

Abstract
The total synthesis of the marine cyclodepsipeptide (-)-doliculide is described. An acyclic (2S,4S,6R)-trimethyl alkanoic acid precursor to the “hydrocarbon” portion of doliculide was synthesized starting with l-ascorbic acid based on a stereocontrolled iterative conjugate addition of lithium dimethylcuprate to acyclic δ-Cmethyl α,β-unsaturated ester derivatives. syn/syn selectivity was achieved by relying on 1,3-induction in preferred folded conformations that avoid 1,5-syn–pentane interactions in the transition states. The nature of the ester moiety seems to play an important role in determining the syn/anti ratios of C-methyl adducts. The 1-methyl-1-cyclopentyl ester group was found to confer the best syn selectivity to the cuprate addition products, especially in seven-carbon enoates.
Polyketide biosynthetic units are ubiquitous among natural products of diverse origin (1–5). Although the more common ones constitute hydroxy stereotriads originating from the condensation of propionyl-CoA biogenetic precursors (6), a small subgroup harbors deoxygenated motifs, commonly known as deoxypropionates. The majority of natural products in this subfamily contain a single deoxypropionate stereotriad. Natural products that contain multiple deoxypropionate units are comparatively fewer and are generally characterized by an all-syn orientation of methyl groups on an acyclic framework, as in the structures of lardolure (7), siphonarienal (6), siphonarienolone (8), pectinatone (9, 10), TMC-151 (11), and branched alkanoic acids isolated from the preen-gland wax of the domestic goose (12). A major cuticular hydrocarbon from a cane beetle species was reported recently to have an unprecedented three sets of anti-disposed deoxypropionates (13).
Despite the presence of a common “chiral hydrocarbon” unit, these compounds exhibit diverse biological properties exemplified by the pheromone lardolure (7) and the cytotoxic TMC-151 (11). Doliculide (14) and borrelidin (15–17) are the only known examples of macrocyclic natural products that contain two and three deoxypropionate units, respectively, within their carbon frameworks (Fig. 1).
Fig. 1.

Doliculide and borrelidin.
It is intriguing that nature should choose to incorporate virtually all-syn deoxypropionate motifs in acyclic and cyclic metabolites, with only an occasional deviation from this seemingly ordered stereochemical array. Insightful proposals have been made in this regard, where control of conformation is dictated by the avoidance of nonbonded interactions between methyl groups in energetically preferred acyclic conformations (18, 19). Within a three-dimensional array, alternating C-methyl groups in an acyclic chain are better accommodated in syn-oriented motifs as compared with anti-oriented motifs, reminiscent of helical isotactic hydrocarbon polymers such as polypropylene (20–24).
In 1994, Yamada and coworkers (14) reported the isolation, structure determination, and total synthesis of doliculide (25), originally obtained from the Japanese sea hare Dolabella auricularia. This unique cyclodepsipeptide exhibited cytotoxicity against HeLa-S3 cells with an IC50 of 0.005 μg/ml (14). Structurally, doliculide is related to a family of cyclodepsipeptides known as geodiamolides (26, 27) and jaspamide (28, 29). It is distinct in many respects, particularly by the presence of three alternating syn-related C-methyl groups as part of the 11-carbon dihydroxy branched carboxylic acid subunit (Fig. 1). Limited structure–activity relationship (SAR) to establish the relative importance of specific functionalities in doliculide has shown that the presence of the deoxypropionate motif and the iodotyrosine appendage are important for activity (14). The “hydrocarbon” portion is probably crucial in conferring a preorganized bioactive conformation. Hamel and coworkers (30) reported that the cytotoxicity of doliculide was caused by interference with the polymerization of actin in developing cells.
A major challenge in the synthesis of doliculide is the elaboration of the syn/syn-trimethyl alkanoic acid portion. Thus, Yamada and coworkers (25) started with (S)-4-methyl-1,3-pentanediol and used an iterative oxazolidinone-based enolate alkylation protocol (31) to introduce the second methyl group in the growing chain. After each iteration, it was necessary to add two steps for the reductive removal of the residual hydroxyl group resulting from each aldol reactions to achieve the correct deoxygenation pattern. The Yamada protocol also necessitated configurational inversion of an intermediate alcohol.
Ghosh and Liu (32) adopted a tactically different approach to elaborate the same acyclic subunit. The sequence started with 4-benzyloxy-3-S-methylbutyronitrile, which was converted to an allylic alcohol and then subjected to a stereoselective Charette cyclopropanation (33). Conversion of the resulting primary alcohol to the iodide and metal-induced ring opening (34) afforded a syn-1,3-dimethyl terminal alkene. Iteration of the same protocol provided the desired syn/syn-trimethyl alkanoic acid, which was elaborated further by using commonly used methods for chain extension.
Rein and coworkers (35) started with a meso-dialdehyde already bearing a propionate triad and effected a desymmetrization via a selective auxiliary-mediated Horner–Wadsworth–Emmons functionalization. Further elaboration of the product afforded an acyclic syn/syn-trimethyl alkanoic acid as a mixture of diastereomers.
General methods that lead to stereoregular and enantioenriched deoxypropionate units rely on a variety of strategies (36–48). However, there are only a few methods that offer the possibility of iteration en route to enantioenriched 2,4,6-trimethyl alkanoic acids with predisposed chiralities. Myers et al. (49) showed that an ephedrine-based enolate alkylation can be adapted to iteration in model systems. Auxiliary-mediated enolate alkylation was also used by Abiko and Masamune (50) and Nicolaou et al. (51) to prepare syn-2,4,6-trimethyl deoxypropionate motifs. Chiral auxiliaries were also used for single-stage conjugate cuprate additions by Oppolzer et al. (52), Breit and Demel (53), Sakai and coworkers (54), and Williams et al. (55) to achieve syn- or anti-dimethyl triads.
(-)-Doliculide: Disconnection and Synthetic Strategy
For the synthesis of (-)-doliculide, we envisioned the disconnection shown in Fig. 2, which logically leads to the desired acyclic syn/syn-2,4,6-trimethyl alkanoic subunit A and a functionalized iodotyrosine B. The hydroxyl group at C9 would be derived from a directed reduction of a ketone C, in turn obtained from the condensation of 2-isopropyl dithiane anion and the ω-iodoalkene D. The latter would result from a series of iterative conjugate additions to α,β-unsaturated esters E and F, obtained from the readily available enoate G, the single stereocenter of which can be related to l-ascorbic acid (Fig. 2). This strategy was predicated upon a successful and stereocontrolled introduction of C-methyl groups by relying on a series of internal 1,3-inductions (F → E → D). In earlier work (56, 57), we showed that a series of iterative 1,2-inductions in cuprate additions and enolate hydroxylations to γ-alkoxy-α,β-unsaturated esters could generate as many as three sets of contiguous methyl-hydroxymethyl triads with high stereocontrol starting with acyclic precursors such as G (58). Thus, a major challenge in the implementation of the strategy shown in Fig. 2 was to test the feasibility of a sequential and stereocontrolled conjugate addition relying on a novel 1,3-induction by an existing C-methyl substituent in acyclic enoate precursors. Added to this daunting task was the need to produce a syn/syn-2,4,6-trimethyl alkanoic acid in enantiopure form corresponding to the required (2S,4S,6R) configuration in (-)-doliculide.
Fig. 2.

Disconnective analysis of (-)-doliculide.
Synthesis of (-)-Doliculide
l-Gulonic γ-lactone, readily available from l-ascorbic acid (59), was converted in two straightforward steps into the enoate 2 (Scheme 1). Three further steps (60) afforded the required starting enoate 3 in 64% overall yield. Addition of lithium dimethylcuprate in the presence of trimethylsilyl chloride as additive (56, 61, 62) proceeded in high yield to give the adduct 4 as the preponderant isomer (14:1). Chain elongation in the usual manner afforded the t-butyl enoate 5, which was subjected to a second cuprate addition. The resulting syn- and anti-diastereomeric adducts were formed in a ratio of 4:1, respectively, as observed in the enantiomeric series (60). We had found previously that the syn/anti ratio of the same cuprate addition was only 1:1 when a methyl rather than a t-butyl ester was used (5, R = Me). A series of esters corresponding to 5 with different alkyl substituents was prepared (see below) and subjected to conjugate addition. We were gratified to find that the 1-methyl-1-cyclopentyl (MCP) ester 6 afforded the desired syn-adduct 7 as the major product (syn/anti, 8:1), thereby doubling the ratio compared with the t-butyl ester 5. With this improvement we proceeded to prepare the enoate 8 by the same three-step protocol used for 5. A third cuprate addition to 8 afforded the syn/syn-3,5,7-trimethyl alkanoate 9 as the major diastereomer in excellent yield (syn/syn:syn/anti, >12:1). The stereochemical identity of 9 was rigorously established from detailed NMR studies of syn/syn and syn/anti isomers as their two-carbon homologated enoates (60). 13C inverse gated analysis of characteristic signals proved to be an excellent direct method for assessing diastereomeric ratios of cuprate adducts in this series (see Supporting Materials and Methods, which is published as supporting information on the PNAS web site). Thus, a 14-step, highly stereocontrolled synthesis of the advanced intermediate 9 from l-gulonic γ-lactone was in hand for further elaboration to the intended final target.
Scheme 1.

Reagents and conditions: a, MeLi·LiBr, CuI, chlorotrimethylsilane, THF, -78°C (95%; anti/syn: 14:1); b, diisobutylaluminum hydride, CH2Cl2, -78°C (80%); c, (ClCO)2, DMSO, Et3N, CH2Cl2, -78°C (96%); d, Ph3PC(H)CO2MCP, CH2Cl2, room temperature (rt) (91%); from 6 to 8: a, 87% (syn/anti: 8:1, 7/7′); repeat b, 80% 7a; repeat c, 88% 7b; repeat d, 81%; from 8 to 9: repeat a, 92% (syn/syn:anti/syn: >12:1); e, DIBAH-H, CH2Cl2, -78°C (92%) 9a; f, (n-Bu)3P, o-nitrophenylselenocyanate, THF, rt, then H2O2,0°C to rt (68%); g, tetrabutylammonium fluoride (TBAF), THF, rt; h, PPh3,I2, imidazole, toluene, 0°C to rt (93%, two steps); i, 2-isopropyl-[1,3]dithiane, t-BuLi, THF/hexamethylphosphoramide, -78°C, then 11 (75%); j, AgNO3, N-chlorosuccinimide, acetone/water, 0°C (79%); k, diisobutylaluminum hydride, toluene, -40°C, 96% (syn/anti: 1:1, 14/14a); l, 16, 1,3-dicyclohexylcarbodiimide, 4-dimethylaminopyridine (DMAP), CH2Cl2, -20°C (77%); m, O3,CH2Cl2/MeOH, -78°C, then PPh3; n, NaClO2,H2O2, MeCN/H2O, 10°C; o, trifluoroacetic acid, CH2Cl2, 0–10°C; p, N,N-bis(2-oxo-3-oxazolidinyl)-phosphinic reagent, DMAP, CH2Cl2, 0°C to rt (49% from 16); q, TBAF, THF, 0°C (95%).
Reduction of the ester group in 9, followed by displacement with o-nitrophenylselenide and elimination (63), afforded the olefin 10, which would serve as a carboxyl equivalent later. Cleavage of the silyl ether and treatment with triphenylphosphine, imidazole, and iodine (64) gave the iodide 11 in excellent overall yield. Displacement with the lithium 2-isopropyl dithiane in a mixture of tetrahydrofuran (THF) containing hexamethylphosphoramide as additive led to the adduct 12 in 75% yield. After treatment with silver nitrate and N-chlorosuccinimide in aqueous acetone (65), the dithiane group was converted to the corresponding ketone 13. We had planned to carry out a stereoselective reduction of the keto group in 13, relying on a directing effect of the resident O-benzyloxymethyl (OBOM) group. Reduction with tetramethylammonium acetoxyborohydride (66) or the Tischenko–Evans (67) SmI2-mediated protocol using β-hydroxy ketones were promising examples of precedents for the desired anti-diol orientation needed in doliculide. However, applying these methods required cleavage of the OBOM group to generate the corresponding β-hydroxy ketone, which in the event of a favorable stereocontrolled reduction would need selective reprotection of the C7 original alcohol. On the other hand, an SmI2-mediated reduction of β-alkoxy ketones by Keck and Wager (68) would suit our needs admirably, particularly because it was reported to be anti-selective. Unfortunately, no reaction took place with SmI2 at ≈7°C for 72 h.
We then returned to more classical methods of chemical reduction. Thus, reduction of 13 with L-Selectride proceeded in good yield, but the desired C9 alcohol was obtained as the minor isomer (syn/anti, 4:1). The relative configuration of the alcohol at C9 was established based on 13C resonance shifts as suggested by Hoffmann and Weidmann (69) for related compounds. Ultimately, reduction with diisobutylaluminum hydride in toluene at -40°C afforded a 1:1 mixture of 14 and the epimeric alcohol, which could be readily separated by chromatography and recycled.
With the desired C9 isomer 14 in hand, we proceeded with the elaboration of the peptidic subunit. Because the possibilities were somewhat limited, we adopted the Ghosh and Liu protocol (32) for the synthesis of the N-Boc glycyl amide precursor 15. Thus, 14 was esterified with the iodotyrosine derivative 15 (32) in the presence of 1,3-dicyclohexylcarbodiimide and 4-dimethylaminopyridine to afford 16 in 77% yield in contrast to the reported 95% (32). Ozonolysis of the double bond, followed by oxidation of the resulting aldehyde with sodium chlorite/H2O2 in aqueous acetonitrile (70), afforded the corresponding acid. Cleavage of the N-Boc group with trifluoroacetic acid, followed by macrolactam formation gave 17, which was subjected to a final deprotection as described by Ghosh and Liu (32) to give (-)-doliculide 1, the spectroscopic data of which were identical to those reported in the literature (14, 32).
The presently described synthesis of (-)-doliculide is considerably shorter than the previously reported ones and validates the notion of iterative 1,3-induction based on resident chirality for the creation of alternating syn/syn-2,4,6-trimethyl alkanoic acid motifs. Among the favorable strategic and practical attributes of our synthesis are the realization that a single stereogenic carbon originating from C5 of l-ascorbic acid was the progenitor of three new centers through a series of highly stereoselective1,2- and 1,3-internal inductions. It is also important to note that the same hydroxyl group in l-ascorbic corresponds to the C7 hydroxyl group in doliculide. Furthermore, the methodology we developed for syn/syn-deoxypropionates by iterative 1,3-syn stereoselective cuprate additions to acyclic enoates does not require additional steps for deoxygenation of residual hydroxyl groups en route to doliculide (25).
Acyclic Stereocontrol in Conjugate Additions: The “Ester” Effect
As explained previously, our synthesis plan was based on exploiting the merits of stereocontrolled conjugate cuprate additions to δ-C-methyl α,β-unsaturated enoates to assess whether any preparatively significant 1,3-induction was possible. As evidenced by the successful completion of the synthesis of (-)-doliculide by using this strategy, the viability of acyclic stereocontrol for a syn/syn-2,4,6-trimethyl deoxypropionate chain is obvious. We recently presented a rationale and a mechanistic model to explain the stereochemical preferences in such conjugate additions in connection with the total synthesis of borrelidin (60). We reasoned that transition-state conformations during cuprate additions could be favored when 1,5-syn–pentane interactions are minimized or avoided. Thus, depending on the ground-state solution conformation of enoates such as 6 and 8, reaction with lithium dimethylcuprate could lead to diastereomeric syn-(desired) and anti-(undesired) dimethyl and trimethyl deoxypropionate arrays in the respective products. The preponderance of the syn adducts 7 and 9 can be rationalized based on the prevalence of enoate conformers of 6 and 8, in which the carbon backbone of the acyclic chain is already folded in an energetically preferred conformation. In Fig. 3A we depict a plausible sequence with enoate 8, in which an enoate/cuprate π complex 8a and the subsequent bulky cuprio(III) intermediate 8b can be favorably accommodated in such a folded conformation before the transfer of a methyl group (71).
Fig. 3.

Proposed intermediates and adducts in the reaction of enoate 8 with lithium dimethylcuprate (A) and superposition of the deoxypropionate subunit in the x-ray structure of TMC-151 on a virtual diamond lattice (B). TBDPS, tert-butyldiphenylsilyl; BOMO, O-benzyloxymethyl; TMSCI, chlorotrimethylsilane; L, ligand (Li+ or trimethylsilyl).
We have used a virtual diamond lattice model (72, 73) to qualitatively visualize the backbone conformations of such acyclic enoates and their putative cuprio intermediates (60). The validity of this visual tool to simulate folded conformations of hydrocarbon chains was also tested in the context of solid-state crystal structures of natural products containing syn/syn-trimethyl deoxypropionate motifs. In Fig. 3B we show the quasiperfect congruence of the carbon backbone in TMC-151 (11) on a diamond lattice. Indeed, the alternating C-methyl groups are disposed in a manner so as to avoid 1,5-syn–pentane interactions in an energetically preferred folded conformation. The solution conformations as determined by detailed NMR studies of 8 and of the two-carbon homologated enoate prepared from 9 (60) closely simulate the molecular shape of the corresponding deoxypropionate chain counterpart in the x-ray structure of TMC-151 (Fig. 3B).
However, the minimization of 1,5-syn–pentane interactions between a resident homoallylic δ-C-methyl group in the ground state of the enoate moiety, or in the β-cuprio(III) adduct before (or after) methyl transfer, may not be fully effective without a preorganization of the acyclic chain.
It is well known that in the gas phase, simple hydrocarbons and their substituted variants populate some of the conformations more than others because of relative energy differences arising from bonded and nonbonded interactions (74, 75). The consequences of minimizing 1,5-syn–pentane interactions in rendering short hydrocarbon chains largely monoconformational have been discussed and reviewed in detail by Hoffmann et al. (76). The notion of using a relatively large substituent as an “anchor” to favor the reactivity of different conformers of a tethered chain has been alluded to by De Clercq (77, 78) in the context of the rate acceleration of intramolecular Diels–Alder reactions. This effect has been of special interest as part of a more generalized phenomenon, also known as the Thorpe–Ingold gem-dialkyl effect (79) or “reactive rotamer effect” (ref. 80 and references therein; ref. 81).
In the case of conjugate additions of organocuprates to enoates such as 5, the ester extremity seems to play an important role in determining syn selectivity, as observed in going from methyl to t-butyl (syn/anti: 1:1 and 4:1, respectively) (60). Although a syn/anti ratio of 10:1 for 9 with the t-butyl enoate 8 was originally acceptable in the third-stage cuprate addition (60), the modest 4:1 ratio obtained for 5 (R = t-butyl) warranted additional study.
We therefore undertook a more systematic investigation of the influence of the ester group on the stereoselectivity of the cuprate addition to the enoate 5 (Table 1). The isopropyl and benzhydryl esters showed slight improvements over the methyl ester but fell short of the t-butyl and neopentyl esters (Table 1, entries 1–5). However, incorporation of an MCP ester moiety doubled the syn/anti selectivity to 8:1 (Table 1, entry 6) (see also Supporting Materials and Methods). As a control, the corresponding cyclopentyl ester gave a ratio similar to the methyl, isopropyl, and benzhydryl esters (Table 1, entry 7). syn/syn selectivity was also improved in the case of the MCP enoate 8 compared with its t-butyl counterpart (60) (10:1 vs. >12:1), albeit not as dramatically as for the shorter homologue 6. Although a rigorous explanation is not possible at this time, the MCP group may offer a quasioptimal spatial disposition in relation to the folded carbon backbone of the enoate 6, hence during the formation of the corresponding π-complex or ensuing β-cuprio intermediate (Table 1) (for a theoretical study of the conformations of α,β-unsaturated esters and their Lewis acid complexes, see ref. 82). The significant erosion of selectivity with the cyclopentyl ester (Table 1, entry 7) and the major improvement compared with the t-butyl ester (Table 1, entries 4 and 6) heightens the relevance of the spatial disposition of the MCP group as an entity in enoate 6.
Table 1. Variations of the ester moiety.
 | ||||
|---|---|---|---|---|
| Entry | R | d.r.* | Yield, % | |
| 1 | Me | 50:50 | 90 | 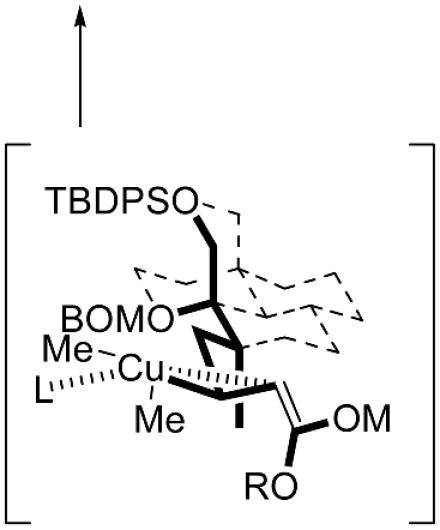 |
| 2 | i-Pr | 63:37 | 91 | |
| 3 | Benzhydryl | 64:36 | 85 | |
| 4 | tert-Bu (5) | 80:20 | 90 | |
| 5 | Neopentyl | 82:18 | 85 | |
| 6 | MCP (6) | 89:11 | 87 | |
| 7 | CP | 54:46 | 90 | L = TMSCI or THF, M = TMS or Li+ |
OBOM or BOMO, O-benzyloxymethyl; TMSCI, chlorotrimethysilane; TBDPS, tert-butyldiphenylsilyl; CP, cyclopentyl.
Diastereomeric ratio determined by inverse-gated homodecoupling 13C NMR
Conclusions
We have described a stereocontrolled total synthesis of (-)-doliculide, starting with l-ascorbic acid by using a strategy that capitalizes on iterative conjugate additions to acyclic α,β-unsaturated esters harboring a δ-C-methyl group with excellent 1,3-induction. The C1–C8 “hydrocarbon” segment in doliculide harboring two syn/syn deoxypropionate triads was synthesized in 14 linear steps, thus shortening the overall operation by at least 10 steps compared with two previous syntheses.
Avoidance of 1,5-syn–pentane interactions during cuprate additions in preferentially folded conformations of δ-C-methyl-α,β-unsaturated esters accounts for the high syn selectivity. The nature of the ester group in the enoates also contributes to the selectivity, possibly because of a preferred spatial disposition in relation to the rest of the carbon backbone.
Supplementary Material
Acknowledgments
We are grateful to Navjot Chahal for technical assistance and Dr. Minh Tan Phan Viet and Sylvie Bilodeau for helpful NMR discussions. We thank the Natural Sciences and Engineering Research Council and Isis Pharmaceuticals (Carlsbad, CA) for generous research support.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: MCP, 1-methyl-1-cyclopentyl; THF, tetrahydrofuran.
References
- 1.Omura, S. (2002) Macrolide Antibiotics: Chemistry, Biology, and Practice (Academic, New York), 2nd Ed.
- 2.Nicolaou, K. C., Vourloumis, D., Winssinger, N. & Baran, P. (2000) Angew. Chem. Int. Ed. Engl. 39, 44-122. [PubMed] [Google Scholar]
- 3.Norcross, R. D. & Paterson, I. (1995) Chem. Rev. (Washington, D.C.) 95, 2041-2114. [Google Scholar]
- 4.O'hagan, D. (1991) The Polyketide Metabolites (Ellis Horwood, Chichester, U.K.).
- 5.Perron, F. & Albizzati, K. F. (1989) Chem. Rev. (Washington, D.C.) 89, 1617-1661. [Google Scholar]
- 6.Norté, M., Fernandez, J. J. & Padilla, A. (1994) Tetrahedron Lett. 35, 3413-3416. [Google Scholar]
- 7.Mori, K. & Kuwahara, S. (1986) Tetrahedron 42, 5545-5550. [Google Scholar]
- 8.Norté, M., Cataldo, F. & Gonzalez, A. G. (1988) Tetrahedron Lett. 29, 2879-2880. [Google Scholar]
- 9.Biskupiak, J. E. & Ireland, C. M. (1983) Tetrahedron Lett. 24, 3055-3058. [Google Scholar]
- 10.Norté, M., Cataldo, F., Gonzalez, A. G., Rodriguez, M. L. & Ruiz-Perez, C. (1990) Tetrahedron 46, 1669-1678. [Google Scholar]
- 11.Kohno, J., Nishio, M., Sakurai, M., Kawano, K., Hajime, H., Kameda, N., Kishi, N., Yamashita, T., Okuda, T. & Komatsubara, S. (1999) Tetrahedron 55, 7771-7786. [Google Scholar]
- 12.Morr, M., Fortkamp, J. & Rühe, S. (1997) Angew. Chem. Int. Ed. Engl. 36, 2460-2462. [Google Scholar]
- 13.Fletcher, M. T., Chow, S., Lambert, L. K., Gallagher, O. P., Cribb, B. W., Allsopp, P. G., Moore, C. J. & Kitching, W. (2003) Org. Lett. 3, 5083-5086. [DOI] [PubMed] [Google Scholar]
- 14.Ishiwata, H., Nemoto, M., Ojika, M. & Yamada, K. (1994) J. Org. Chem. 59, 4710-4711. [Google Scholar]
- 15.Lumb, M., Macey, P. E., Spyvee, J., Withmarsh, J. M. & Wright, R. D. (1965) Nature 206, 263-265. [DOI] [PubMed] [Google Scholar]
- 16.Keller-Schierlein, W. (1967) Helv. Chim. Acta 50, 731-753. [DOI] [PubMed] [Google Scholar]
- 17.Anderson, B. F., Herlt, A. J., Rickards, R. W. & Robertson, G. B. (1989) Aust. J. Chem. 42, 717-730. [Google Scholar]
- 18.Hoffmann, R. W. (2000) Angew. Chem. Int. Ed. Engl. 39, 2054-2070. [PubMed] [Google Scholar]
- 19.Anderson, J. E. (1992) in The Chemistry of Alkanes and Cycloalkanes, eds. Patai, S. & Rapoport, Z. J. (Wiley, New York), pp. 45-133.
- 20.Leitereg, T. J. & Cram, D. J. (1968) J. Am. Chem. Soc. 90, 4019-4026. [Google Scholar]
- 21.Natta, G. & Corradini, P. (1959) J. Polym. Sci. 39, 29-46. [Google Scholar]
- 22.Pucci, S., Aglietto, M., Luisi, P. L. & Pino, P. (1967) J. Am. Chem. Soc. 89, 2787-2788. [Google Scholar]
- 23.Nolte, R. J. M. (1994) Chem. Soc. Rev. 23, 11-19. [Google Scholar]
- 24.Okamoto, Y. & Nakano, T. (1994) Chem. Rev. (Washington, D.C.) 94, 349-372. [Google Scholar]
- 25.Ishiwata, H., Sone, H., Kizoshi, H. & Yamada, K. (1994) Tetrahedron 50, 12853-12882. [Google Scholar]
- 26.Chan, W. R., Tinto, W. F., Manchand, P. S. & Todaro, L. J. (1987) J. Org. Chem. 52, 3091-3093. [Google Scholar]
- 27.Dilip de Silva, E., Anderson, R. J. & Allen, T. M. (1990) Tetrahedron Lett. 31, 489-492. [Google Scholar]
- 28.Zabriskie, T. M., Klocke, J. M., Ireland, C. M., Marcus, A. H., Molinski, T. F., Faulkner, D. J., Xu, C. & Clardy, J. (1986) J. Am. Chem. Soc. 108, 3123-3124. [Google Scholar]
- 29.Crews, P., Manes, L. V. & Boehler, M., (1986) Tetrahedron Lett. 27, 2797-2800. [Google Scholar]
- 30.Bai, R., Covell, D. G., Lui, C. F., Ghosh, A. K. & Hamel, E. (2002) J. Biol. Chem. 277, 32165-32171. [DOI] [PubMed] [Google Scholar]
- 31.Evans, D. A., Bartroli, J. & Shih, T. L. (1981) J. Am. Chem. Soc. 103, 2127-2129. [Google Scholar]
- 32.Ghosh, A. K. & Liu, C. (2001) Org. Lett. 3, 635-638. [DOI] [PubMed] [Google Scholar]
- 33.Charette, A. B. & Juteau, H. (1994) J. Am. Chem. Soc. 116, 2651-2652. [Google Scholar]
- 34.Charette, A. B. & Naud, J. (1998) Tetrahedron Lett. 39, 7259-7262. [Google Scholar]
- 35.Tullis, J. S., Helquist, P. & Rein, T. (1999) Phosphorus Sulfur Silicon Relat. Elem. 144–146, 165-168. [Google Scholar]
- 36.Tomooka, K., Nagasawa, A., Wei, S.-Y. & Nakai, T. (1996) Tetrahedron Lett. 37, 8895-8898. [Google Scholar]
- 37.Evans, D. A. & Morrissey, M. M. (1984) J. Am. Chem. Soc. 106, 3866-3868. [Google Scholar]
- 38.Marshall, J. A. & Blough, B. F. (1990) J. Org. Chem. 55, 1540-1547. [Google Scholar]
- 39.Brown, J. M. (1987) Angew. Chem. Int. Ed. Engl. 99, 169-182. [Google Scholar]
- 40.Kaino, M., Naruse, Y., Ishihara, K. & Yamamoto, H. (1990) J. Org. Chem. 55, 5814-5815. [Google Scholar]
- 41.Gambacorta, A., Tofani, D., Lupattelli, P. & Tafi, A. (2002) Tetrahedron Lett. 43, 2195-2198. [Google Scholar]
- 42.Lautens, M., Colucci, J. T., Hiebert, S., Smith, N. D. & Bouchain, G. (2002) Org. Lett. 4, 1879-1882. [DOI] [PubMed] [Google Scholar]
- 43.Hiratake, J., Inagaki, M., Yamamoto, Y. & Oda, T. (1987) J. Chem. Soc. Perkin Trans. 1, 1053-1058.
- 44.Fujita, K. & Mori, K. (2001) Eur. J. Org. Chem. 493-502.
- 45.Datel, D. V., VanMiddlesworth, F., Donaubauer, J., Garnett, P. & Sih, C. J. (1986) J. Am. Chem. Soc. 108, 4603-4614. [Google Scholar]
- 46.Sefkow, M., Neidlein, A., Sommerfeld, T., Sternfeld, F., Maestro, N. A. & Seebach, D. (1994) Liebigs Ann. Chem. 719-729.
- 47.Mori, I., Bartlett, P. A. & Heathcock, C. H. (1990) J. Org. Chem. 55, 5966-5977. [Google Scholar]
- 48.Wilson, S. R. & Price, M. F. (1982) J. Am. Chem. Soc. 104, 1124-1126. [Google Scholar]
- 49.Myers, A. G., Yang, B. H., Chen, H. & Kopecky, D. (1997) Synlett, 457-459.
- 50.Abiko, A. & Masamune, S. (1996) Tetrahedron Lett. 37, 1081-1084. [Google Scholar]
- 51.Nicolaou, K. C., Yue, E. W., Naniwa, Y., De Riccardis, F., Nadin, A., Leresche, J. E., La Greca, S. & Yang, Z. (1994) Angew. Chem. Int. Ed. Engl. 33, 2184-2187. [Google Scholar]
- 52.Oppolzer, W., Maretti, R. & Bernardelli, G. (1986) Tetrahedron Lett. 27, 4713-4716. [Google Scholar]
- 53.Breit, B. & Demel, P. (2000) Tetrahedron 56, 2833-2846. [Google Scholar]
- 54.Ogawa, T., Suemune, H. & Sakai, K. (1993) Chem. Pharm. Bull. 41, 1652-1654. [Google Scholar]
- 55.Williams, D. R., Kissel, W. S., Li, J. J. & Mullins, R. J. (2002) Tetrahedron Lett. 43, 3723-3727. [Google Scholar]
- 56.Hanessian, S. & Sumi, K. (1991) Synthesis, 1083-1089.
- 57.Hanessian, S., Gai, Y. & Wang, W. (1996) Tetrahedron Lett. 37, 7473-7476. [Google Scholar]
- 58.Hanessian, S., Wang, W., Gai, Y. & Olivier, E. (1997) J. Am. Chem. Soc. 119, 10034-10041. [Google Scholar]
- 59.Hubschwerlen, C. (1986) Synthesis, 962-964.
- 60.Hanessian, S., Yang, Y., Giroux, S., Mascitti, V., Ma, J. & Raeppel, F. (2003) J. Am. Chem. Soc. 125, 13784-13792. [DOI] [PubMed] [Google Scholar]
- 61.Corey, E. J. & Boaz, N. W. (1985) Tetrahedron Lett. 26, 6015-6018. [Google Scholar]
- 62.Corey, E. J. & Boaz, N. W. (1985) Tetrahedron Lett. 26, 6019-6022. [Google Scholar]
- 63.Grieco, P. A., Gilman, S. & Nishizawa, M. (1976) J. Org. Chem. 41, 1485-1486. [Google Scholar]
- 64.Garreg, Per J. & Samuelsson, B. (1979) J. Chem. Soc. Chem. Commun., 978-980.
- 65.Corey, E. J. & Erickson, B. W. (1971) J. Org. Chem. 36, 3553-3560. [Google Scholar]
- 66.Evans, D. A., Gauchet-Prunet, J. A., Carreira, E. M. & Charette, A. B. (1991) J. Org. Chem. 56, 741-750. [Google Scholar]
- 67.Evans, D. A. & Hoveyda, A. H. (1990) J. Am. Chem. Soc. 112, 6447-6449. [Google Scholar]
- 68.Keck, G. E. & Wager, C. A. (2000) Org. Lett. 2, 2307-2310. [DOI] [PubMed] [Google Scholar]
- 69.Hoffmann, R. W. & Weidmann, U. (1985) Chem. Ber. 118, 3980-3992. [Google Scholar]
- 70.Dalcanale, E. & Montanari, F. (1986) J. Org. Chem. 51, 567-569. [Google Scholar]
- 71.Woodward, S. (2000) Chem. Soc. Rev. 29, 393-401. [Google Scholar]
- 72.Dale, J. (1966) Angew. Chem. Int. Ed. Engl. 5, 1000-10021. [Google Scholar]
- 73.Egan, R. S., Perun, T. J., Martin, J. R. & Mitscher, L. A. (1973) Tetrahedron 29, 2525-2538. [Google Scholar]
- 74.Alder, R. W., Allen, P. R., Hnyk, D., Rankin, D. W. H., Robertson, H. E., Smart, B. A., Gillespie, R. J. & Bytheway, I. (1999) J. Org. Chem. 64, 4226-4232. [Google Scholar]
- 75.Tsuzuki, S., Schäfer, L., Goto, H., Jemmis, E. D., Hosoya, H., Siam, K. & Osawa, E. (1991) J. Am. Chem. Soc. 113, 4665-4671. [Google Scholar]
- 76.Hoffmann, R. W., Göttlich, R. & Schopfer, U. (2001) Eur. J. Org. Chem. 1865-1871.
- 77.Cauwberghs, S., De Clercq, P. J., Tinant, B. & Declercq, J. P. (1988) Tetrahedron Lett. 29, 2493-2496. [Google Scholar]
- 78.De Corte, F., Nuyttens, F., Cauwberghs, S. & De Clercq, P. (1993) Tetrahedron Lett. 34, 1831-1832.J. P. [Google Scholar]
- 79.Beesley, R. M., Ingold, C. K. & Thorpe, J. F. (1915) J. Chem. Soc. 107, 1080-1106. [Google Scholar]
- 80.Jung, M. E. & Gervay, J. (1991) J. Am. Chem. Soc. 113, 224-232. [Google Scholar]
- 81.Curtin, M. L. & Okamura, W. H. (1990) J. Org. Chem. 55, 5278-5287. [Google Scholar]
- 82.Loncharich, R. J., Schwartz, T. R. & Houk, K. N. (1987) J. Am. Chem. Soc. 109, 14-23. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}