

Abstract
Substantial structural transformations of much use in complex chemical synthesis can be achieved by channeling the reactivity of highly unsaturated molecules. This report describes the direct conversion of an acyclic polyunsaturated diketone to the HIV-1 Rev/Rev-responsive element inhibitor harziphilone under mild reaction conditions.
Keywords: nucleophilic catalysis, Baylis–Hillman reaction, conjugate additions, electrocyclizations, domino reactions
The reactivity of activated polyenes is the basis for some of the most impressive structural transformations in nature and chemical synthesis. The biosyntheses of the complex structures of the polycyclic triterpenes from squalene and 2,3-oxidosqualene may be viewed as paragons for processes that build polycyclic molecular complexity directly from reactive polyenes (1–3). In connection with our efforts to channel the reactivity of activated polyunsaturated molecules, we sought mild reaction conditions under which compound 1 could be isomerized to the bicyclic structure of harziphilone (2), a naturally occurring inhibitor of the binding interaction between the HIV-1 Rev protein and the Rev-responsive element (RRE) of viral mRNA (4) (Scheme 1). This report describes an enantioselective synthesis of (+)-harziphilone based on the general idea expressed in Scheme 1.
Scheme 1.

Can (+)-harziphilone (2) be formed by a cycloisomerization of acyclic diketone 1?
The structure of (+)-harziphilone (2) comprises fifteen carbon atoms, two six-membered ring systems, one carbocyclic and the other heterocyclic, a keto group, two contiguous hydroxyl-bearing stereocenters, and a pentadienyl side chain. All of the polar groupings of harziphilone are confined to the six-membered carbocycle, whereas the hydrophobic pentadienyl moiety is displayed as a side chain on the α-pyran ring. Our approach to the problem of synthesizing this natural product was guided by the retrosynthetic analysis shown in Scheme 2.
Scheme 2.

A retrosynthetic analysis of harziphilone (2).
The independent early investigations of Büchi (5) and Marvell (6, 7) provided a sound basis for the proposal that the oxacyclic ring of harziphilone, with its particular arrangement of alkenes, could arise by a 6π-electrocyclization of a compound of type 3 (for selected examples of this type of valence isomerization in synthesis see refs. 8–11). With several electrophilic sites and a high degree of unsaturation, 3 was perceived to be a challenging objective for synthesis, and we elected to explore the possibility of producing this potentially transient intermediate in situ from a doubly activated, polyunsaturated acyclic compound of type 1. To implement this concept, we would seek a suitable heteroatom nucleophile that could attack the unsubstituted enone moiety in 1. Although almost certainly reversible, such a site-selective, intermolecular conjugate addition reaction could trigger an intramolecular conjugate addition (see arrows in 4), resulting in the production of a key carbon–carbon bond and the carbocyclic ring of (+)-harziphilone (for selected tandem conjugate addition/Michael cyclization reactions see refs. 12–17). A β-elimination of the heteroatom nucleophile might then generate 3 and set the stage for a final cycloisomerization to (+)-harziphilone (2; P = H).
Materials and Methods
Experimental procedures and characterization data for the compounds described in this article are provided in the supporting text, which is published as supporting information on the PNAS web site.
Results and Discussion
Aspects of our design for synthesis call to mind the Morita–Baylis–Hillman reaction (18, 19), and we reasoned that nucleophiles known to be effective at instigating this useful reaction might also bring about the type of bicyclization we were seeking. In the course of searching for a mild method for transforming doubly activated acyclic compounds to the bicyclic architecture of harziphilone, we reacted enone ynone 5† with 10 mol % 1,4-diazabicyclo[2.2.2]-octane (DABCO) in chloroform at room temperature (Scheme 3). After 10 days, compound 5 was completely converted into the harziphilone-like compound 6. By contrast, when acyclic diol 7 was treated with 10 mol % DABCO under the same conditions, bicyclic diol 8 was produced in nearly quantitative yield after only 20 h. On steric grounds, the unsubstituted enone β-carbons of compounds 5 and 7 are both susceptible to nucleophilic attack; however, it may well be that an internally hydrogen-bonded carbonyl in 7 heightens the reactivity of the enone β-carbon toward the nucleophilic catalyst and is the origin of the 12-fold difference in rate between the cycloisomerizations of compounds 5 and 7 (for examples of intramolecular catalysis through hydrogen bonding see refs. 21–24).
Scheme 3.

Cycloisomerizations of compounds 5 and 7 catalyzed by DABCO. TFA, trifluoroacetic acid; rt, room temperature.
With these promising preliminary results reinforcing our confidence in the concepts shown in Schemes 1 and 2, we turned our attention to the problem of synthesizing compound 1. Our preferred pathway commences with an asymmetric reduction of commercially available 2-methyl-2-cyclopenten-1-one (9) (Scheme 4) by the powerful method of Corey, Bakshi, and Shibata (CBS) (25). Silylation of the resulting secondary alcohol was then followed by an efficient and highly diastereoselective dihydroxylation (26) of the ring alkene to give diol 10. After protection of the vicinal diol system in the form of an acetonide, the silyl ether was cleaved with fluoride ion and the resulting alcohol was oxidized to optically active cyclopentanone 11.
Scheme 4.

Step a: (S)-2-methyl-CBS-oxazaborolidine (0.2 eq), BH3·SMe2 (0.6 eq), tetrahydrofuran (THF), 0°C, enantiomeric excess = 90%. Step b: t-BuMe2SiCl, imidazole, 4-dimethylaminopyridine (4-DMAP), CH2Cl2, 73% yield over two steps. Step c: OsO4 (0.03 eq), 4-methylmorpholine N-oxide (2.5 eq), i-PrOH, 0°C, 86% yield, diastereomeric excess = 98%. Step d: 2-methoxypropene, 10-camphorsulfonic acid (0.1 eq), CH2Cl2, 98% yield. Step e: n-Bu4NF, THF, 99% yield. Step f: SO3·pyridine, DMSO, Et3N, CH2Cl2, 85% yield. Step g: Ac2O, pyridine, 4-DMAP, 80°C, 84% yield. Step h: K2OsO2(OH)4 (0.1 eq), NaIO4, t-BuOH/H2O. Step i: Me3SiCHN2, MeOH/benzene, 56% yield over two steps. Step j: K2CO3, MeOH, CH3COC(N2)PO(OMe)2, 62% yield. Step k: lithium diisopropylamide, THF, -78°C; sorbaldehyde, -78°C to rt, 65% yield. Step I: Et3SiCl, imidazole, CH2Cl2, rt, 94%. Step m: LiN(OMe)Me, THF, -78°C, 80% yield. Step n: vinylMgBr, THF, 68% yield. Step o: n-Bu4NF, AcOH, THF, 80% yield. Step p: Dess-Martin periodinane, NaHCO3, CH2Cl2, 72% yield. Step q: 1:1 vol/vol trifluoroacetic acid/H2O, 0°C, 85% yield. TBS, t-BuMe2Si; TES, Et3Si.
Our aim was to transform 11 to a bifunctional acyclic structure with differentiated oxidation states at the terminal carbons through an oxidative ring cleavage. To this end, 11 was enolized and acetylated on oxygen. Oxidative cleavage of the intermediate enol acetate afforded an aldehyde carboxylic acid, which was esterified with (trimethylsilyl)diazomethane to give the corresponding aldehyde methyl ester. A chemoselective reaction of the aldehyde group with Ohira's diazoketophosphonate (27) in basic methanol then gave rise to alkynyl ester 12.
The lithium acetylide formed by a low-temperature deprotonation of alkyne 12 was then reacted with commercially available sorbaldehyde, after which silylation of the resulting secondary alcohol afforded compound 13 in 61% overall yield. To construct the required enone moiety, the methyl ester of 13 was converted to a Weinreb amide (28), which reacted smoothly with vinyl-magnesium bromide to give the corresponding enone. After cleavage of the triethylsilyl ether with fluoride ion, a Dess–Martin oxidation (29) yielded the desired diketone. Finally, an acidic hydrolysis of the acetonide protecting group afforded compound 1 and set the stage for the pivotal bicyclization reaction.
In relation to compounds 5 and 7 (Scheme 3), compound 1 possesses two additional electron-deficient carbon atoms that could predispose it toward undesired reactivity. Fortunately, however, it was possible to transform 1 directly to (+)-harziphilone (2) under the conditions shown in Scheme 5 in 70% yield. To rationalize the course of this assisted bicycloisomerization, we suggest that DABCO adds reversibly to the unsubstituted and potentially activated enone system of 1, affording a Baylis–Hillman-like zwitterion 14. An intramolecular carbon–carbon bond formation could then generate an allenolate ion and subsequently the putative zwitterion 15 through a proton transfer. A simple β-elimination reaction could then return the nucleophilic catalyst to the reaction medium and afford 16, thus enabling a final 6π-electrocyclization to (+)-harziphilone (2).‡ Alternatively, the oxacyclic ring of 2 could arise from zwitterion 15 by an intramolecular displacement of a neutral molecule of DABCO.
Scheme 5.

A DABCO-catalyzed cycloisomerization of compound 1 to (+)-harziphilone (2). Conditions: Compound 1 (0.1 M in CHCl3), DABCO (0.1 eq), rt, 24 h, 70% yield of compound 2. EC, electrocyclization.
Under mild reaction conditions, the nucleophilic catalyst DABCO is capable of inducing unsaturated acyclic diketones to convert directly into the bicyclic architecture of the naturally occurring HIV-1 Rev/RRE inhibitor harziphilone. The total synthesis and DABCO-catalyzed bicyclizations described herein are the basis for an effort to create manifold side-chain variants of this natural product. Efforts are also underway to elucidate the binding interactions between harziphilone and HIV-1 constructs by NMR spectroscopy.
Supplementary Material
Acknowledgments
We thank Drs. D.-H. Huang and L. Pasternack for NMR assistance and Dr. G. Siuzdak for mass spectrometric assistance. This work was supported by National Institute for General Medical Sciences Grant GM65483 (to E.J.S.), The Skaggs Foundation at The Scripps Research Institute, the Deutscher Akademischer Austauschdienst (K.P.), a Skaggs Predoctoral Fellowship (to L.M.S.), the Achievement Rewards for College Scientists Foundation (L.M.S.), a Beckman Young Investigator Award (to E.J.S.), AstraZeneca, Eli Lilly, Pfizer, Merck Research Laboratories, Bristol–Myers Squibb, and a Camille Dreyfus Teacher–Scholar Award (to E.J.S.).
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: RRE, Rev-responsive element; DABCO, 1,4-diazabicyclo[2.2.2]octane; rt, room temperature.
Footnotes
See supporting information for a scheme showing a synthesis of compound 5 from propargyl alcohol by a pathway featuring a Sharpless asymmetric dihydroxylation reaction. For a review on the Sharpless asymmetric dihydroxylation see ref. 20.
An optical rotation for naturally occurring harziphilone was not reported in ref. 4. Our sample of synthetic (+)-harziphilone (2) showed an 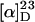 of +67° (c = 1.1 mg/ml, CHCl3). The spectroscopic data (1H NMR, 13C NMR, and IR) obtained for 2 matched the data reported for natural harziphilone in ref. 4. We also synthesized (–)-harziphilone by the chemistry described herein for evaluation of both enantiomers of harziphilone in an HIV-1 Rev/RRE-binding assay.
of +67° (c = 1.1 mg/ml, CHCl3). The spectroscopic data (1H NMR, 13C NMR, and IR) obtained for 2 matched the data reported for natural harziphilone in ref. 4. We also synthesized (–)-harziphilone by the chemistry described herein for evaluation of both enantiomers of harziphilone in an HIV-1 Rev/RRE-binding assay.
References
- 1.Wendt, K. U., Schulz, G. E., Corey, E. J. & Liu, D. R. (2000) Angew. Chem. Int. Ed. 39, 2812-2833. [PubMed] [Google Scholar]
- 2.Johnson, W. S. (1991) Tetrahedron 47, xi-1. [Google Scholar]
- 3.Sorensen, E. J. (2003) Bioorg. Med. Chem. 11, 3225-3228. [DOI] [PubMed] [Google Scholar]
- 4.Qian-Cutrone, J., Huang, S., Chang, L.-P., Pirnik, D. M., Klohr, S. E., Dalterio, R. A., Hugill, R., Lowe, S., Alam, M. & Kadow, K. F. (1996) J. Antibiot. 49, 990-997. [DOI] [PubMed] [Google Scholar]
- 5.Büchi, G. & Yang, N. C. (1957) J. Am. Chem. Soc. 79, 2318-2323. [Google Scholar]
- 6.Marvell, E. N., Caple, G., Gosink, T. A. & Zimmer, G. (1966) J. Am. Chem. Soc. 88, 619-620. [Google Scholar]
- 7.Marvell, E. N., Chadwick, T., Caple, G., Gosink, T. & Zimmer, G. (1972) J. Org. Chem. 37, 2992-2997. [Google Scholar]
- 8.Li, C., Johnson, R. P. & Porco, J. A., Jr. (2003) J. Am. Chem. Soc. 125, 5095-5106. [DOI] [PubMed] [Google Scholar]
- 9.Shoji, M., Yamaguchi, J., Kakeya, H., Osada, H. & Hayashi, Y. (2002) Angew. Chem. Int. Ed. 41, 3192-3194. [DOI] [PubMed] [Google Scholar]
- 10.Li, C., Bardhan, S., Pace, E. A., Liang, M.-C., Gilmore, T. D. & Porco, J. A., Jr. (2002) Org. Lett. 4, 3267-3270. [DOI] [PubMed] [Google Scholar]
- 11.Malerich, J. P. & Trauner, D. (2003) J. Am. Chem. Soc. 125, 9554-9555. [DOI] [PubMed] [Google Scholar]
- 12.Ernest, I. (1976) Angew. Chem. Int. Ed. 15, 207-214. [DOI] [PubMed] [Google Scholar]
- 13.Uyehara, T., Shida, N. & Yamamoto, Y. (1989) J. Chem. Soc. Chem. Commun. 2, 113-114. [Google Scholar]
- 14.Saito, S., Hirohara, Y., Narahara, O. & Moriwake, T. (1989) J. Am. Chem. Soc. 111, 4533-4535. [Google Scholar]
- 15.Klimko, P. G. & Singleton, D. A. (1992) J. Org. Chem. 57, 1733-1740. [Google Scholar]
- 16.Wang, L.-C., Luis, A. L., Agapiou, K., Jang, H.-Y. & Krische, M. J. (2002) J. Am. Chem. Soc. 124, 2402-2403. [DOI] [PubMed] [Google Scholar]
- 17.Frank, S. A., Mergott, D. J. & Roush, W. R. (2002) J. Am. Chem. Soc. 124, 2404-2405. [DOI] [PubMed] [Google Scholar]
- 18.Ciganek, E. (1997) Org. React. 51, 201-350. [Google Scholar]
- 19.Basavaiah, D., Rao, A. J. & Satyanarayana, T. (2003) Chem. Rev. 103, 811-891. [DOI] [PubMed] [Google Scholar]
- 20.Kolb, H. C., Van Nieuwenhze, M. S. & Sharpless, K. B. (1994) Chem. Rev. 94, 2483-2547. [Google Scholar]
- 21.Choy, W., Reed, L. A., III, & Masamune, S. (1983) J. Org. Chem. 48, 1137-1139. [Google Scholar]
- 22.Masamune, S., Reed, L. S., III, Davis, J. T. & Choy, W. (1983) J. Org. Chem. 48, 4441-4444. [Google Scholar]
- 23.Allen, J. G. & Danishefsky, S. J. (2001) J. Am. Chem. Soc. 123, 351-352. [DOI] [PubMed] [Google Scholar]
- 24.Cox, C. D., Siu, T. & Danishefsky, S. J. (2003) Angew. Chem. Int. Ed. 42, 5625-5629. [DOI] [PubMed] [Google Scholar]
- 25.Corey, E. J. & Helal, C. J. (1998) Angew. Chem. Int. Ed. 37, 1986-2012. [DOI] [PubMed] [Google Scholar]
- 26.VanRheenen, V., Kelly, R. C. & Cha, D. Y. (1976) Tetrahedron Lett. 17, 1973-1976. [Google Scholar]
- 27.Ohira, S. (1989) Synth. Commun. 19, 561-564. [Google Scholar]
- 28.Basha, A., Lipton, M. & Weinreb, S. M. (1977) Tetrahedron Lett. 18, 4171-4174. [Google Scholar]
- 29.Dess, D. B. & Martin, J. C. (1983) J. Org. Chem. 48, 4155-4156. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}