

Abstract
Securamine A is a structurally intriguing alkaloid possessing a pyrroloindole core joined via a modified isoprene subunit to a functionalized imidazole ring. Recent synthetic efforts in this laboratory have resulted in the efficient construction of key lactone 36, which undergoes tandem azide reduction/ring expansion to macrolactam 37. Macrolactam 37 possesses the complete macrocyclic core of securamine A.
The bryozoans Flustra foliacea and Chartella papyracea have proven to be a rich source of structurally unprecedented halogenated indole-alkaloids. A series of investigations resulted in the isolation of a host of novel natural products, including the flustramines (1–3), chartellines (4–6), and chartellamides (7). Additionally, two reports describe the securamines (8, 9), which are characterized by a central tricyclic pyrroloindole core and a highly substituted imidazole ring linked via a modified isoprene subunit and a macrocyclic cis-enamide (Fig. 1). Interestingly, pyrroloindole securamine A (1) exists in a synthetically exploitable solvent-dependent equilibrium with ring-opened isomer securine A (2) (8, 9).
Fig. 1.

Representative akaloids from C. papyracea.
Despite synthetic work toward both the flustramines (10–12) and chartellines/chartellamides (13, 14), no efforts toward the construction of the securamine/securine skeleton have yet been reported (15, 16). Intrigued by the densely functionalized heterocycles characterizing the securamines, in addition to reports that securine A serves as a biogenic precursor for a variety of other natural products (8, 9), we have focused our efforts on the efficient construction of 1 and 2.
Retrosynthetically, securine A was visualized as the union of two heterocyclic subunits (pyrroloindole and imidazole) joined into a macrocycle via two tethers (the isoprene and enamide) (Fig. 2). We envisioned that the elimination-prone C(10) neopentyl chloride moiety (13, 14) could be installed at a late stage via direct chlorination of the corresponding alcohol, whereas the sensitive enamide-moiety could be generated from a C(2)-C(3) amido-alcohol (5). Orthogonal diol 5 would arise via macrocyclization of the corresponding amino alcohol, which could be accessed directly from a C(2)-C(3) olefin (17, 18). Key indole 6 could be accessed from internal alkyne 7, which would be generated from elaboration of 8. Imidazole 8 could be derived from the condensation of two equivalents of formamide with α-bromo ketone 9 (19).
Fig. 2.

Securine A retrosynthetic analysis.
Methods
Unless otherwise stated, reactions were performed in flamedried glassware under a nitrogen atmosphere by using freshly distilled solvents. Experimental and spectral data pertaining to compounds 7, 11–15, 18–20, 22, and 25–40 can be found in Supporting Text, which is published as supporting information on the PNAS web site.
Results
Exposure of 10 to bromine and acetic acid gave smooth conversion to the corresponding α-bromoketone. Subsequent dissolution in neat formamide and prolonged heating gave imidazole 8 in 80% overall yield after two recrystallizations (19). Benzylation at N(5) and bromination at C(4) proceeded in a highly regioselective manner to afford versatile bromide 11. Subsequent installation of the requisite C(2)-C(3) terminal olefin, two-step adjustment of the C(10) oxidation state, and addition of propargyl magnesium bromide furnished key neopentyl alcohol 13 in excellent overall yield (27% yield, eight steps) from 10 (Scheme 1).
Scheme 1.

Imidazole construction and model chlorine installation. DMF, N,N-dimethylformamide; LAH, lithium aluminum hydride; NBS, N-bromosuccinimide; THF, tetrahydrofuran.
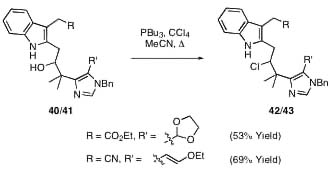
With 13 in hand we explored chlorination under a variety of conditions. In most cases, the desired neopentyl chloride was accompanied by variable amounts of 15, arising via an imidazole-assisted elimination pathway (Scheme 2). Similar participation via intermediate cyclpropanes has been reported in other homobenzyllic systems (20, 21). Interestingly, phosphine cone angle seemed to correlate directly with the ratio of 14 to 15, and we found 14 could be accessed in reasonable yield by using tributylphosphine and carbon tetrachloride (Scheme 1). Although 14 proved relatively uncooperative toward further advancement to 1, we were pleased to find that a variety of related hindered alcohols, including those possessing fully elaborated indoles (Scheme 9, which is published as supporting information on the PNAS web site), also proved suitable for direct phosphine-mediated introduction of the neopentyl chlorine. Resolved to install the C(10) chlorine at a later stage, we proceeded to test our plans for dehydrative enamide installation.
Scheme 2.

Imidazole-assisted rearrangement.
Efforts to regioselectively functionalize the C(2)-C(3) olefin of a variety of model imidazoles (12–14) with standard aminohydroxylation conditions proved problematic (17, 18). However, in a reaction sequence inspired by Khuong-Huu and coworkers (22), we found that addition of the silyl ether derived from 12 to a premixed solution of ICl and sodium acetate in cold acetonitrile furnished primary iodide 18 in excellent overall yield. Subsequent iodide displacement with sodium azide affords azido acetate 19, possessing the proper C(2)/C(3) functionalization for advancement to 1. Tandem tributyltinhydride-mediated azide reduction-acetate migration followed by hydrogenation afforded model amido alcohol 20. We were pleased to find that the bromide and chloride derived from 20 underwent smooth elimination exclusively to the desired cis-enamide 22 upon treatment with a variety of bases (K2CO3, 1,8-diazabicyclo[5.4.0]-undec-7-ene, basic amberlist, Et3N, Ag2CO3) at low temperatures (0°C to room temperature) (23, 24). The mild conditions suitable for elimination are consistent with participation of the pendant imidazole functionality (Scheme 3).
Scheme 3.

C2-C3 functionalization and model enamide installation. TBSO, Me2tBuSiO-; DMF, N,N-dimethylformamide.
With working model systems for the introduction of the most sensitive functionality in place, we turned toward the completion of the securamine carbocyclic skeleton. In contemplating a suitable end-game scenario, we considered the enticing possibility of simultaneous installation of the C(10) chlorine and enamide functionalities (Scheme 4). In our hands neopentyl chloride 14 (in addition to 42 and 43, see Scheme 9) proved resistant to elimination upon exposure to a variety of bases [K2CO3, 1,8-diazabicyclo[5.4.0]-undec-7-ene, NH4OH(aq)] suitable for the conversion of 20 to 22; we hoped that the imidazole functionality could be used to effect the low temperature selective elimination of a C(3) chloride in the presence of a C(10) chloride (Scheme 4).
Scheme 4.

Tandem chloride installation/enamide formation.
Turning toward the advancement of 13, Sonagashira coupling with iodoaniline 28 in the presence of catalytic palladium (II) afforded 7 (Scheme 6). A variety of methods aimed at simultaneous palladium-mediated indole formation/(C20) alkylation were explored initially. Optimization of a tandem three-component coupling based on the Cacchi indole synthesis proved quite promising. Unfortunately, attempted oxidations of 26/27 proved problematic (see ref. 15) (Scheme 5). However, we ultimately found that the most practical synthetic access to 31 was routed through the C(13) unsubstituted indole 29, obtained via prolonged exposure of 7 to Pd(PPh3)4 (25) (Scheme 6). Installation of the requisite C(21)-C(22) chain via alkylation of 29 initially proved problematic, with a variety of well-documented protocols giving low recovered yields of alkylated indole 30 (Scheme 6). Extensive optimization with regard to base, solvent, and electrophile revealed that n-butyl α-iodoacetate proved unique in its ability to function as a suitable electrophile, giving rise to an excellent yield of 31 (26).
Scheme 6.

Indole formation/C20 alkylation. DMF, N,N-dimethylformamide.
Scheme 5.

Tandem indole formation/C20 alkylation.
With 31 in hand we turned toward the functionalization of the C(2)-C(3) olefin in analogy with model olefin 12 (Scheme 3). The C(10) hydroxyl and indole were necessarily protected as a silyl ether and carbamate, respectively (Scheme 7). Subsequent exposure to iodine monochloride/sodium acetate followed by sodium azide afforded azido acetate 33 in excellent overall yield from 32 as a roughly 6:1 mixture of diastereomers. Simultaneous hydrolysis of the C(2) acetate, C(22) butyl ester, and indole carbamate was effected upon prolonged exposure to dilute lithium hydroxide to afford acid 34 in quantitative yield.
Scheme 7.

C2/C3 functionalization and macrocyclization. TBSCl, Me2tBuSiCl; TBSO, Me2tBuSiO-; DMF, N,N-dimethylformamide; THF, tetrahydrofuran; DMAP, 4-(dimethylamino)pyridine; TBAF, Bu4NF.
Exhaustive reduction of 34 (H2,Pd/C, MeOH/EtOAc) served to simultaneously cleave the N(5) benzyl group and reduce the C(2) azide. However, the corresponding amino acid proved exceedingly difficult to handle and could not be coaxed into undergoing smooth macrolactamization under a variety of standard conditions. Recalling the propensity of a C(3) acetate to migrate upon reduction of 19, we planned to circumvent this problem via construction of the key securamine macrolactam via ring expansion of the corresponding (n-2) azido-lactone. Further, we were pleased to find that hydroxy acid 34 proved an excellent substrate for macrolactonization, giving rise to 35 cleanly as a single diastereromic product upon treatment with Yamaguchi's reagent. Desilylation of 35 proceeded without incident to afford 36 in excellent yield.
Prolonged exposure of 36 to tributyltinhydride/2,2′-azobisisobutyronitrile in refluxing benzene afforded the corresponding ring-expanded macrolactam 37. Hydrogenation of 37 gave key diol 38, primed for attempts at tandem chloride/enamide installation. Screening of a variety of conditions previously suitable for chlorination and/or enamide formation revealed a tendency of 37 and 38 to undergo a tandem chlorination/elimination reaction. However, isomer 39 remains the only isolable product under all of the conditions explored thus far, and the desired regioisomeric enamide remains undetectable as a component of the relatively clean reaction mixtures (Scheme 8).
Scheme 8.

Tandem azide reduction/ring expansion. AIBN, 2,2′-azobisisobutyronitrile.
Conclusions
Despite the unanticipated tendency of diol 38 to undergo undesirable reaction pathways, our approach toward the securamine A macrocycle remains quite promising. Key azido lactone 36, possessing the complete securamine A carbon skeleton in the correct oxidation states, has been accessed in an extremely efficient and high-yielding sequence (17 steps, 6.3% overall yield, 85% average yield per step) from commercially available starting materials. Our approach to 1 includes a highly efficient indole alkylation (29→31) and a tandem azide reduction-ring expansion of azido-lactone 36 to afford the complete securamine macrocyclic skeleton (37).
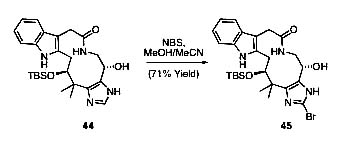
Model system work has indicated the feasibility of the phosphine-mediated installation of the extremely hindered securamine neopentyl chlorine (13→14, 40→42, 41→43) as well as a dehydrative approach to the requisite securamine cis-enamide (20→22). Additionally, preliminary data suggest securamine A might arise quite efficiently from selective C(6) bromination of a des-bromo analog (Scheme 10, which is published as supporting information on the PNAS web site). Utilization of our current strategy for the exploration of a variety of end-game scenarios continues, as the C(10) and C(2) hydroxyls remain orthogonal in a variety of our key synthetic intermediates (33–36, 44, and 45).
Supplementary Material
Acknowledgments
J.B.S. thanks the National Science Foundation, the American Chemical Society Division of Medicinal Chemistry, and Pfizer, Inc. for research fellowships. J.L.W. acknowledges Bristol-Myers Squibb, Eli Lilly, GlaxoSmithKline, Yamanouchi, and AstraZeneca for financial support through their Faculty Awards Programs and the National Institutes of Health for Grant GM 93591.
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Carle, J. & Christophersen, C. (1979) J. Am. Chem. Soc. 101, 4012-4013. [Google Scholar]
- 2.Carle, J. & Christophersen, C. (1980) J. Org. Chem. 45, 1586-1589. [Google Scholar]
- 3.Laycock, M., Wright, J., Findlay, J. & Patil, A. (1986) Can. J. Chem. 64, 1312-1316. [Google Scholar]
- 4.Chevolot, L., Chevolot, A., Gajhede, M., Larsen, C., Anthoni, U. & Christophersen, C. (1985) J. Am. Chem. Soc. 107, 4542-4543. [Google Scholar]
- 5.Anthoni, U., Chevolot, L., Larsen, C., Nielsen, P. & Christophersen, C. (1987) J. Org. Chem. 52, 4709-4712. [Google Scholar]
- 6.Nielsen, P., Anthoni, U. & Christophersen, C. (1988) Acta Chem. Scand. B 42, 489-491. [Google Scholar]
- 7.Anthoni, U., Bock, K., Chevolot, L., Larsen, C., Nielsen, P. & Christophersen, C. (1987) J. Org. Chem. 52, 5638-5639. [Google Scholar]
- 8.Rahbaek, L., Anthoni, U., Chrisophersen, C., Nielsen, P. & Petersen, B. (1996) J. Org. Chem. 61, 887-889. [Google Scholar]
- 9.Rahbaek, L. & Christophersen, C. (1997) J. Nat. Prod. 60, 175. [Google Scholar]
- 10.Hino, T., Tanaka, T., Matsuki, K. & Nakagawa, M. (1983) Chem. Pharm. Bull. 31, 1806-1808. [Google Scholar]
- 11.Muthusubramanian, P., Carle, J. & Christophersen, C. (1983) Acta Chem. Scand. B 37, 803-807. [Google Scholar]
- 12.Jensen, J., Anthoni, U., Christophersen, C. & Nielsen, P. (1995) Acta Chem. Scand. 49, 68-71. [Google Scholar]
- 13.Lin, X. & Weinreb, S. (2001) Tetrahedron Lett. 42, 2631-2633. [Google Scholar]
- 14.Pinder, J. & Weinreb, S. (2003) Tetrahedron Lett. 44, 4141-4143. [Google Scholar]
- 15.Chaffee, S. C. (1999) Ph.D. dissertation (Yale Univ., New Haven, CT).
- 16.Korakas, P. (2003) Ph.D. dissertation (Yale Univ., New Haven, CT).
- 17.Chang, H. & Sharpless, K. (1996) Angew. Chem. Int. Ed. 35, 182-186. [Google Scholar]
- 18.Nesterenko, V., Byers, J. & Hergenrother, P. (2003) Org. Lett. 5, 281-284. [DOI] [PubMed] [Google Scholar]
- 19.Jonsson, A. (1954) Acta Chem. Scand. 8, 1389-1393. [Google Scholar]
- 20.Battersby, A., Nicoletti, M., Staunton, J. & Vieggaar, R. (1980) J. Chem. Soc. Perkin Trans. 1, 43-51. [DOI] [PubMed]
- 21.Cram, D. (1949) J. Am. Chem. Soc. 71, 3863-3870. [Google Scholar]
- 22.Tchissamboca, L., Benechie, M. & Khuonghuu, F. (1982) Tetrahedron Lett. 38, 2687-2695. [Google Scholar]
- 23.Xiao, D., East, S. & Joullie, M. (1998) Tetrahedron Lett. 39, 9631-9632. [Google Scholar]
- 24.Aizpurua, J. & Cossio, F. (1986) Tetrahedron Lett. 27, 4359-4362. [Google Scholar]
- 25.Ma, C., Yu, S., He, X., Liu, X. & Cook, J. (2000) Tetrahedron Lett. 41, 2781-2785. [Google Scholar]
- 26.Schreiber, S., Ragan, J. & Standaert, R. (1991) Strategies Tactics Org. Synth. 3, 417-461. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}