

Abstract
The syntheses of isoroquefortine C and a related heterocycle were achieved by implementation of both intra- and intermolecular vinyl amidation reactions. These accomplishments represent a significant advance in the use of these strategies in the generation of complex molecules.
Metabolites produced by fungi represent one of the largest classes of natural products. Such compounds vary widely in their structural composition and have found application in pharmaceuticals such as antibiotics, immunosuppressants, antifungal agents, and growth promoters (1). Other congeners within this class display biological properties harmful to humans and other animals. These natural products have been classified as mycotoxins and are produced as secondary metabolites by an array of soilborne and airborne fungi (2). Mycotoxins have been isolated as contaminants of a variety of grain products and have been a topic of great interest for scientists concerned with veterinary health and agricultural safety.
In most cases, eradication of the fungal infection responsible for mycotoxin production is the most reasonable method for elimination of these compounds from commercial foodstuff. However, an interesting exception to this generalization is found in the roquefortine family of natural products (Fig. 1). Roquefortine C (1) was isolated independently by researchers in Japan (3, 4) and France (5, 6) from the Penicillium roqueforti Thom strain. This finding was significant due to the fact that this fungus is essential for the production of Roquefort and a number of other blue-veined cheeses. In subsequent communications, roquefortines have been found as metabolites of additional P. roqueforti strains as well as other Penicillium species isolated from a variety of contaminated food products such as feed grain (7, 8), wine (9), and beer (10). The presumed biosynthetic precursor of roquefortine C (1), the dihydro compound roquefortine D (2), has also been isolated from cultures of the P. roqueforti fungal species (11).
Fig. 1.

The roquefortine class of natural products.
Roquefortine C has received attention because of its neurotoxic properties. Wagener and coworkers (12) described paralytic activities in cockerels, and Frayssinet and Frayssinet found the LD50 in mice to be 15–20 mg/kg after i.p. injection (5). Symptoms included prostration, seizures, and death. Support for these findings was provided by Ohmomo (13), who used a similar mouse assay and reported an LD50 of 20 mg/kg. However, Arnold et al. (14) were unable to reproduce this activity and found the lethal dose to be an order of magnitude larger than this value. A nonalkaloid natural product also produced by P. roqueforti, PR toxin, was found to possess greater toxicity.
A subsequent publication by Häggblom (8) further complicated the biological activity profile of roquefortine C. In this finding, roquefortine C, but not PR toxin or other mycotoxins, was isolated from a grain sample that produced paralytic symptoms in cows. These effects disappeared as soon as the cows were no longer fed moldy grain. Another report investigated the effects of this metabolite on bacterial growth and showed it to possess inhibitory properties toward Gram-positive bacteria containing hemins but no potency toward Gram-negative organisms (15). Additional studies led to the finding that roquefortine C inhibited bacterial RNA synthesis but only modestly affected DNA and protein production (16).
The lack of consistent toxicological data, along with the ubiquitous nature of P. roqueforti in human and animal food products, establishes roquefortine C as a natural product worthy of synthetic investigation. The compound is also interesting from a synthetic chemistry standpoint, because it possesses distinctive structural characteristics. Roquefortine C contains the unusual E-dehydrohistidine moiety, a system that typically undergoes facile isomerization under acidic, basic, or photochemical conditions (17–21). This functional group is found in only two natural products, the other being oxaline (22). The total syntheses of these compounds have yet to be accomplished.
Isoroquefortine C (3), or the 3,12 double-bond isomer of roquefortine C (Fig. 2), was obtained by photochemical irradiation of the natural product (23). The biological activity of isoroquefortine C has yet to be investigated. The goals of the current synthetic efforts have been to develop a strategy applicable to both roquefortine C and isoroquefortine C. A successful route to either of these compounds would allow for investigation of their interconversion and stability.
Fig. 2.

Photochemical isomerization of roquefortine C to isoroquefortine C.
We previously reported the synthesis of roquefortine D (24) as well as the generation of isoroquefortine C using Wittig–Horner olefination to establish the enamide stereochemistry (25). This latter method was not applicable to the synthesis of roquefortine C. We now disclose recent efforts toward generation of the dehydrohistidine system using copper-catalyzed vinyl amidation chemistry. This method has allowed for the synthesis of both isoroquefortine C and a polycyclic heterocycle (26). Studies on the stability of isoroquefortine C and efforts to accomplish isomerization to the natural product are described also.
Due to the questionable stability of the dehydrohistidine moiety in the roquefortine system, palladium- or coppercatalyzed vinyl amidation offered great promise (Fig. 3). The transformation proceeds with stereospecificity and under relatively mild conditions (27, 28). A large number of catalyst systems have been reported for the analogous nitrogen—carbon bond-forming reactions involving amines or amides with aryl halides or triflates. In contrast, the use of vinyl systems with amines (29, 30) or amides (31, 32) has only recently received attention. Therefore, investigation offered the potential for significant extension of this methodology.
Fig. 3.

Retrosynthetic analysis of roquefortine C involving intramolecular vinyl amidation.
Materials and Methods
Experimental data for compounds 5–9, 11–14, 16, 17, 19, 20, and 23–26 are available in Supporting Materials and Methods, which is published as supporting information on the PNAS web site.
Trityl-hexahydropyrroloindoleimidazolidinone (21). Vinyl bromide 20 (0.175 g, 0.238 mmol), cuprous iodide (0.0045 g, 0.024 mmol), and finely powdered potassium carbonate (0.066 g, 0.476 mmol) were added to a thick-walled pressure tube, and the vessel was fitted with a rubber septum. The tube was evacuated and back-filled with argon three times. Dioxane (2 ml) and N, N′-dimethylethylenediamine (0.0052 ml, 0.048 mmol) were added, and the reaction mixture was flushed with argon and sealed by replacement of the septum with a Teflon screw cap. The reaction was heated to 100°C for 14 h and then cooled to room temperature, diluted with ethyl acetate (10 ml), and filtered through a plug of silica. The silica plug was flushed with additional ethyl acetate (10 ml), and the filtrate then was concentrated under reduced pressure. The crude product was purified by column chromatography (1–2% methanol/methylene chloride) to yield 21 as a white foam (0.111 g, 74%). Rf = 0.48 (5% methanol/methylene chloride); 1H NMR (500 MHz, CDCl3) δ 0.90 (s, 3H), 1.10 (s, 3H), 2.59 (dd, J = 15.0, 9.6 Hz, 1H), 31.4 (dd, J = 14.0, 6.6 Hz, 1H), 4.18 (dd, J = 9.6, 6.8 Hz, 1H), 5.09 (d, J = 17.3 Hz, 1H), 5.16 (d, J = 10.7 Hz, 1H), 5.39 (s, 1H), 5.47 (broad s, 2H), 6.01 (dd, J = 17.3, 10.7 Hz, 1H), 6.83 (broad s, 1H), 7.00–7.05 (m, 2H), 7.12–7.40 (m, 17H), 7.52 (s, 1H), 8.21 (s, 1H); 13C NMR (125 MHz, CDCl3) δ 23.0, 23.1, 39.1, 41.0, 59.1, 62.3, 76.2, 86.8, 114.7, 114.9, 123.5, 125.5, 125.6, 128.2, 128.3, 129.2, 129.7, 133.4, 134.3, 138.4, 141.9, 143.3, 151.8, 169.5, 172.9; IR (neat) 3,331 (w, broad), 3,170 (w, broad), 2,966 (w), 1,690 (s), 1,661 (m), 1,595 (w), 1,472 (m), 1,449 (m), 1,384 (m) cm-1; high-resolution MS (electrospray) m/z calculated for C41H37N5O2Na (M + Na)+: 654.284495; found: 654.283253;  (c 0.55, CHCl3).
(c 0.55, CHCl3).
Hexahydropyrroloindoleimidazolidinone (22). A solution of trityl-protected imidazole 21 (0.020 g, 0.032 mmol) and hydroxybenzotriazole (0.0129 g, 0.095 mmol) in trifluoroethanol (0.5 ml) was stirred at room temperature for 48 h. The solution was diluted with water and extracted with ethyl acetate (10 ml). The organic layer was dried over sodium sulfate and concentrated under reduced pressure. The crude product was purified by column chromatography (80% acetone/hexanes) to yield 22 as a white foam (0.0099 g, 80%). Rf = 0.22 (10% methanol/methylene chloride); 1H NMR (500 MHz, CDCl3) δ 1.01 (s, 3H), 1.13 (s, 3H), 2.64 (s, 1H), 2.69 (dd, J = 14.0, 9.6 Hz, 1H), 3.25 (dd, J = 14.0, 6.4 Hz, 1H), 4.32 (dd, J = 9.6, 6.4 Hz, 1H), 5.11 (dd, J = 17.6, 1.1 Hz, 1H), 5.19 (dd, J = 11.0, 1.1 Hz, 1H), 5.47 (s., 1H), 5.52 (broad s, 1H), 6.02 (dd, J = 17.6, 11.0 Hz, 1H), 6.79 (broad s, 1H), 6.94 (d, J = 7.7 Hz, 1H), 6.99 (s, 1H), 7.09 (dt, J = 7.7, 1.0 Hz, 1H), 7.26 (m, 1H), 7.29 (m, 1H), 7.34 and 7.43 (m, 1H, conformers), 7.69 and 7.94 (m, 1H, conformers); 13C NMR (125 MHz, CDCl3) δ 23.0, 23.1, 39.2, 41.0, 58.9, 62.2, 87.6, 114.8, 115.0, 116.8, 124.0, 125.8, 125.9, 129.4, 134.1, 134.4, 136.6, 137.2, 143.1, 151.7, 170.4, 171.9; IR (neat) 3,203 (m, broad), 2,956 (m), 2,923 (m), 1,693 (s), 1,682 (s), 1,642 (s), 1,593 (m), 1,392 (s) cm-1; high-resolution MS (electrospray) m/z calculated for C22H24N5O2 (M + H)+: 390.193000; found: 390.193970;  (c 0.56, CHCl3).
(c 0.56, CHCl3).
(2S,3aR,8aS)-10b-(1,1-Dimethylallyl)-3(Z)-(1H-imidazol-4-ylmethylene)-2,3,6,10b,11,11a-hexahydro-5aH-pyrazino[1′,2′:1,5]pyrrolo[2,3-b]indole-1,4-dione (3) (Isoroquefortine C). Tosylated imidazole 26 (0.040 g, 0.074 mmol) was dissolved in tetrahydrofuran (2.5 ml) and 1 M aqueous sodium hydroxide (0.4 ml) was added. The reaction was stirred at room temperature for 12 h, concentrated under reduced pressure, and diluted with water (5 ml). The solution was acidified to pH 3 by the addition of 0.2 M aqueous hydrogen chloride and then extracted with ethyl acetate (3 × 10 ml). The combined organic layer was washed with brine, dried over sodium sulfate, and concentrated under reduced pressure. The crude product was purified by column chromatography (2–3% methanol/methylene chloride) to yield isoroquefortine C (3) as a white foam (0.0157 g, 55%). All physical data were in agreement with that reported in the literature (23, 33). Rf = 0.55 (10% methanol/methylene chloride); 1H NMR (500 MHz, CDCl3) δ 1.04 (s, 3H), 1.15 (s, 3H), 2.48 (dd, J = 12.2, 11.5 Hz, 1H), 2.60 (dd, J = 12.2, 5.7 Hz, 1H), 4.11 (dd, J = 11.5, 5.7 Hz, 1H), 4.98 (broad s, 1H), 5.09 (d, J = 17.4 Hz, 1H), 5.13 (d, J = 10.8 Hz, 1H), 5.67 (s, 1H), 6.00 (dd, J = 17.4, 10.8 Hz, 1H), 6.59 (d, J = 7.5 Hz, 1H), 6.71 (s, 1H), 6.76 (t, J = 7.5 Hz, 1H), 7.09 (s, 1H), 7.10 (t, J = 7.5 Hz, 1H), 7.17 (m, 1H), 7.68 (s, 1H), 10.44 (broad s, 1H), 11.72 (broad s, 1H); 13C NMR (125 MHz, CDCl3) δ 22.5, 23.0, 37.3, 40.9, 59.1, 61.6, 78.0, 105.4, 109.0, 114.5, 117.6, 118.8, 125.2, 126.3, 128.9, 135.4, 137.2, 143.5, 150.4, 158.4, 165.6; IR (neat) 3,210 (m, broad), 2,962 (m), 1,682 (s), 1,665 (s), 1,622 (s), 1,435 (s), 1,384 (m) cm-1; 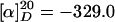 (c 0.8, CHCl3).
(c 0.8, CHCl3).
Results
Implementation of the intramolecular vinyl amidation reaction required the synthesis of two fragments: a hexahydropyrroloindole moiety and a dehydrohistidine derivative (Fig. 3). The synthesis of the former compound can be found in Scheme 1. Sequential protection of the primary and secondary amines of l-tryptophan methyl ester (4) provided the fully protected amino acid derivative (6). A two-step procedure developed by Danishefsky and coworkers (34, 35) was then used for conversion of the resulting compound to the prenylated, tricyclic system. The first step of this sequence involves formation of a 3-selenenylated pyrroloindole (7a and 7b) by treatment with N-phenylselenophthalimide (36). This inseparable set of diastereomers then was treated with methyl triflate and prenyl tributylstannane to provide the alkylated compound (8). As desired, ammonolysis of the methyl ester resulted in simultaneous removal of the fluorenylmethoxycarbonyl protecting group, producing the requisite amino amide (9).
Scheme 1.

Synthesis of the hexahydropyrroloindole moiety by a selenation/prenylation sequence. Reagents and conditions: a, fluorenylmethoxycarbonyl-Cl, Na2CO3, dioxane, H2O, 99%; b, (tert-butyloxycarbonyl)2O, dimethylallyl monophosphate, CH3CN, 83%; c, N-(phenylseleno)phthalimide, pyridinium p-toluenesulfonate, CH2Cl2, 72%; d, prenyl tributylstannane, methyl triflate, 2,6-di-tert-butyl-4-methylpyridine, CH2Cl2, 60%; e, NH3, MeOH, 89%.
Our attention then turned to formation of the vinyl halide dehydrohistidine fragment. Although a number of methods were investigated, elaboration of commercially available urocanic acid (10) proved most efficient (Scheme 2). Methylation and trityl protection of the imidazole nitrogen provided the ester 12 in good yields (37). A previously reported bromination/elimination sequence then was used to introduce the halogen substituent (38). Bromination of this enone in carbon tetrachloride resulted in formation of the enantiomeric dibromides (13a and 13b), although the compounds were unstable to silica gel column chromatography. Treatment of the crude product with 1,8-diazabicyclo[5.4.0]undec-7-ene, however, successfully promoted hydrogen bromide elimination and provided a separable mixture of E and Z vinyl bromides in a 6:1 ratio of desired (14) to undesired (15) compound. The structures of these compounds were confirmed by x-ray crystallography. Saponification of the methyl ester produced the unsaturated carboxylic acid (16).
Scheme 2.

Synthesis of the bromo-dehydrohistidine fragment. Reagents and conditions: a, H2SO4, Na2SO4, MeOH, 99%; b, trityl chloride, triethylamine, dimethylformamide, 91%; c, Br2, CCl4; d, diazabicycloundecane, CH2Cl2, 74%, two steps; e, LiOH, tetrahydrofuran, H2O, 94%.
With the two desired components in hand, efforts then focused on amide bond formation and generation of the vinyl amidation precursor (Scheme 3). Initial attempts led to production of the desired amide (17) in only low yields, and extensive investigation of alternative procedures resulted in no improvement. Despite this setback, sufficient material was available for investigation of the key diketopiperazine ring closure. A large number of palladium catalysts, phosphine ligands, bases, and solvents were screened for this transformation. Two methods seemed to produce the desired compound (18) successfully with crude yields of 13% and 45%, as indicated by high-resolution MS and NMR data. However, attempts to purify this product by a variety of methods led to decomposition. Removal of the acid-labile protecting groups also proved unsuccessful. Modification of the pyrroloindole fragment by use of the sterically less demanding methoxycarbonyl protecting group did not result in any improvements in the amide bond-forming reaction. Because of the difficulties obtaining the required starting material and the apparent instability of the intermediate produced by this route, an alternative method was examined.
Scheme 3.

Diketopiperazine ring closure by palladium-catalyzed intramolecular vinyl amidation. Reagents and conditions: a, N,N-bis(2-oxo-3-oxazolidinyl)-phosphinic chloride, triethylamine, CH2Cl2, trace; b, Pd(OAc)2 (10 mol %), (+/-)-2,2′-bis(diphenylphosphino)-1,1′-binaphthyl (15 mol %), K2CO3, toluene, 13% or Pd2(dba)3 (12.5 mol %), P(o-tolyl)3 (37.5 mol %), K2CO3, toluene, 45% crude yields.
The problems associated with generation of the amidation precursor were believed to be due to an unfavorable steric interaction with the indoline protecting group. Therefore, we examined amide bond formation with a fully unprotected compound (Scheme 4). Removal of the tert-butoxycarbonyl moiety provided the diamine intermediate (19). Amide bond formation proceeded smoothly to yield the primary amide 20. Reaction of this compound under the previously optimized palladium conditions led only to the recovery of starting material and decomposition products. Use of a copper catalyst with the protocol developed by Buchwald and coworkers (32), however, led to formation of a cyclized product (21) in good yields. Examination of this product by 15N NMR (26) was extremely helpful in determining its structure to be the imidazolidinone shown, resulting from preferential attack of the indoline nitrogen rather than the amide nitrogen. This finding was of interest in that it represents only the second report of reaction of a substrate containing both amide and amine functional groups under vinyl or aryl amidation conditions. The sole other example to date involves preferential intermolecular reaction of the primary amide of 4-aminobenzamide (32). In the intramolecular reaction of 20, the preference for a six-membered over a seven-membered metallocycle intermediate may be used to rationalize the results. Removal of the triphenylmethyl protecting group by traditional acid-catalyzed methods proved problematic; however, this transformation was accomplished in excellent yield by using trifluoroethanol and hydroxybenzotriazole (39). This heterocycle (22) possesses an unknown tetracyclic ring system.
Scheme 4.

Formation of an imidazolidinone by intramolecular copper-catalyzed vinyl amidation. Reagents and conditions: a, iodotrimethylsilane, CH3CN, 70%; b, N,N-bis(2-oxo-3-oxazolidinyl)-phosphinic chloride, triethylamine, CH2Cl2, 74%; c, CuI (10 mol %), N,N′-dimethylethylenediamine (20 mol %), K2CO3, dioxane, 74%; d, hydroxybenzotriazole, trifluoroethanol, 80%.
With the reactivity of the unprotected amidation precursor (20) (Scheme 4) clearly established, efforts were made to protect the aniline nitrogen after N,N-bis(2-oxo-3-oxazolidinyl)-phosphinic chloride-mediated coupling. Unfortunately, none of a wide variety of protecting groups and reaction conditions proved successful. In all cases, recovery of starting material (triethylsilyl, triisopropylsilyl, methyl carbamate), protection of the amide nitrogen only [(2-(trimethylsilyl)ethoxymethyl, methoxymethyl, tosyl], or decomposition (trifluoroacetyl, formyl) were observed. Use of strong bases (NaH) or increased temperatures also invariably led to decomposition and recovery of compounds lacking the reverse prenyl moiety. These results led us to explore the intermolecular variant of the copper-catalyzed amidation.
The hexahydropyrroloindole (23) and tosyl-protected dehydrohistidine compound (24) were prepared by methods analogous to those described above (Scheme 5). Closure of the diketopiperazine ring proceeded under copper catalysis to provide enamide 25, although reaction required increased catalyst loading and afforded a lower yield than the intramolecular variant. Removal of both indoline and imidazole protecting groups provided isoroquefortine C (3). Treatment of a sample of roquefortine C under these deprotection conditions failed to yield any isoroquefortine C. We concluded that the vinyl amidation procedure led to isomerization of the enamide double bond after coupling. As additional evidence, treatment of primary amide 23 with the isomeric brominated dehydrohistidine 15 (Scheme 2) also led to the formation of compound 25.
Scheme 5.

Formation of isoroquefortine C by intermolecular copper-catalyzed vinyl amidation. Reagents and conditions: a, CuI (20 mol %), N,N′-dimethylethylenediamine (40 mol %), K2CO3, toluene, 41%; b, iodotrimethylsilane, 2,6-lutidine, CH2Cl2, CH3CN, 68%; c, NaOH, tetrahydrofuran, H2O, 55%.
With an efficient method developed for the synthesis of isoroquefortine C, examination of its potential isomerization to the natural product proceeded. A wide variety of conditions (thermal, Lewis and protic acid, basic, photochemical) failed to produce the desired compound and without exception resulted in the recovery of starting material or the generation of decomposition products. Although unsuccessful in producing roquefortine C, the results demonstrate the stability of the isoroquefortine C conformation toward a wide variety of reagents.
The syntheses of isoroquefortine C (3) and a related heterocycle (22) were achieved by implementation of both intra- and intermolecular vinyl amidation reactions. These accomplishments represent a significant advance in the use of these strategies in the generation of complex molecules. The stability of the dehydrohistidine moiety of isoroquefortine C to a variety of conditions has been demonstrated. This result supports the hypothesis that the biosynthesis of the natural product roquefortine C (1) is achieved by dehydrogenation of roquefortine D (2) and does not involve dehydrohistidine isomerization (40).
Supplementary Material
Acknowledgments
This work was supported by National Science Foundation Grant CHE 01-30958.
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Jang, Z. & An, Z. (2000) in Bioactive Natural Products (Part C), ed. Atta-Ur-Rahman (Elsevier, New York), Vol. 22, pp. 245-272. [Google Scholar]
- 2.Bennett, J. W. & Klich, M. (2003) Clin. Microbiol. Rev. 16, 497-516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ohmomo, S., Sato, T., Utagawa, T. & Abe, M. (1975) Agric. Biol. Chem. 39, 1333-1334. [Google Scholar]
- 4.Ohmomo, S., Utagawa, S. & Abe, M. (1977) Agric. Biol. Chem. 41, 2097-2098. [Google Scholar]
- 5.Scott, P. M., Merrien, M.-A. & Polonsky, J. (1976) Experientia 32, 140-142. [Google Scholar]
- 6.Scott, P. M. & Kennedy, B. P. C. (1976) J. Agric. Food Chem. 24, 868-868. [DOI] [PubMed] [Google Scholar]
- 7.Ohmomo, S. & Kitamoto, H. K. (1994) J. Sci. Food Agric. 64, 211-215. [Google Scholar]
- 8.Häggblom, P. (1990) Appl. Environ. Microbiol. 56, 2924-2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Möller, T., Åkerstrand, K. & Massoud, T. (1997) Nat. Toxins 5, 86-89. [DOI] [PubMed] [Google Scholar]
- 10.Cole, R. J., Dorner, J. W., Cox, R. H. & Raymond, L. W. (1983) J. Agric. Food Chem. 31, 657-659. [DOI] [PubMed] [Google Scholar]
- 11.Ohmomo, S., Oguma, K., Ohashi, T. & Abe, M. (1978) Agric. Biol. Chem. 42, 2387-2389. [Google Scholar]
- 12.Wagener, R. E., Davis, N. D. & Diener, U. L. (1980) Appl. Environ. Microbiol. 39, 882-887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ohmomo, S. (1982) J. Antibact. Antifung. Agents 10, 253-264. [Google Scholar]
- 14.Arnold, D. L., Scott, P. M., McGuire, P. F., Harwig, J. & Nera, E. A. (1978) Food Cosmet. Toxicol. 16, 369-371. [DOI] [PubMed] [Google Scholar]
- 15.Kopp, B. & Rehm, H. J. (1979) Eur. J. Appl. Microbiol. Biotechnol. 6, 397-401. [Google Scholar]
- 16.Kopp, B. & Rehm, H. J. (1981) Eur. J. Appl. Microbiol. Biotechnol. 13, 232-235. [Google Scholar]
- 17.Wild, J. (1982) Ph.D. thesis (University of Stuttgart, Stuttgart).
- 18.Poisel, H. & Schmidt, U. (1975) Chem. Ber. 108, 2547-2553. [Google Scholar]
- 19.Schmidt, U., Griesser, H., Leitenberger, V., Lieberknecht, A., Mangold, R., Meyer, R. & Riedl, H. (1992) Synthesis, 487-490.
- 20.Kim, D., Li, Y., Horenstein, B. A. & Nakanishi, K. (1990) Tetrahedron Lett. 49, 7119-7122. [Google Scholar]
- 21.Horenstein, B. A. & Nakanishi, K. (1989) J. Am. Chem. Soc. 111, 6242-6246. [Google Scholar]
- 22.Nagel, D. W., Pachler, K. G. R., Steyn, P. S., Vleggaar, R. & Wessels, P. L. (1976) Tetrahedron 32, 2625-2631. [Google Scholar]
- 23.Scott, P. M., Polonsky, J. & Merrien, M.-A. (1979) J. Agric. Food Chem. 27, 201-202. [Google Scholar]
- 24.Chen, W.-C. & Joullié, M. M. (1998) Tetrahedron Lett. 39, 8401-8404. [Google Scholar]
- 25.Schiavi, B. M., Richard, D. J. & Joullié, M. M. (2002) J. Org. Chem. 67, 620-624. [DOI] [PubMed] [Google Scholar]
- 26.Hadden, C. E., Richard, D. J., Joullié, M. M. & Martin, G. E. (2003) J. Heterocycl. Chem. 40, 359-362. [Google Scholar]
- 27.Wolfe, J. P., Wagaw, S., Marcouw, J.-F. & Buchwald, S. L. (1998) Acc. Chem. Res. 31, 805-818. [Google Scholar]
- 28.Hartwig, J. F. (1998) Angew. Chem. Int. Ed. Engl. 37, 2046-2067. [DOI] [PubMed] [Google Scholar]
- 29.Lebedev, A. Y., Izmer, V. V., Kazyul'kin, D. N., Beletskaya, I. P. & Voskoboynikov, A. Z. (2002) Org. Lett. 4, 623-626. [DOI] [PubMed] [Google Scholar]
- 30.Willis, M. C. & Brace, G. N. (2002) Tetrahedron Lett. 43, 9085-9088. [Google Scholar]
- 31.Shen, R. & Porco, J. A., Jr. (2000) Org. Lett. 2, 1333-1336. [DOI] [PubMed] [Google Scholar]
- 32.Jiang, L., Job, G. E., Klapars, A. & Buchwald, S. L. (2003) Org. Lett. 5, 3667-3669. [DOI] [PubMed] [Google Scholar]
- 33.Vleggaar, R. & Wessels, P. L. (1980) J. Chem. Soc. Chem. Commun., 160-162.
- 34.Marsden, S. P., Depew, K. M. & Danishefsky, S. J. (1994) J. Am. Chem. Soc. 116, 11143-11144. [Google Scholar]
- 35.Depew, K. M., Marsden, S. P., Zatorska, D., Zatorski, A., Bornmann, W. G. & Danishefsky, S. J. (1999) J. Am. Chem. Soc. 121, 11953-11963. [Google Scholar]
- 36.Nicolaou, K. C., Claremon, D. A., Barnette, W. E. & Seitz, S. P. (1979) J. Am. Chem. Soc. 101, 3704-3706. [Google Scholar]
- 37.Pirrung, M. C. & Pei, T. (2000) J. Org. Chem. 65, 2229-2230. [DOI] [PubMed] [Google Scholar]
- 38.Cloninger, M. J. & Frey, P. A. (1998) Bioorg. Chem. 26, 323-333. [Google Scholar]
- 39.Bodanszky, M., Bednarek, M. A. & Bodanszky, A. (1982) Int. J. Pept. Protein Res. 20, 387-395. [DOI] [PubMed] [Google Scholar]
- 40.Ohmomo, S., Ohashi, T. & Abe, M. (1979) Agric. Biol. Chem. 10, 2035-2038. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}