a ruthenium hydride, which serves as homogeneous catalyst saturating the double bond (30, 41, 42). The analytical and spectroscopic data of (-)-isooncinotine 1 thus obtained from diene 13 in 76% over three steps in “one pot” are in excellent agreement with those reported in the literature (6). Moreover, this total synthesis firmly establishes the (R)-configuration of the alkaloid, which was only deduced previously by comparison of the chiroptical properties of degradation products with those of (-)-N-methyl-coniine (6).
Conclusions
The total synthesis of the spermidine alkaloid (-)-isooncinotine 1 in optically active form is reported. The approach is largely catalysis-based and scrutinizes methodology developed previously in our laboratories. Thus, it shows that cheap and nontoxic iron salts warrant consideration as substitutes for established palladium and nickel catalysts in alkyl–aryl cross-coupling reactions of (hetero)aryl chlorides (24, 25, 30). Likewise, the potential of an entry into optically active piperidines by asymmetric hydrogenation of substituted pyridines (23) is demonstrated by the conversion of 9 into compound 10. Not only does this heterogeneous asymmetric hydrogenation deliver the desired product with an excellent ee, but it also integrates the cleavage of the auxiliary and the removal of a lateral protecting group into a practical and scalable one-pot operation. An equally highly integrated maneuver accounts for the end game of the synthesis in which the 22-membered ring is forged and saturated in another one-pot process comprising an RCM reaction followed by the hydrogenation of the resulting cycloalkene by using a single ruthenium precatalyst. The indenylidene complex 14 (36) described previously as a readily available and equipotent substitute for the classical Grubbs catalyst serves this twofold purpose very well. Together with the copper-catalyzed installation of the oxazolidin-2-one group (7 → 9) via a procedure recently outlined for the N-arylation of amides (31), all but one key steps of the chosen route rely on metal-catalyzed transformations. The only major exception concerns the formation of alkene 12, which had to be done by a standard Wittig reaction using overstoichiometric amounts of the reagent. This contrast highlights a major shortcoming in the present armamentarium of preparative organic chemistry and substantiates the notion that catalytic methods for carbonyl olefination are on urgent demand.
Acknowledgments
Generous financial support by the Deutsche Forschungsgemeinschaft (Leibniz award to A.F.), the Merck Research Council, and the Arthur C. Cope Scholar Funds (administered by the American Chemical Society) is gratefully acknowledged. B.S. thanks the Fonds der Chemischen Industrie for a Kekulé stipend, and F.G. thanks the Fonds der Chemischen Industrie for a Liebig fellowship.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: THF, tetrahydrofuran; HMPA, hexamethylphosphoramide; DMF, dimethylformamide; EI, electron impact; HRMS, high-resolution MS; ee, enantiomeric excess; RCM, ring-closing metathesis.
Footnotes
For example, compounds 2 and 3 are inseparable by chromatography (6). A pure sample of 2 was obtained only after 3 was transformed into 1 by base-induced ring expansion.
Slow addition of the Grignard reagent over 30 min is essential, resulting in the exclusive formation of the monosubstitution product, whereas more rapid addition affords product mixtures.
References
- 1.Morris, D. R. & Marton, L. J. (1981) Polyamines in Biology and Medicine (Dekker, New York).
- 2.Guggisberg, A. & Hesse, M. (1998) Alkaloids 50, 219-256. [Google Scholar]
- 3.Karigiannis, G. & Papaioannou, D. (2000) Eur. J. Org. Chem., 1841-1863.
- 4.Frydman, B., Bhattacharya, S., Sarkar, A., Drandarov, K., Chesnov, S., Guggisberg, A., Popaj, K., Yurdakul, A., Hesse, M., Basu, H. S., et al. (2004) J. Med. Chem. 47, 1051-1059. [DOI] [PubMed] [Google Scholar]
- 5.Badawi, M. M., Guggisberg, A., van den Broek, P., Hesse, M. & Schmid, H. (1968) Helv. Chim. Acta 51, 1813-1817. [DOI] [PubMed] [Google Scholar]
- 6.Guggisberg, A., Badawi, M. M., Hesse, M. & Schmid, H. (1974) Helv. Chim. Acta 57, 414-434. [DOI] [PubMed] [Google Scholar]
- 7.Doll, M. K.-H., Guggisberg, A. & Hesse, M. (1996) Helv. Chim. Acta 79, 973-981. [Google Scholar]
- 8.Kuksa, V., Buchan, R. & Lin, P. K. T. (2000) Synthesis 1189-1207.
- 9.Schneider, F., Bernauer, K., Guggisberg, A., van den Broek, P., Hesse, M. & Schmid, H. (1974) Helv. Chim. Acta 57, 434-440. [DOI] [PubMed] [Google Scholar]
- 10.Guggisberg, A., van den Broek, P., Hesse, M., Schmid, H., Schneider, F. & Bernauer, K. (1976) Helv. Chim. Acta 59, 3013-3025. [DOI] [PubMed] [Google Scholar]
- 11.Bienz, S., Guggisberg, A., Wälchli, R & Hesse, M. (1988) Helv. Chim. Acta 71, 1708-1718. [Google Scholar]
- 12.Doll, M. K.-H., Guggisberg, A. & Hesse, M. (1996) Helv. Chim. Acta 79, 1379-1386. [Google Scholar]
- 13.Ina, H., Ito, M. & Kibayashi, C. (1996) J. Org. Chem. 61, 1023-1029. [Google Scholar]
- 14.Fürstner, A. & Rumbo, A. (2000) J. Org. Chem. 65, 2608-2611. [DOI] [PubMed] [Google Scholar]
- 15.Fürstner, A. (2003) Angew. Chem. Int. Ed. 42, 3582-3603. [DOI] [PubMed] [Google Scholar]
- 16.Fürstner, A. (1999) Synlett, 1523-1533.
- 17.Trnka, T. M. & Grubbs, R. H. (2001) Acc. Chem. Res. 34, 18-29. [DOI] [PubMed] [Google Scholar]
- 18.Fürstner, A. (2000) Angew. Chem. Int. Ed. 39, 3012-3043. [PubMed] [Google Scholar]
- 19.Schuster, M. & Blechert, S. (1997) Angew. Chem. Int. Ed. Engl. 36, 2037-2056. [Google Scholar]
- 20.Fürstner, A. & Langemann, K. (1997) Synthesis, 792-803.
- 21.Fürstner, A., Thiel, O. R. & Lehmann, C. W. (2002) Organometallics 21, 331-335. [Google Scholar]
- 22.Buffat, M. G. P. (2004) Tetrahedron 60, 1701-1729. [Google Scholar]
- 23.Glorius, F., Spielkamp, N., Holle, S., Goddard, R. & Lehmann, C. W. (2004) Angew. Chem. Int. Ed. 43, 2850-2852. [DOI] [PubMed] [Google Scholar]
- 24.Fürstner, A., Leitner, A., Méndez, M. & Krause, H. (2002) J. Am. Chem. Soc. 124, 13856-13863. [DOI] [PubMed] [Google Scholar]
- 25.Fürstner, A. & Leitner, A. (2002) Angew. Chem. Int. Ed. 41, 609-612. [Google Scholar]
- 26.Diederich, F. & Stang, P. J., eds. (1998) Metal-Catalyzed Cross-Coupling Reactions (Wiley–VCH, Weinheim, Germany).
- 27.Kochi, J. K. (1974) Acc. Chem. Res. 7, 351-360. [Google Scholar]
- 28.Fürstner, A. & Méndez, M. (2003) Angew. Chem. Int. Ed. 42, 5355-5357. [DOI] [PubMed] [Google Scholar]
- 29.Fürstner, A., De Souza, D., Parra-Rapado, L. & Jensen, J. T. (2003) Angew. Chem. Int. Ed. 42, 5358-5360. [DOI] [PubMed] [Google Scholar]
- 30.Fürstner, A. & Leitner, A. (2003) Angew. Chem. Int. Ed. 42, 308-311. [DOI] [PubMed] [Google Scholar]
- 31.Klapars, A., Huang, X. & Buchwald, S. L. (2002) J. Am. Chem. Soc. 124, 7421-7428. [DOI] [PubMed] [Google Scholar]
- 32.Kan, T. & Fukuyama, T. (2004) Chem. Commun., 353-359. [DOI] [PubMed]
- 33.Kan, T., Fujiwara, A., Kobayashi, H. & Fukuyama, T. (2002) Tetrahedron 58, 6267-6276. [Google Scholar]
- 34.Fukuyama, T., Cheung, M., Jow, C.-K., Hidai, Y. & Kan, T. (1997) Tetrahedron Lett. 38, 5831-5834. [Google Scholar]
- 35.Shin, I. & Park, K. (2002) Org. Lett. 4, 869-872. [DOI] [PubMed] [Google Scholar]
- 36.Fürstner, A., Guth, O., Düffels, A., Seidel, G., Liebl, M., Gabor, B. & Mynott, R. (2001) Chem. Eur. J. 7, 4811-4820. [DOI] [PubMed] [Google Scholar]
- 37.Fürstner, A., Radkowski, K., Wirtz, C., Goddard, R., Lehmann, C. W. & Mynott, R. (2002) J. Am. Chem. Soc. 124, 7061-7069. [DOI] [PubMed] [Google Scholar]
- 38.Fürstner, A., Jeanjean, F. & Razon, P. (2002) Angew. Chem. Int. Ed. 41, 2097-2101. [PubMed] [Google Scholar]
- 39.Schwab, P., Grubbs, R. H. & Ziller, J. W. (1996) J. Am. Chem. Soc. 118, 100-110. [Google Scholar]
- 40.Fürstner, A., Grabowski, J. & Lehmann, C. W. (1999) J. Org. Chem. 64, 8275-8280. [DOI] [PubMed] [Google Scholar]
- 41.Louie, J., Bielawski, C. W. & Grubbs, R. H. (2001) J. Am. Chem. Soc. 123, 11312-11313. [DOI] [PubMed] [Google Scholar]
- 42.Schmidt, B. (2004) Eur. J. Org. Chem., 1865-1880.