

Abstract
An efficient total synthesis of (+)-vincristine has been accomplished through a stereoselective coupling of demethylvindoline and the eleven-membered carbomethoxyverbanamine presursor. Demethylvindoline was prepared by oxidation of 17-hydroxy-11-methoxytabersonine, followed by regioselective acetylation with mixed anhydride method. Although an initial attempt of coupling by using demethylvindoline formamide was not successful and resulted in recovery of the starting compounds, the reaction using demethylvindoline took place smoothly to furnish the desired bisindole product with the correct stereochemistry at C18′. After formation of the piperidine ring by sequential removal of the protective groups and intramolecular nucleophilic cyclization, the total synthesis of vincristine was completed by formylation of N1.
Vinblastine (1) was first discovered as an unexpected myelosuppressive agent in 1958 by Noble and coworkers during search for an antidiabetic agent in Catharanthus roseus (1). Independently, researchers at Eli Lilly found that extracts of C. roseus possessed activity against P-1534 leukemia in mice and isolated vinblastine as its active entity in 1959 (2). The structure of a related compound, vincristine methiodide, was then determined by an x-ray crystallography in 1965 (3). Finally, structural studies on these alkaloids (4) revealed that both vinblastine (1) and vincristine (2) (5) are bisindole alkaloids, containing vindoline (3) attached to a tetracyclic indole, carbomethoxyvelbanamine (6) (Fig. 1).
Fig. 1.

Structures of vinblastine, vincristine, and vindoline.
These bisindole alkaloids inhibit assembly of tubulin units into microtubules, resulting in the arrest of cell division at metaphase. Although these compounds are very similar in structure and exhibit the same mode of action, they have different effects on the body (6). Vinblastine (1) is mainly useful for treating lymphocytic lymphoma, histiocytic lymphoma, Kaposi's sarcoma, and advanced testicular and breast cancer. Vincristine (2), on the other hand, displays curative effects in acute leukemia, rhabdomyosarcoma, neuroblastoma, and other lymphomas. Clinical treatment with these alkaloids is commonly accompanied by toxic side effects, and the typical dose-limiting factor for the former is bone marrow damage and the dose-limiting factor for the latter is neurotoxicity. Thus, development of more active and less toxic congeners is still an active area of research, where establishment of an efficient synthetic route would be required to carry out systematic studies on the structure–activity relationships.
Numerous synthetic studies have been directed toward Vinca alkaloids (7–14), and to date four total syntheses of vinblastine (1) have been reported (15–18). In contrast, vincristine (2), which is more difficult to obtain than vinblastine (1) because of very low content in the plant (≈0.0003% yield from C. roseus), has not yet been synthesized by means of total synthesis. It has only been prepared by derivatization of natural vinblastine (1) by direct oxidation of the N-methyl group (19–21) or by formylation of demethylvinblastine isolated from nature or prepared from vinblastine (1) by microbiological demethylation (22). Our own efforts in this area have recently led us to accomplish a total synthesis of vinblastine (1) (23) through an efficient total synthesis of vindoline (3) (23, 24). In this work, we describe a total synthesis of vincristine (2) through the stereoselective coupling of demethylvindoline and the eleven-membered carbomethoxyverbanamine presursor. This is a previously unreported example of de novo total synthesis of vincristine (2) without dependence on either derivatization of vinblastine (1)or semisynthesis with vindoline (3).
Materials and Methods
Synthetic Materials and Methods. All nonaqueous reactions were carried out under an inert atmosphere of argon in oven-dried glassware unless otherwise noted. Dichloromethane, toluene, and benzene were distilled from calcium hydride. Dehydrated tetrahydrofuran, diethyl ether, acetonitrile, N,N-dimethylformamide, methanol, and ethanol were purchased from Kanto Chemical Co. (Tokyo) and stored over molecular sieves 3A or 4A. All other reagents were commercially available and used without further purification. 1H and 13C NMR spectra were recorded on a JEOL LA 400-MHz spectrometer. IR spectra were recorded on a FT/IR-410 Fourier Transform Infrared Spectrophotometer (Jasco, Tokyo). Mass spectra and high-resolution mass spectra (HRMS) were obtained on a JEOL JMS-GCmate MS-DIP20. Fast atom bombardment (FAB) mass spectra were obtained with 3-nitrobenzyl alcohol (for low-resolution mass spectra) or polyethylene glycol (for HRMS) as the matrix. Optical rotations were measured on a Jasco DIP-1000 Digital Polarimeter at room temperature with the sodium D line. Melting points, determined on a Micro Melting Point Apparatus (Yanaco LID Co., Kyoto), are uncorrected.
Deacetyldemethylvindoline (11). A stirred two-phase suspension of 10 (54.1 mg, 0.141 mmol) in 10% methanol/dichloromethane (3.1 ml) and saturated aqueous sodium bicarbonate (2.2 ml) was cooled in an ice bath; to this suspension was added m-chloroperbenzoic acid (65 mg, ≈75%, ≈0.28 mol) dissolved in a small amount of 10% methanol/dichloromethane at 0°C for 2 min. After stirring for additional 10 min, methanolic solution of sodium cyanoborohydride (27 mg, 0.42 mmol) containing methyl orange was added, and 10% methanolic hydrogen chloride was added dropwise until the color of the solution turned red. The resulting solution was stirred for 5 min before additional methanolic solution of sodium cyanoborohydride (27 mg, 0.42 mmol) was added. After stirring for 3 min, the reaction mixture was allowed to warm up to room temperature and stirred for 30 min, keeping pH at ≈3 by addition of 10% methanolic hydrogen chloride. Then, sodium carbonate was added until the pH of the mixture increased to ≈10. Then, sodium bisulfite (381 mg) was added. The resulting suspension was stirred for 3 h. The mixture was then partitioned between dichloromethane and 3% aqueous ammonia. The aqueous layer was thoroughly extracted with dichloromethane. The combined organic extracts were dried over anhydrous magnesium sulfate, filtered, and concentrated in vacuo. The residue was purified with preparative TLC (5% methanol in dichloromethane) to give 11 (33.5 mg, 0.0837 mmol, 59.1%) as a white solid. 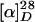 -4.9 (c 1.0, chloroform); IR (film) 3383, 2960, 1728, 1618, 1502, 1459, 1258, 1160, 1032, 752 cm-1; 1H HMR (400 MHz, CDCl3) δ 0.61 (3H, t, J = 7.4 Hz), 1.04–1.12 (1H, m), 1.34–1.42 (1H, m), 2.24–2.52 (5H, m), 2.82 (1H, dt, J = 1.5, 15.9 Hz), 3.35 (1H, td, J = 2.9, 18.1 Hz), 3.46 (1H, ddd, J = 1.5, 4.6, 15.9 Hz), 3.75 (3H, s), 3.83 (3H, s), 4.10 (1H, d, J = 2.9 Hz), 4.16 (1H, br s), 4.55 (1H, d, J = 2.4 Hz), 5.65 (1H, dt, J = 1.5, 9.7 Hz), 5.85 (1H, ddd, J = 1.5, 4.6, 9.7 Hz), 6.13 (1H, d, J = 2.2 Hz), 6.24 (1H, dd, J = 2.2, 8.2 Hz), 6.90 (1H, d, J = 8.2 Hz), 9.32 (1H, br s); 13C NMR (100 MHz, CDCl3) δ 7.8, 32.4, 42.3, 44.0, 51.1, 52.3, 52.4, 52.6, 55.2, 69.3, 73.5, 74.5, 80.6, 94.8, 103.9, 123.2, 123.9, 125.3, 130.5, 150.0, 160.7, 174.0; HRMS (FAB) calcd for C22H29N2O5 [M + H+] 401.2076, found 401.2077.
-4.9 (c 1.0, chloroform); IR (film) 3383, 2960, 1728, 1618, 1502, 1459, 1258, 1160, 1032, 752 cm-1; 1H HMR (400 MHz, CDCl3) δ 0.61 (3H, t, J = 7.4 Hz), 1.04–1.12 (1H, m), 1.34–1.42 (1H, m), 2.24–2.52 (5H, m), 2.82 (1H, dt, J = 1.5, 15.9 Hz), 3.35 (1H, td, J = 2.9, 18.1 Hz), 3.46 (1H, ddd, J = 1.5, 4.6, 15.9 Hz), 3.75 (3H, s), 3.83 (3H, s), 4.10 (1H, d, J = 2.9 Hz), 4.16 (1H, br s), 4.55 (1H, d, J = 2.4 Hz), 5.65 (1H, dt, J = 1.5, 9.7 Hz), 5.85 (1H, ddd, J = 1.5, 4.6, 9.7 Hz), 6.13 (1H, d, J = 2.2 Hz), 6.24 (1H, dd, J = 2.2, 8.2 Hz), 6.90 (1H, d, J = 8.2 Hz), 9.32 (1H, br s); 13C NMR (100 MHz, CDCl3) δ 7.8, 32.4, 42.3, 44.0, 51.1, 52.3, 52.4, 52.6, 55.2, 69.3, 73.5, 74.5, 80.6, 94.8, 103.9, 123.2, 123.9, 125.3, 130.5, 150.0, 160.7, 174.0; HRMS (FAB) calcd for C22H29N2O5 [M + H+] 401.2076, found 401.2077.
Demethylvindoline (12). Deacetyldemethylvindoline (11) (40.3 mg, 0.101 mmol) and sodium acetate (41.3 mg, 0.503 mmol) were dissolved in acetic anhydride (1.0 ml) at 0°C. After stirring for 12 h, the resulting suspension was diluted with dichloromethane, quenched with 15% aqueous ammonia, and diluted with water. After removal of the ice bath, the organic layer was separated. The aqueous layer was extracted with dichloromethane five times. The combined organic extracts were dried over anhydrous sodium sulfate, filtered, and concentrated in vacuo. The residue was purified with flash-column chromatography on neutral SiO2 (methanol/hexane/dichloromethane, 2:10:78) to give 39.2 mg (0.0886 mmol, 87.7%) of demethylvindoline (12) as a colorless solid. mp 101–103°C (ether); 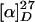 -36.6 (c 1.35, chloroform); IR (film) 3368, 2962, 1740, 1618, 1503, 1459, 1372, 1254, 1160, 1033, 753 cm-1; 1H NMR (400 MHz, CDCl3) δ 0.48 (3H, t, J = 7.3 Hz), 1.15–1.27 (1H, m), 1.49–1.61 (1H, m), 2.10 (3H, s), 2.29–2.53 (4H, m), 2.81 (1H, dt, J = 1.5, 16.1 Hz), 3.37 (1H, td, J = 3.2, 8.5 Hz), 3.51 (1H, ddd, J = 1.5, 4.6, 16.1 Hz), 3.75 (3H, s), 3.77 (3H, s), 4.14 (1H, d, J = 2.8 Hz), 4.58 (1H, d, J = 2.8 Hz), 5.27 (1H, dt, J = 1.5, 10.0 Hz), 5.56 (1H, s), 5.83 (1H, ddd, J = 1.5, 4.6, 10.0 Hz), 6.15 (1H, d, J = 2.2), 6.26 (1H, dd, J = 2.2, 8.3 Hz), 6.92 (1H, d, J = 8.3 Hz), 9.48 (1H, br s); 13C NMR (100 MHz, CDCl3) δ 7.8, 20.9, 31.4, 42.3, 43.5, 51.1, 52.4, 52.6 (2 carbons), 55.3, 68.3, 74.0, 76.4, 79.7, 95.0, 104.3, 123.1, 123.8, 125.2, 130.1, 150.1, 160.8, 170.6, 172.9; HRMS (FAB) calcd for C24H31N2O6 [M + H+] 443.2182, found 443.2182.
-36.6 (c 1.35, chloroform); IR (film) 3368, 2962, 1740, 1618, 1503, 1459, 1372, 1254, 1160, 1033, 753 cm-1; 1H NMR (400 MHz, CDCl3) δ 0.48 (3H, t, J = 7.3 Hz), 1.15–1.27 (1H, m), 1.49–1.61 (1H, m), 2.10 (3H, s), 2.29–2.53 (4H, m), 2.81 (1H, dt, J = 1.5, 16.1 Hz), 3.37 (1H, td, J = 3.2, 8.5 Hz), 3.51 (1H, ddd, J = 1.5, 4.6, 16.1 Hz), 3.75 (3H, s), 3.77 (3H, s), 4.14 (1H, d, J = 2.8 Hz), 4.58 (1H, d, J = 2.8 Hz), 5.27 (1H, dt, J = 1.5, 10.0 Hz), 5.56 (1H, s), 5.83 (1H, ddd, J = 1.5, 4.6, 10.0 Hz), 6.15 (1H, d, J = 2.2), 6.26 (1H, dd, J = 2.2, 8.3 Hz), 6.92 (1H, d, J = 8.3 Hz), 9.48 (1H, br s); 13C NMR (100 MHz, CDCl3) δ 7.8, 20.9, 31.4, 42.3, 43.5, 51.1, 52.4, 52.6 (2 carbons), 55.3, 68.3, 74.0, 76.4, 79.7, 95.0, 104.3, 123.1, 123.8, 125.2, 130.1, 150.1, 160.8, 170.6, 172.9; HRMS (FAB) calcd for C24H31N2O6 [M + H+] 443.2182, found 443.2182.
Coupling Reaction. To a solution of indoles 13 (85.0 mg, 0.105 mmol) in dichloromethane (0.70 ml) was added t-butyl hypochlorite (12.9 μl, 0.114 mmol) at 0°C and the reaction mixture was stirred at 0°C for 10 min. The excess amount of t-butyl hypochlorite was removed by passage through a silica gel column to afford chloroindolenines 5 as a yellow oil. The chloroindolenines (5) and demethylvindoline (12) (38.8 mg, 0.0877 mmol) was dissolved in dichloromethane (1.0 ml), and the solution was cooled to 0°C. To the solution was added trifluoroacetic acid (67 μl, 0.88 mmol), and the reaction mixture was stirred at room temperature for 20 min before it was quenched with saturated aqueous sodium bicarbonate. The organic layer was separated, and the aqueous layer was extracted with dichloromethane. Then, the organic extracts were combined, dried over anhydrous sodium sulfate, filtered, and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel (50–100% ethyl acetate-hexane) to afford 82.0 mg (0.0656 mmol, 74.8%) of 14 as a slightly yellow solid. 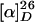 +35.4 (c 1.95, chloroform); IR (film) 3414, 2952, 1778, 1737, 1618, 1547, 1460, 1370, 1256, 1174, 749 cm-1; 1H NMR (400 MHz, CDCl3) δ 0.43 (3H, t, J = 7.3 Hz), 0.64 (3H, t, J = 7.6 Hz), 1.26 (3H, t, J = 7.1 Hz), 1.54 (2H, br s), 1.76–2.04 (4H, m), 2.11 (4H, br s), 2.41 (3H, s), 2.41–2.47 (2H, m), 2.60–2.75 (3H, m), 2.78–2.93 (2H, m), 2.99 (1H, d, J = 16.1 Hz), 3.13–3.34 (3H, m), 3.40 (3H, s), 3.45 (1H, m), 3.69 (3H, s), 3.59–3.66 (1H, m), 3.69 (3H, s), 3.74 (3H, s), 3.74–3.93 (3H, m), 4.19 (1H, d, J = 3.4 Hz), 4.74 (1H, d, J = 3.4 Hz), 5.42 (1H, d, J = 10.0 Hz), 5.69 (1H, s), 5.79 (1H, dd, J = 10.0, 3.2 Hz), 6.01 (1H, s), 6.94 (1H, t, J = 7.3 Hz), 7.10 (2H, m), 7.31–7.37 (3H, m), 7.52 (1H, s), 7.64 (1H, dd, J = 8.1, 1.2 Hz), 7.70 (1H, td, J = 7.7, 1.2 Hz), 7.75–7.83 (4H, m), 9.50 (1H, br s), 10.83 (1H, br s); 13C NMR (100 MHz, CDCl3) δ 6.9, 8.49, 20.9, 21.5, 25.0, 29.7, 30.6, 32.2, 37.6, 39.3, 41.7, 43.7, 50.2, 50.9, 52.5, 52.6, 52.7, 52.8, 54.9, 55.4, 59.8, 69.2, 72.8, 74.1, 75.7, 79.4, 93.5, 94.7, 108.0, 111.2, 113.6, 117.0, 118.8, 120.3, 121.3, 121.7, 123.7, 124.5, 125.3, 127.3, 128.0 (2 carbons), 129.1, 129.4, 129.8 (2 carbons), 131.1, 131.5, 132.3, 121.7, 134.5, 135.6, 144.9, 148.8, 149.4, 155.6, 158.0, 170.5, 173.4, 177.4; HRMS (FAB) calcd for C60H67F3N5O17S2 [M + H+] 1250.3926, found 1250.3992.
+35.4 (c 1.95, chloroform); IR (film) 3414, 2952, 1778, 1737, 1618, 1547, 1460, 1370, 1256, 1174, 749 cm-1; 1H NMR (400 MHz, CDCl3) δ 0.43 (3H, t, J = 7.3 Hz), 0.64 (3H, t, J = 7.6 Hz), 1.26 (3H, t, J = 7.1 Hz), 1.54 (2H, br s), 1.76–2.04 (4H, m), 2.11 (4H, br s), 2.41 (3H, s), 2.41–2.47 (2H, m), 2.60–2.75 (3H, m), 2.78–2.93 (2H, m), 2.99 (1H, d, J = 16.1 Hz), 3.13–3.34 (3H, m), 3.40 (3H, s), 3.45 (1H, m), 3.69 (3H, s), 3.59–3.66 (1H, m), 3.69 (3H, s), 3.74 (3H, s), 3.74–3.93 (3H, m), 4.19 (1H, d, J = 3.4 Hz), 4.74 (1H, d, J = 3.4 Hz), 5.42 (1H, d, J = 10.0 Hz), 5.69 (1H, s), 5.79 (1H, dd, J = 10.0, 3.2 Hz), 6.01 (1H, s), 6.94 (1H, t, J = 7.3 Hz), 7.10 (2H, m), 7.31–7.37 (3H, m), 7.52 (1H, s), 7.64 (1H, dd, J = 8.1, 1.2 Hz), 7.70 (1H, td, J = 7.7, 1.2 Hz), 7.75–7.83 (4H, m), 9.50 (1H, br s), 10.83 (1H, br s); 13C NMR (100 MHz, CDCl3) δ 6.9, 8.49, 20.9, 21.5, 25.0, 29.7, 30.6, 32.2, 37.6, 39.3, 41.7, 43.7, 50.2, 50.9, 52.5, 52.6, 52.7, 52.8, 54.9, 55.4, 59.8, 69.2, 72.8, 74.1, 75.7, 79.4, 93.5, 94.7, 108.0, 111.2, 113.6, 117.0, 118.8, 120.3, 121.3, 121.7, 123.7, 124.5, 125.3, 127.3, 128.0 (2 carbons), 129.1, 129.4, 129.8 (2 carbons), 131.1, 131.5, 132.3, 121.7, 134.5, 135.6, 144.9, 148.8, 149.4, 155.6, 158.0, 170.5, 173.4, 177.4; HRMS (FAB) calcd for C60H67F3N5O17S2 [M + H+] 1250.3926, found 1250.3992.
Alcohol (15). To a solution of trifluoroacetate 14 (13.3 mg, 0.0106 mmol) in methanol (0.50 ml) was added triethylamine (27 μl) at room temperature. The reaction mixture was stirred at room temperature for 40 min before it was evaporated under reduced pressure to afford alcohol 15 (12.1 mg, 0.0105 mmol, 98.5%) as a pale yellow solid. 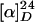 +52.5 (c 1.81, chloroform); IR (film) 3412, 2951, 1736, 1618, 1546, 1459, 1356, 1257, 1175, 748 cm-1; 1H NMR (400 MHz, CDCl3) δ 0.54 (3H, t, J = 7.2 Hz), 0.68 (3H, t, J = 7.3 Hz), 0.97 (1H, m), 1.07 (1H, m), 1.26 (1H, br s), 1.33 (1H, m), 1.42 (1H, d, J = 14.9 Hz), 1.53 (1H, m), 1.95 (1H, m), 2.06–2.10 (1H, m), 2.12 (3H, s), 2.44 (3H, s), 2.44 (1H, m), 2.46–2.56 (2H, m), 2.61–2.72 (2H, m), 2.77–2.90 (3H, m), 3.25–3.37 (2H, m), 3.39 (3H, s), 3.43–3.48 (1H, m), 3.65 (1H, dd, J = 9.2, 2.4 Hz), 3.74 (4H, s), 3.72 (1H, m), 3.98 (1H, dd, J = 14.6, 5.1 Hz), 4.18 (1H, d, J = 3.0 Hz), 4.74 (1H, d, J = 3.0 Hz), 5.45 (1H, d, J = 10.5 Hz), 5.72 (1H, s), 5.81 (1H, dd, J = 10.2, 4.2 Hz), 5.97 (1H, s), 7.01 (1H, t, J = 7.5 Hz), 7.14–7.20 (2H, m), 7.34 (1H, d, J = 8.0 Hz), 7.42 (1H, d, J = 8.3 Hz), 7.59 (1H, s), 7.64–7.69 (2H, m), 7.73–7.82 (4H, m), 11.2 (1H, br s); 13C NMR (100 MHz, CDCl3) δ 6.8, 8.6, 21.0, 21.6, 26.5, 28.9, 32.3, 35.7, 38.4, 40.1, 41.9, 43.8, 50.1, 50.7, 52.2, 52.5, 52.6, 54.2, 55.4, 64.6, 68.6, 73.3 (2 carbons), 74.1, 74.3, 75.9, 79.5, 93.1, 106.6, 111.7, 117.2, 119.5, 121.1, 121.7, 122.1, 123.6, 124.8, 125.3, 127.4, 128.0 (2 carbons), 129.5, 129.8 (2 carbons), 130.4, 131.0, 131.1, 132.8, 133.8, 134.1, 138.1, 144.7, 148.9, 149.4, 157.3, 170.6, 173.5, 176.7; HRMS (FAB) calcd for C58H68N5O16S2 [M + H+] 1154.4102, found 1154.3998.
+52.5 (c 1.81, chloroform); IR (film) 3412, 2951, 1736, 1618, 1546, 1459, 1356, 1257, 1175, 748 cm-1; 1H NMR (400 MHz, CDCl3) δ 0.54 (3H, t, J = 7.2 Hz), 0.68 (3H, t, J = 7.3 Hz), 0.97 (1H, m), 1.07 (1H, m), 1.26 (1H, br s), 1.33 (1H, m), 1.42 (1H, d, J = 14.9 Hz), 1.53 (1H, m), 1.95 (1H, m), 2.06–2.10 (1H, m), 2.12 (3H, s), 2.44 (3H, s), 2.44 (1H, m), 2.46–2.56 (2H, m), 2.61–2.72 (2H, m), 2.77–2.90 (3H, m), 3.25–3.37 (2H, m), 3.39 (3H, s), 3.43–3.48 (1H, m), 3.65 (1H, dd, J = 9.2, 2.4 Hz), 3.74 (4H, s), 3.72 (1H, m), 3.98 (1H, dd, J = 14.6, 5.1 Hz), 4.18 (1H, d, J = 3.0 Hz), 4.74 (1H, d, J = 3.0 Hz), 5.45 (1H, d, J = 10.5 Hz), 5.72 (1H, s), 5.81 (1H, dd, J = 10.2, 4.2 Hz), 5.97 (1H, s), 7.01 (1H, t, J = 7.5 Hz), 7.14–7.20 (2H, m), 7.34 (1H, d, J = 8.0 Hz), 7.42 (1H, d, J = 8.3 Hz), 7.59 (1H, s), 7.64–7.69 (2H, m), 7.73–7.82 (4H, m), 11.2 (1H, br s); 13C NMR (100 MHz, CDCl3) δ 6.8, 8.6, 21.0, 21.6, 26.5, 28.9, 32.3, 35.7, 38.4, 40.1, 41.9, 43.8, 50.1, 50.7, 52.2, 52.5, 52.6, 54.2, 55.4, 64.6, 68.6, 73.3 (2 carbons), 74.1, 74.3, 75.9, 79.5, 93.1, 106.6, 111.7, 117.2, 119.5, 121.1, 121.7, 122.1, 123.6, 124.8, 125.3, 127.4, 128.0 (2 carbons), 129.5, 129.8 (2 carbons), 130.4, 131.0, 131.1, 132.8, 133.8, 134.1, 138.1, 144.7, 148.9, 149.4, 157.3, 170.6, 173.5, 176.7; HRMS (FAB) calcd for C58H68N5O16S2 [M + H+] 1154.4102, found 1154.3998.
Deprotection of o-Nitrobenzenesulfonyl Group. To a solution of alcohol 15 (13.3 mg, 11.5 μmol) and 1,8-diazabicyclo[5.4.0]undec-7-ene (5.1 μl, 35 μmol) in acetone (0.50 ml) was added a solution of 2-mercaptoethanol (12 μmol) in acetone (80 μl) at room temperature. An additional five portions of 2-mercaptoethanol (2.3 μmol in 16 μl of acetone) were added during the period of 150 min. Then, the reaction mixture was concentrated under reduced pressure and purified with preparative TLC (7.5% methanol in dichloromethane) to afford the secondary amine (6.8 mg, 7.0 μmol, 61%) as a white amorphous solid. The unstable secondary amine thus obtained was used immediately for the next step.
(+)-Demethylvinblastine (16). A solution of the secondary amine (6.8 mg, 7.0 μmol) and sodium bicarbonate (20 mg) in isopropyl alcohol/water (1:1, 2.0 ml) was stirred for 20 h before it was partitioned between dichloromethane and brine. The aqueous layer was thoroughly extracted with dichloromethane. The combined organic layers were dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure. The residue was purified by preparative TLC (10% methanol in dichloromethane) to give 3.2 mg (4.0 μmol, 57%) of (+)-demethylvinblastine (16) as a white solid. 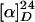 +10 (c 0.73, chloroform); IR (film) 3470, 2925, 2853, 1739, 1618, 1499, 1460, 1257, 1038, 744 cm-1; 1H NMR (400 MHz, CDCl3) δ 0.81 (3H, t, J = 7.3 Hz), 0.90 (3H, t, J = 7.4 Hz), 1.24–1.50 (7H, m), 1.60–1.73 (1H, m), 1.91–1.99 (1H, m), 2.13 (3H, s), 2.15–2.32 (2H, m), 2.39–2.48 (2H, m), 2.54 (1H, s), 2.78–2.84 (3H, m), 3.14 (1H, m), 3.22–3.45 (4H, m), 3.61 (3H, s), 3.75 (3H, s), 3.78 (3H, s), 3.95 (1H, t, J = 14.3 Hz), 4.13 (1H, d, J = 3.0 Hz), 4.60 (1H, d, J = 3.0 Hz), 5.31 (1H, d, J = 10.2 Hz), 5.51 (1H, s), 5.85 (1H, dd, J = 10.3, 4.6 Hz), 6.22 (1H, s), 6.65 (1H, s), 7.08–7.18 (3H, m), 7.52 (1H, d, J = 7.6 Hz), 8.02 (1H, br s), 9.85 (1H, br s); 13C NMR (100 MHz, CDCl3) δ 7.0, 8.4, 21.2, 28.8, 30.4, 31.2, 34.4, 34.5, 41.6, 43.0, 43.5, 48.3, 50.6, 51.1, 52.5, 52.8, 53.3, 55.8, 55.8, 55.9, 64.4, 66.7, 69.8, 74.2, 76.6, 79.8, 94.0, 110.4, 116.9, 118.6, 118.9, 120.8, 122.3, 122.3, 122.8, 124.2, 124.7, 129.5, 129.9, 131.7, 135.1, 149.2, 158.0, 171.0, 172.8, 175.1; HRMS (FAB) calcd for C45H57N4O9 [M + H+] 797.4125, found 797.4201.
+10 (c 0.73, chloroform); IR (film) 3470, 2925, 2853, 1739, 1618, 1499, 1460, 1257, 1038, 744 cm-1; 1H NMR (400 MHz, CDCl3) δ 0.81 (3H, t, J = 7.3 Hz), 0.90 (3H, t, J = 7.4 Hz), 1.24–1.50 (7H, m), 1.60–1.73 (1H, m), 1.91–1.99 (1H, m), 2.13 (3H, s), 2.15–2.32 (2H, m), 2.39–2.48 (2H, m), 2.54 (1H, s), 2.78–2.84 (3H, m), 3.14 (1H, m), 3.22–3.45 (4H, m), 3.61 (3H, s), 3.75 (3H, s), 3.78 (3H, s), 3.95 (1H, t, J = 14.3 Hz), 4.13 (1H, d, J = 3.0 Hz), 4.60 (1H, d, J = 3.0 Hz), 5.31 (1H, d, J = 10.2 Hz), 5.51 (1H, s), 5.85 (1H, dd, J = 10.3, 4.6 Hz), 6.22 (1H, s), 6.65 (1H, s), 7.08–7.18 (3H, m), 7.52 (1H, d, J = 7.6 Hz), 8.02 (1H, br s), 9.85 (1H, br s); 13C NMR (100 MHz, CDCl3) δ 7.0, 8.4, 21.2, 28.8, 30.4, 31.2, 34.4, 34.5, 41.6, 43.0, 43.5, 48.3, 50.6, 51.1, 52.5, 52.8, 53.3, 55.8, 55.8, 55.9, 64.4, 66.7, 69.8, 74.2, 76.6, 79.8, 94.0, 110.4, 116.9, 118.6, 118.9, 120.8, 122.3, 122.3, 122.8, 124.2, 124.7, 129.5, 129.9, 131.7, 135.1, 149.2, 158.0, 171.0, 172.8, 175.1; HRMS (FAB) calcd for C45H57N4O9 [M + H+] 797.4125, found 797.4201.
(+)-Vincristine (2). A mixture of acetic anhydride and formic acid (11:5, vol/vol) was heated at 50°C for 1 h and cooled to room temperature. (+)-Demethylvinblastine (16) (3.0 mg, 3.8 μmol) was dissolved in 0.30 ml of this acidic mixture at room temperature and the resulting mixture was stirred for 1.5 h. Then, the mixture was diluted with dichloromethane and quenched with 20% aqueous ammonia. After removal of the organic layer, the aqueous layer was thoroughly extracted with dichloromethane. The combined organic extracts were dried over anhydrous sodium sulfate, filtered, and concentrated in vacuo. The residue was purified with preparative TLC (30% methanol in diethyl ether) to give (+)-vincristine (2) (2.7 mg, 3.3 μmol, 87%) as a white solid. This material was obtained as a mixture of two rotamers. 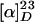 +45 (c 0.89, chloroform) for synthetic vincristine;
+45 (c 0.89, chloroform) for synthetic vincristine; 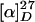 +54 (c 0.95, chloroform) for natural vincristine; IR (film) 3445, 2924, 2852, 1740, 1681, 1597, 1498, 1458, 1369, 1230, 1033, 747 cm-1; 1H NMR (400 MHz, CDCl3) δ 0.59 (1H, m), 0.82–0.94 (6H, m), 1.20–1.48 (6H, m), 1.60–1.76 (2H, m), 2.06 ((3/5)3H, s), 2.10 ((2/5)3H, s), 2.17 (1H, s), 2.36 (2H, m), 2.60 (1H, m), 2.79 (2H, br s), 2.87–2.93 (2H, m), 3.15 (2H, m), 3.28 (1H, d, J = 14.5 Hz), 3.32–3.40 (2H, m), 3.67 (3H, s), 3.72 ((3/5)3H, s), 3.79 ((2/5)3H, s), 3.88 ((3/5)3H, s), 3.89 ((2/5)3H, s), 4.01 (1H, m), 4.51 ((2/5)H, s), 4.74 ((3/5)H, s), 5.23 (1H, d, J = 14.5 Hz), 5.41 (1H, d, J = 10.5 Hz), 5.92 (1H, m), 6.79 ((3/5)H, s), 6.88 ((2/5)H, s), 6.93 (1H, s), 7.10–7.21 (3H, m), 7.55 (1H, d, J = 7.8 Hz), 7.74 ((2/5)H, s), 8.05 ((3/5)H, s), 8.16 ((2/5)H, s), 8.76 ((3/5)H, s), 9.40 ((3/5)H, br s), 9.57 ((2/5)H, br s); HRMS (FAB) calcd for C46H57N4O10 [M + H+] 825.4074, found 825.4110.
+54 (c 0.95, chloroform) for natural vincristine; IR (film) 3445, 2924, 2852, 1740, 1681, 1597, 1498, 1458, 1369, 1230, 1033, 747 cm-1; 1H NMR (400 MHz, CDCl3) δ 0.59 (1H, m), 0.82–0.94 (6H, m), 1.20–1.48 (6H, m), 1.60–1.76 (2H, m), 2.06 ((3/5)3H, s), 2.10 ((2/5)3H, s), 2.17 (1H, s), 2.36 (2H, m), 2.60 (1H, m), 2.79 (2H, br s), 2.87–2.93 (2H, m), 3.15 (2H, m), 3.28 (1H, d, J = 14.5 Hz), 3.32–3.40 (2H, m), 3.67 (3H, s), 3.72 ((3/5)3H, s), 3.79 ((2/5)3H, s), 3.88 ((3/5)3H, s), 3.89 ((2/5)3H, s), 4.01 (1H, m), 4.51 ((2/5)H, s), 4.74 ((3/5)H, s), 5.23 (1H, d, J = 14.5 Hz), 5.41 (1H, d, J = 10.5 Hz), 5.92 (1H, m), 6.79 ((3/5)H, s), 6.88 ((2/5)H, s), 6.93 (1H, s), 7.10–7.21 (3H, m), 7.55 (1H, d, J = 7.8 Hz), 7.74 ((2/5)H, s), 8.05 ((3/5)H, s), 8.16 ((2/5)H, s), 8.76 ((3/5)H, s), 9.40 ((3/5)H, br s), 9.57 ((2/5)H, br s); HRMS (FAB) calcd for C46H57N4O10 [M + H+] 825.4074, found 825.4110.
Results and Discussion
Synthetic Plan. The synthetic plan of vincristine (2), based on our total synthesis of vinblastine (1) (23), is depicted in Fig. 2. To construct the stereochemistry of C18′, we planned to exploit the coupling reaction using the chloroindolenine intermediate 5 bearing an eleven-membered ring. Conformational preference of the eleven-membered ring of the active species such as 8 would be a key factor to construct the desired stereochemistry. Thus, semiempirical calculations (mopac pm3) of a model compound 8 showed that lowest-energy conformation of the eleven-membered ring possessed a chair-like shape (Fig. 3). Therefore, nucleophilic attack of the vindoline derivative 6 would occur from the sterically less hindered α-face to construct the desired stereochemistry. This conformational analysis was also supported by a model reaction using the simplified upper unit with natural vindoline as reported by Schill and coworkers (25). After the stereoselective introduction of the vindoline moiety, vincristine would be obtained by the formation of piperidine ring in the upper indole unit. The desired demethylvindoline segment 6, on the other hand, would be synthesized by a modification of our efficient synthesis of vindoline (24).
Fig. 2.

Retrosynthetic analysis of vincristine.
Fig. 3.

Conformation of the eleven-membered ring (MOPAC PM3).
Synthesis of Demethylvindoline Formamide (6). 11-Methoxytabersonine (7) was synthesized from 7-hydroxyquinoline (9) (26) through our 2,3-disubstituted indole synthesis by radical cyclization of 2-alkenylthioanilide (27) as a key process (Fig. 4) (23, 24). After oxidation of allylic position with benzene seleninic anhydride (10), further oxidation of enamine moiety of 10 was extensively investigated. To this end, deacetyldemethylvindoline (11) was successfully obtained by treatment with m-chloroperbenzoic acid under buffered conditions (10, 13, 24), followed by reduction of the resultant imine with sodium cyanoborohydride. To perform the subsequent selective acetylation and formylation, a careful choice of reaction conditions was necessary. Thus, on treatment of the diol 11 with acetic anhydride in the presence of sodium acetate at 0°C (7), regioselective acetylation took place to give demethylvindoline (12) exclusively. Finally, the corresponding formamide 6 was obtained by a mixed anhydride method with formic acid and acetic anhydride.
Fig. 4.

Synthesis of demethylvindoline formamide.
Crucial Coupling Reaction and Total Synthesis of (+)-Vincristine (2). With the requisite demethylvindoline formamide (6) in hand, we next examined the crucial coupling reaction of the two segments. The indole segment 13 bearing an eleven-membered ring, a precursor of chloroindolenine species, was efficiently synthesized from quinoline according to our reported sequence (23). After chlorination of compound 13 with tert-butyl hypochlorite, the resultant chloroindolenine 5 and demethylvindoline formamide (6) were dissolved in dichloromethane and treated with trifluoroacetic acid (Fig. 5). To our disappointment, the desired coupling product could not be obtained, and the starting compounds were recovered. At this point, we considered that this failure of the coupling reaction might be due to the lower nucleophilicity of demethylvindoline formamide (6) than that of vindoline (3), and therefore decided to perform the coupling reaction with demethylvindoline (12), which should possess higher nucleophilicity than the corresponding formamide. As expected, the chloroindolenine 5 and 12 were treated with trifluoroacetic acid to give coupling product 14 in 75% yield as a sole isomer (Fig. 6). Subsequent selective methanolysis of the trifluoroacetate 14 gave tertiary alcohol 15, which was treated with 2-mercaptoethanol and 1,8-diazabicyclo[5.4.0]undec-7-ene in acetone to remove 2-nitrobenzenesulfonyl group (28–30) to yield the corresponding secondary amine. The piperidine ring of the upper part was constructed by intramolecular SN2 reaction to furnish demethylvinblastine (16). Finally, formylation of N1 position according to the procedure described (19) afforded (+)-vincristine (2). The spectral data of the synthetic samples were identical with those of natural vincristine.
Fig. 5.

Unsuccessful coupling reaction. TFA, trifluoroacetic acid or trifluoroacetyl.
Fig. 6.

Total synthesis of (+)-vincristine (2). TFA, trifluoroacetic acid or trifluoroacetyl; DBU, 1,8-diazabicyclo[5.4.0]undec-7-ene.
Conclusions
We have accomplished a total synthesis of (+)-vincristine (2) through an efficient synthesis of both indole segments and their stereoselective coupling. The success of the crucial coupling reaction depended not only on the appropriate conformation of the upper eleven-membered carbomethoxyverbanamine precursor, but also on the fine tuning of the nucleophilic nature of the lower vindoline derivative. We believe that the present synthetic pathway would be useful for the development of an efficient anticancer agent, because it could be applied to the synthesis of a range of vincristine analogues, including those not accessible by chemical modification of natural compounds.
Acknowledgments
We thank Drs. Michael J. Martinelli and Kevin Lydon (Eli Lilly & Co.) for providing samples of natural vincristine, Dr. Yoshihiko Hirose (Amano Pharmaceutical) for providing lipase PS, and Dr. Yoichi Nakao and Mr. Masaki Fujita (University of Tokyo) for mass spectrometric analysis. This work was financially supported in part by Core Research for Evolution Science and Technology, Japan Science and Technology Agency, the Mitsubishi Foundation, and the Uehara Memorial Foundation.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: HRMS, high-resolution mass spectra; FAB, fast atom bombardment.
References
- 1.Noble, R. L., Beer, C. T. & Cutts, J. H. (1958) Ann. N. Y. Acad. Sci. 76, 882-894. [DOI] [PubMed] [Google Scholar]
- 2.Svoboda, G. H., Neuss, N. & Gorman, M. (1959) J. Am. Pharm. Assoc. Sci. Ed. 48, 659-666. [DOI] [PubMed] [Google Scholar]
- 3.Moncrief, J. W. & Lipscomb, W. N. (1965) J. Am. Chem. Soc. 87, 4963-4964. [DOI] [PubMed] [Google Scholar]
- 4.Neuss, N., Gorman, M., Boaz, H. E. & Cone, N. J. (1962) J. Am. Chem. Soc. 84, 1509-1510. [Google Scholar]
- 5.Svoboda, G. H. (1961) Lloydia 24, 173-178. [Google Scholar]
- 6.Brossi, A. & Suffness, M., eds. (1990) Antitumor Bisindole Alkaloids from Catharanthus roseus (L.) (Academic, San Diego), Vol. 37.
- 7.Ando, M., Büchi, G. & Ohnuma, T. (1975) J. Am. Chem. Soc. 97, 6880-6881. [Google Scholar]
- 8.Ban, Y., Sekine, Y. & Oishi, T. (1978) Tetrahedron Lett., 151-154.
- 9.Kutney, J. P., B.-Trepp, U., Chan, K. K., de Souza, J. P., Fujise, Y., Honda, T., Katsube, J., Klein, F. K., Leutwiler, A., Morehead, S., et al. (1978) J. Am. Chem. Soc. 100, 4220-4224. [Google Scholar]
- 10.Danieli, B., Lesma, G., Palmisano, G. & Riba, R. (1984) J. Chem. Soc. Chem. Commun., 909-911.
- 11.Andriamialisoa, R. Z., Langlois, N. & Langlois, Y. (1985) J. Org. Chem. 50, 961-967. [DOI] [PubMed] [Google Scholar]
- 12.Feldman, P. L. & Rapoport, H. (1987) J. Am. Chem. Soc. 109, 1603-1604. [Google Scholar]
- 13.Kuehne, M. E., Podhorez, D. E., Mulamba, T. & Bornmann, W. G. (1987) J. Org. Chem. 52, 347-353. [Google Scholar]
- 14.Kutney, J. P., Beck, J., Bylsma, F., Cook, J., Cretney, W. J., Fuji, K., Imhof, R. & Treasurywala, A. M. (1975) Helv. Chim. Acta 58, 1690-1719. [DOI] [PubMed] [Google Scholar]
- 15.Mangeney, P., Andriamialisoa, R. Z., Langlois, N., Langlois, Y. & Potier, P. (1979) J. Am. Chem. Soc. 101, 2243-2245. [DOI] [PubMed] [Google Scholar]
- 16.Kutney, J. P., Choi, L. S. L., Nakano, J., Tsukamoto, H., McHugh, M. & Boulet, C. A. (1988) Heterocycles 27, 1845-1856. [Google Scholar]
- 17.Kuehne, M. E., Matson, P. A. & Bornmann, W. G. (1991) J. Org. Chem. 56, 513-528. [Google Scholar]
- 18.Magnus, P., Mendoza, J. S., Stamford, A., Ladlow, M. & Willis, P. (1992) J. Am. Chem. Soc. 114, 10232-10245. [Google Scholar]
- 19.Jovanovics, K., Szasz, K., Fekete, G., Bittner, E., Dezseri, E. & Elex, J. (1975) U.S. Patent 3,899,493–750,812; (1975) Chem. Abstr. 83, 179360. [Google Scholar]
- 20.Conrad, R. A. (1982) Eur. Patent 37,290 A1 811,007; (1982) Chem. Abstr. 96, 104583. [Google Scholar]
- 21.Pearce, H. L. (1982) Eur. Patent 37,289 A1 811,007; (1982) Chem. Abstr. 96, 85834. [Google Scholar]
- 22.Brannon, D. R. & Neuss, N. (1975) Ger. Patent 2,440,931; (1975) Chem. Abstr. 83, 7184. [Google Scholar]
- 23.Yokoshima, S., Ueda, T., Kobayashi, S., Sato, A., Kuboyama, T., Tokuyama, H. & Fukuyama, T. (2002) J. Am. Chem. Soc. 124, 2137-2139. [DOI] [PubMed] [Google Scholar]
- 24.Kobayashi, S., Ueda, T. & Fukuyama, T. (2000) Synlett, 883-886.
- 25.Schill, G., Priester, C. U., Windhövel, U. F. & Fritz, H. (1987) Tetrahedron 43, 3765-3786. [Google Scholar]
- 26.Tokuyama, H., Sato, M., Ueda, T. & Fukuyama, T. (2001) Heterocycles 54, 105-108. [Google Scholar]
- 27.Tokuyama, H., Yamashita, T., Reding, M. T., Kaburagi, Y. & Fukuyama, T. (1999) J. Am. Chem. Soc. 121, 3791-3792. [Google Scholar]
- 28.Fukuyama, T., Jow, C.-K. & Cheung, M. (1995) Tetrahedron Lett. 36, 6373-6375. [Google Scholar]
- 29.Fukuyama, T., Cheung, M., Jow, C.-K., Hidai, Y. & Kan, T. (1997) Tetrahedron Lett. 38, 5831-5834. [Google Scholar]
- 30.Kan, T. & Fukuyama, T. (2001) J. Syn. Org. Chem. Jpn. 59, 779-789. [Google Scholar]