

Abstract
Background:
Neuromyelitis optica spectrum disorders (NMOSDs) represent 20% of all demyelinating disorders in South India. No studies have determined the seroprevalence to both antibodies against aquaporin-4* and antimyelin oligodendrocyte glycoprotein antibody (anti-MOG+) in this population.
Objective:
To identify and characterize seropositive patients for anti-aquaporin-4 antibody (anti-AQP4+) and anti-MOG+ in South India.
Materials and Methods:
We included 125 consecutive patients (15 children) who were serologically characterized using live transfected cells to human M23-AQP4 or full-length MOG.
Results:
Among a total of 125 patients, 30.4% of patients were anti-AQP4+, 20% were anti-MOG+, and 49.6% were seronegative. No patient was positive for both. Anti-MOG+ patients represented 28.7% (25/87) of seronegative NMOSD. In comparison to anti-AQP4+ patients, anti-MOG+ patients were commonly male, had less frequent attacks and milder disability on expanded disability status score scale. Seronegative patients were also predominantly male, 36% (9/25) had monophasic longitudinally extensive transverse myelitis and disability was comparable with anti-AQP4+ patients. Lumbar cord involvement was common in anti-MOG+ and seronegatives, whereas anti-AQP4+ patients had more cervical lesions.
Conclusion:
Anti-AQP4+/anti-MOG + patients accounted for nearly half of the patients suspected of having NMOSD in South India, indicating that antibody testing may be useful on the management of subgroups with different prognosis.
Keywords: Antibody, aquaporin-4, India, myelin oligodendrocyte glycoprotein, neuromyelitis optica
Introduction
Neuromyelitis optica spectrum disorder (NMOSD) is an autoimmune inflammatory central nervous system disease predominantly targeting the optic nerve and spinal cord. Prevalent worldwide the disease shows similarity in the age of onset and female preponderance in different populations.[1] The term NMOSD currently encompasses a variety of clinical phenotypes including NMO, recurrent optic neuritis (ROPN), recurrent transverse myelitis (RTM), and isolated transverse myelitis with longitudinally extensive transverse myelitis (LETM) and more. While anti-aquaporin-4 antibody (anti-AQP4) is considered important for the NMOSD diagnosis, 10–40% of patients are seronegative.[2] Recently, autoantibodies targeting myelin oligodendrocyte glycoprotein (anti-MOG) has been reported in seronegative patients with NMOSD.[2,3,4,5,6,7]
In India, the seropositivity for anti-AQP4+ (using commercial cell-based assays) is below 50% in studies that included consecutive patients diagnosed with NMOSD.[8] Some patients may have been falsely negative due to the limitation of assays[9,10] used and/or samples collected after treatments, but others may have antigenic targets other than AQP4. The frequency and clinical presentation of anti-MOG+ patients in India have not been previously studied, and seropositivity for anti-AQP4+ has not been evaluated using cell-based assay (CBA) with live transfected cells. Therefore, we performed this study to determine seropositive status for anti-AQP4 and anti-MOG antibody and to compare the clinical and imaging features between these groups and those that were seronegative.
Materials and Methods
Patients and sample collection
A total of 125 consecutive patients from the Mangalore demyelinating disease registry suspected of having NMOSD by virtue of their clinical and radiological presentations were included in this study. Fifty-one patients fulfilled 2006 Wingerchuk criteria for NMO,[11] 51 patients with isolated LETM, 21 ROPN, and two patients with recurrent brainstem attacks and recurrent tumefactive brain lesions.[12,13] All study patients had details of clinical attacks, expanded disability status score (EDSS), and visual functional score (VFS) of the EDSS obtained during each hospital visit. Lumbar puncture was not done in all patients, and hence data were not included in the study. Anti-nuclear antibody was the only serum autoantibody routinely tested for all patients. We (LP, SM) reviewed all magnetic resonance imaging (MRI) of brain and spinal cord retrospectively in a blinded manner. Anti-AQP4 and anti-MOG CBAs used live transfected cells with AQP4-M23 isoform and full-length MOG as described previously[14] and two observers (DKS and TT) analyzed the samples blindly.
Statistical analysis
Categorical variables including disease phenotype, gender, and age of onset (recoded after calculating median) were calculated by Chi-square test. Clinical data were compared between 3 groups (anti-AQP4+, anti-MOG+ and seronegative groups) using Kruskal–Wallis one-way analysis of variance test. A P < 0.05 was considered statistically significant. Statistical analysis was performed using SPSS 20.0 (IBM corporation, Armonk, NY, USA).
Standard protocol approvals, registrations, and patient consents
The conduct of the study was as per the Helsinki protocol. This study was approved by the Institutional Ethics Committee, and all patients signed an informed consent form.
Results
Among a total of 125 patients tested for anti-MOG and anti-AQP4 antibody, 30.4% (38/125) patients were anti-AQP4+. Moreover, 20% (25/125) were anti-MOG + and 49.6% (62/125) were seronegatives. No patient was positive for both antibodies. Serum antinuclear antibody was positive in 31.6% (12/38) of anti-AQP4+, 4% (1/25) anti-MOG+, and 15% (9/62) of seronegative patients (P < 0.001).
Clinical characteristics
Demographics
Anti-AQP4+ patients were predominantly females (34/38, 89.5%). In contrast, anti-MOG+ patients (16/25, 64%) and seronegative patients (35/62, 56.5%) were predominantly males. Age at onset of disease was similar between groups (P = 0.94).
Disease phenotype
Relapsing disease was seen in all anti-AQP4+ patients, 64% of anti-MOG+ and 56.5% of seronegative patients. NMO fulfilling Wingerchuk 2006 criteria were seen in 84.2% of anti-AQP4+ patients. Only 8% (2/25) of anti-MOG+ and 27.4% (17/62) of seronegative patients had a disease phenotype compatible with NMO. RTM was seen in 15.8% (6/38) of anti-AQP4+, 12% (3/25) of anti-MOG+, and 9.7% (6/62) of seronegative patients. In contrast, ROPN was seen in 44% (11/25) of anti-MOG+ patients, followed by 16.1% (10/62) of seronegative patients but this phenotype was absent in anti-AQP4+ group. A single attack of LETM predominated in seronegative 27/62 (43.5%) patients. Among anti-MOG+ patients, 9/25 (36%) had a single attack of LETM. Two patients were labeled as “other” in the seronegative group included one patient with recurrent tumefactive demyelinating brain lesions and another with recurrent brainstem demyelination.
Disease course and severity
In 28.9% (11/38) of the anti-AQP4+ patients, we observed a preceding/ongoing clinical event (fever - 7, diarrhea - 1, postpartum - 1, pregnancy - 1, mumps - 1) associated with clinical attacks. For the anti-MOG+ group, we identified a single patient who developed symptoms after 3 months postpartum. Two patients with seronegative monophasic transverse myelitis had preceding fever and one patient had preceding varicella-zoster infection.
LETM was the initial attack in 63.1% (24/38) of anti-AQP4+, 56% (14/25) of anti-MOG+, and 77.4% (48/62) of seronegative patients. Unilateral OPN was the initial event in 26.3% (10/38) of anti-AQP4+, 40% (10/25) of anti-MOG+, and 16.1% (10/62) of seronegative patients. Brainstem attack as an initial event was seen in 7.9% (3/38) of anti-AQP4+, two of whom had nausea, vomiting, and hiccups preceding the onset of disease. None of the anti-MOG + or seronegative patients had initial involvement of the brainstem. Bilateral OPN was seen in one patient each from all three subgroups of patients. Lhermitte's sign was noted in 15.8% (6/38) anti-AQP4+, 4% (1/25) of the anti-MOG+, and in none of the seronegative patients.
Duration of disease [Table 1] was similar between anti-AQP4+ and anti-MOG + patients (P = 0.27). However, the disease course was distinct between the two antibody-positive groups [Figure 1]. High attack frequency (P < 0.0001), poor VFS (P < 0.01), and high EDSS score (P < 0.001) were seen in anti-AQP4 + patients compared to anti-MOG+ patients. A notable exception was one male patient from the anti-MOG+ group who had unilateral blindness and paraplegia after recurrent OPN and myelitis. The seronegative group had significant disability measured by EDSS, comparable with anti-AQP4+ patients (P = 0.10). However, VFSs were similar to anti-MOG+ group (P = 0.43).
Table 1.
Clinical course of seropositive and seronegative neuromyelitis optica spectrum disorders
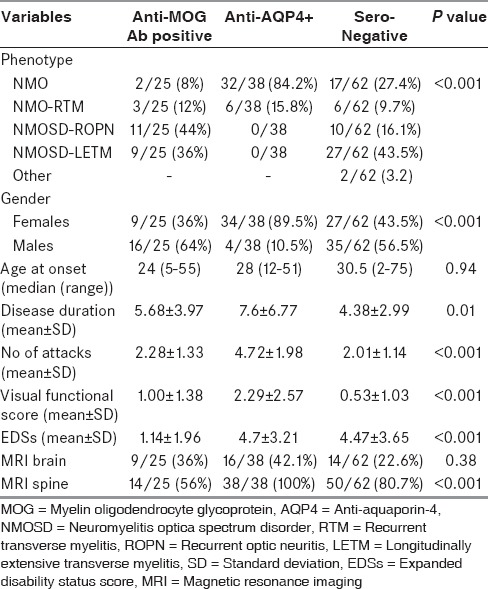
Figure 1.

Comparison of disability (expanded disability status score) between anti-myelin oligodendrocyte glycoprotein and anti-aquaporin-4 antibody patients
Young onset neuromyelitis optica spectrum disorder
There were 15 children in this study cohort with a median age of onset of 10.5 ± 4.7 (range 5–16 years). There was a single patient (15 years) with anti-AQP4 + NMO. There were eight anti-MOG+ patients including four ROPN, two each of RTM, and isolated ataxia telangiectasia mutated (ATM) patients. The six seronegative patients were equally divided among NMO, ATM, and ROPN. Encephalopathy was not a presenting feature in this subgroup of our study. The clinical subtype was also not age-specific among anti-MOG+ and seronegative patients.
Magnetic resonance imaging of the brain and spine
Brain abnormalities were seen in all three groups of patients. The frequency of lesions ranged from 22.6% to 42.1% [Table 1] and was not significantly different between the groups (P = 0.38). Distribution of brain lesions in the subcortex, periventricular region, thalamus, corpus callosum, brainstem, and cerebellum was compared between anti-AQP4 + and anti-MOG+ patients, but there was no significant difference between the two groups. Subcortical atypical white matter lesions were the most common abnormality, and it was seen in 24% (6/25) of anti-MOG+ and 26.3% (10/38) of anti-AQP4+ patients. Brain lesions were symptomatic in 8% (2/25) of anti-MOG+ and 10.5% (4/38) of anti-AQP4+ patients.
Unilateral or bilateral thickening of optic nerve with T2-weighted hyperintense signals and/or patchy enhancement was seen in 61.5% (8/13) of anti-MOG+ and 68.8% (22/32) of anti-AQP4+ patients (P = 0.24) who had OPN (bilateral involvement was seen in 15.4% [2/13] of anti-MOG+ as opposed to 50% [16/32] in anti-AQP4+ patients [P < 0.03]). Optic chiasm was partially involved in 15.4% (2/13) and 37.5% (12/32) of anti-MOG+ and anti-AQP4+ groups, respectively (P = 0.15) [Figure 2a].
Figure 2.
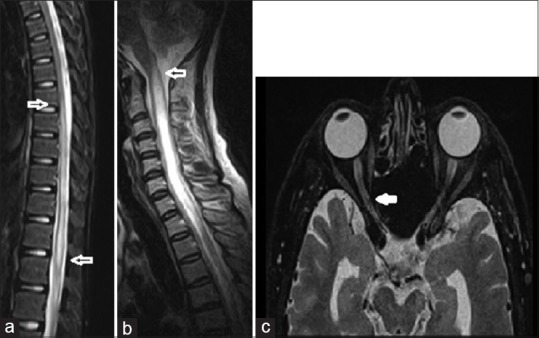
Magnetic resonance imaging of the spinal cord and optic nerve in anti-myelin oligodendrocyte glycoprotein patients. (a) T2-weighted sagittal image of the cord in a 16-year-old male with anti-myelin oligodendrocyte glycoprotein and isolated longitudinally extensive transverse myelitis involving dorsal cord with extension to the conus (block arrow), (b) 42-year-old woman with anti-aquaporin-4 antibody and longitudinally extensive transverse myelitis in the cervical cord extending into the caudal brainstem (block arrow), (c) axial images of the orbit showing long segment hyperintense signals in the optic nerve (block arrow) and optic atrophy in the other in the same patient
MRI of the spinal cord was abnormal in all anti-AQP4+ and 80.7% of the seronegative patients, whereas only 56% of anti-MOG+ patients had spinal cord lesions (P < 0.001). There was a significant difference (P < 0.001) in the location of spinal cord lesions. Dorsal and lumbar cord lesions predominated in anti-MOG+ [Figure 2b] and seronegative patients, whereas cervical and dorsal cord lesions were frequent in anti-AQP4+ patients. Cervical cord lesions extending into the medulla was seen in 34.2% (13/38) of anti-AQP4+ patients [Figure 2c] and none in the other two groups. Segmental cord atrophy was seen in 10.5% (4/38) of anti-AQP4+ and 4% (1/25) of the anti-MOG+ patients.
Treatment
Patients were diagnosed to have relapsing NMOSD after a mean period of 3.9 ± 5.5 years from disease onset for anti-AQP4+ patients, 3.1 ± 3.8 years for anti-MOG+ patients and 4.1 ± 4.6 years for seronegative patients. All patients who relapsed were treated with immunosuppressants irrespective of their antibody status choice of immunosuppressive agent was decided by the patients financial status (azathioprine [AZA] being the least expensive). In the anti-AQP4+ group, 7.9% (3/38) of patients were not on long-term immunosuppressive therapy. Among the remaining patients, 42.2% (16/38) took AZA, 50% (19/38) mycophenolate mofetil (MMF), and 7.89% (3/38) rituximab. Among seronegative patients, 53.2% (33/62) had a recurrent disease, 72.7% (24/33) received MMF, and 27.3% (9/33) AZA. Four patients with seronegative ROPN and two patients with anti-MOG+ ROPN remained asymptomatic after discontinuation of immunosuppressants (median follow-up of 22 months). Among anti-MOG+ patients, one patient diagnosed to have RTM and three who had ROPN opted to take parenteral steroids during relapse but were not on long-term immunosuppressants. They remained well after the last attack (median follow-up of 26 months). In the anti-AQP4+ cohort, 36.8% (14/38) of patients relapsed while on immunosuppressive therapy, but poor drug compliance was observed in 57.1% (8/14) of those patients under treatment. One patient each from the anti-MOG+ and anti-AQP4 + group had been treated elsewhere with beta interferon for 12 and 18 months respectively, with clinical worsening.
Death occurred in 10.5% (4/38) of the anti-AQP4+, 4% (1/25) of the anti-MOG+, and 12.9% (8/62) of the seronegative patients. One patient who was anti-AQP4+ died of respiratory failure while others had discontinued immunosuppressive therapy and died of complications from a chronic bedridden state.
Discussion
The discovery of anti-AQP4 (also known as NMO-IgG)[15] enabled the specific diagnosis of a disorder with a predictable course that requires early initiation of immunosuppressants. Since worsening of disability in NMOSD is attack-related, this strategy has worked well in the case of anti-AQP4+ cases and remarkably reduced morbidity and mortality associated with the disease.[15] The recent discovery of anti-MOG+ cases is a strong indicator that anti-AQP4+ and negative cases may have different disease mechanisms.
In our study from Southern India, we have for the first time investigated the detection of anti-AQP4 and anti-MOG antibody using CBAs with live transfected cells. None of our patients were positive for both antibodies. Seropositivity for anti-AQP4+ among patients diagnosed with NMO by Wingerchuk 2006 criteria was relatively high (84.2%) in our patients when compared to reports from other Asian countries were it ranges from 55% to 60%.[16,17] Anti-MOG+ cases constituted 7.4% (16/215) of all NMOSD in one study,[5] whereas it was 20% in our study (25/125) and a similar rate was reported from Thailand.[17] The common clinical presentation among anti-MOG+ patients was ROPN followed by myelitis with involvement of dorsal and lumbar segments of the spinal cord. Majority of patients with anti-AQP4+ presented as relapsing NMO and had myelitis with involvement of the cervical cord often extending into the caudal brainstem. The coexistence of other serum autoantibodies was higher in the anti-AQP4 group and may be useful to differentiate among the three groups. The severity of disease in the anti-AQP4+ group was likely to have been influenced by the delay in diagnosis, delay in initiation of immunosuppressive therapy, and noncompliance with therapy mostly arising from financial constraints.
We have for the first time compared brain lesion distribution between anti-MOG+ and anti-AQP4+ patients and found no significant difference between the two groups. Orbital MRI showed bilateral optic nerve with chiasmal involvement more commonly in anti-AQP4+ patients. We found two patients with anti-MOG+ NMOSD showing optic chiasmal involvement on MRI in contrary to other published series.[14,17] The data from our cohort study support the view that beta interferon therapy may worsen anti-AQP4+[18] and also anti-MOG+ disease.
Our study characterized the clinical profile of seronegative patients after extracting both anti-AQP4+ and anti-MOG+ patients from the group of high-risk NMOSD patients. Nearly, half of our patients were seronegative which is much higher than reported from elsewhere in Asia.[5] A substantial number (43.5%) of them had monophasic LETM. They were mostly men who had relative sparing of vision but disability levels comparable to the anti-AQP4+ patients indicating that they may not have a good recovery. Male predominance in seronegative LETM has been highlighted in earlier studies too.[19] These patients may represent a heterogeneous group, and some patients may have other unidentified antigenic targets in the central nervous system.
To summarize, anti AQP4+ patients are predominantly women have an NMO phenotype disorder commonly with high attack frequency and significant visual dysfunction. Anti-MOG+ patients may be more male dominant. They commonly present as monophasic LETM or ROPN and may be more frequently seen in pediatric age group. Anti-MOG+ patients also have a relatively benign course compared to anti-AQP4+ patients.
Identification of these subgroups in NMOSD is important from the point of diagnosis and management of patients with NMOSD. While it is clear that monophasic NMOSD associated with anti-AQP4+ requires vigorous immunosuppression, it is unclear if the same approach is to be recommended for anti-MOG+ and seronegative patients. Evaluating a larger cohort of patients with a longer follow-up may yield a better understanding of the clinical course and disease outcome in anti-MOG+ and seronegative NMOSD.
Financial support and sponsorship
Nil.
Conflicts of interest
Authors Lekha Pandit, Sharik Mustafa, Anitha D’Cunha, Chaithra Malli, Akshatha Sudhir, and Toshiyuki Takahashi have no conflict of interest to disclose.
Dr. Sato has received a scholarship from the Ministry of Education, Culture, Sports, Science, and Technology of Japan, grant-in-aid for scientific research from the Japan Society for the Promotion of Science (KAKENHI 15K19472), research support from CAPES/Brasil (CSF-PAJT - 88887.091277/2014-00), and speaker honoraria from Novartis.
Dr. Fujihara serves on scientific advisory boards for Bayer Schering Pharma, Biogen Idec, Mitsubishi Tanabe Pharma Corporation, Novartis Pharma, Chugai Pharmaceutical, Ono Pharmaceutical, Nihon Pharmaceutical, Merck Serono, Alexion Pharmaceuticals, Medimmune, and Medical Review; has received funding for travel and speaker honoraria from Bayer Schering Pharma, Biogen Idec, Eisai Inc., Mitsubishi Tanabe Pharma Corporation, Novartis Pharma, Astellas Pharma Inc., Takeda Pharmaceutical Company Limited, Asahi Kasei Medical Co., Daiichi Sankyo, and Nihon Pharmaceutical, serve as an editorial board member of Clinical and Experimental Neuroimmunology (2009-present) and an advisory board member of Sri Lanka Journal of Neurology has received research support from Bayer Schering Pharma, Biogen Idec Japan, Asahi Kasei Medical, The Chemo-Sero-Therapeutic Research Institute, Teva Pharmaceutical, Mitsubishi Tanabe Pharma, Teijin Pharma, Chugai Pharmaceutical, Ono Pharmaceutical, Nihon Pharmaceutical, and Genzyme Japan, is funded as the secondary investigator (#22229008, 2010–2015) by the grants-in-aid for scientific research from the Ministry of Education, Science, and Technology of Japan and as the secondary investigator by the grants-in-aid for scientific research from the Ministry of Health, Welfare, and Labor of Japan (2010-present).
References
- 1.Pandit L, Asgari N, Apiwattanakul M, Palace J, Paul F, Leite MI, et al. Demographic and clinical features of neuromyelitis optica: A review. Mult Scler. 2015;21:845–53. doi: 10.1177/1352458515572406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jarius S, Wildemann B. AQP4 antibodies in neuromyelitis optica: Diagnostic and pathogenetic relevance. Nat Rev Neurol. 2010;6:383–92. doi: 10.1038/nrneurol.2010.72. [DOI] [PubMed] [Google Scholar]
- 3.Kitley J, Woodhall M, Waters P, Leite MI, Devenney E, Craig J, et al. Myelin-oligodendrocyte glycoprotein antibodies in adults with a neuromyelitis optica phenotype. Neurology. 2012;79:1273–7. doi: 10.1212/WNL.0b013e31826aac4e. [DOI] [PubMed] [Google Scholar]
- 4.Woodhall M, Çoban A, Waters P, Ekizoglu E, Kürtüncü M, Shugaiv E, et al. Glycine receptor and myelin oligodendrocyte glycoprotein antibodies in Turkish patients with neuromyelitis optica. J Neurol Sci. 2013;335:221–3. doi: 10.1016/j.jns.2013.08.034. [DOI] [PubMed] [Google Scholar]
- 5.Sato DK, Callegaro D, Lana-Peixoto MA, Waters PJ, de Haidar Jorge FM, Takahashi T, et al. Distinction between MOG antibody-positive and AQP4 antibody-positive NMO spectrum disorders. Neurology. 2014;82:474–81. doi: 10.1212/WNL.0000000000000101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ramanathan S, Reddel SW, Henderson A, Parratt JD, Barnett M, Gatt PN, et al. Antibodies to myelin oligodendrocyte glycoprotein in bilateral and recurrent optic neuritis. Neurol Neuroimmunol Neuroinflamm. 2014;1:e40. doi: 10.1212/NXI.0000000000000040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pröbstel AK, Rudolf G, Dornmair K, Collongues N, Chanson JB, Sanderson NS, et al. Anti-MOG antibodies are present in a subgroup of patients with a neuromyelitis optica phenotype. J Neuroinflammation. 2015;12:46. doi: 10.1186/s12974-015-0256-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pandit L, Mustafa S, Kunder R, Shetty R, Misri Z, Pai S, et al. Optimizing the management of neuromyelitis optica and spectrum disorders in resource poor settings: Experience from the Mangalore demyelinating disease registry. Ann Indian Acad Neurol. 2013;16:572–6. doi: 10.4103/0972-2327.120474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Waters PJ, McKeon A, Leite MI, Rajasekharan S, Lennon VA, Villalobos A, et al. Serologic diagnosis of NMO: A multicenter comparison of aquaporin-4-IgG assays. Neurology. 2012;78:665–71. doi: 10.1212/WNL.0b013e318248dec1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marignier R, Bernard-Valnet R, Giraudon P, Collongues N, Papeix C, Zéphir H, et al. Aquaporin-4 antibody-negative neuromyelitis optica: Distinct assay sensitivity-dependent entity. Neurology. 2013;80:2194–200. doi: 10.1212/WNL.0b013e318296e917. [DOI] [PubMed] [Google Scholar]
- 11.Wingerchuk DM, Lennon VA, Pittock SJ, Lucchinetti CF, Weinshenker BG. Revised diagnostic criteria for neuromyelitis optica. Neurology. 2006;66:1485–9. doi: 10.1212/01.wnl.0000216139.44259.74. [DOI] [PubMed] [Google Scholar]
- 12.Wingerchuk DM, Lennon VA, Lucchinetti CF, Pittock SJ, Weinshenker BG. The spectrum of neuromyelitis optica. Lancet Neurol. 2007;6:805–15. doi: 10.1016/S1474-4422(07)70216-8. [DOI] [PubMed] [Google Scholar]
- 13.Wingerchuk DM, Banwell B, Bennett JL, Cabre P, Carroll W, Chitnis T, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology. 2015;85:177–89. doi: 10.1212/WNL.0000000000001729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Akaishi T, Sato DK, Nakashima I, Takeshita T, Takahashi T, Doi H, et al. MRI and retinal abnormalities in isolated optic neuritis with myelin oligodendrocyte glycoprotein and aquaporin-4 antibodies: A comparative study. J Neurol Neurosurg Psychiatry. 2016;87:446–8. doi: 10.1136/jnnp-2014-310206. [DOI] [PubMed] [Google Scholar]
- 15.Lennon VA, Wingerchuk DM, Kryzer TJ, Pittock SJ, Lucchinetti CF, Fujihara K, et al. A serum autoantibody marker of neuromyelitis optica: Distinction from multiple sclerosis. Lancet. 2004;364:2106–12. doi: 10.1016/S0140-6736(04)17551-X. [DOI] [PubMed] [Google Scholar]
- 16.Tanaka K, Tani T, Tanaka M, Saida T, Idezuka J, Yamazaki M, et al. Anti-aquaporin 4 antibody in selected Japanese multiple sclerosis patients with long spinal cord lesions. Mult Scler. 2007;13:850–5. doi: 10.1177/1352458507076976. [DOI] [PubMed] [Google Scholar]
- 17.Siritho S, Sato DK, Kaneko K, Fujihara K, Prayoonwiwat N. The clinical spectrum associated with myelin oligodendrocyte glycoprotein antibodies (anti-MOG-Ab) in Thai patients. Mult Scler. 2015:pii: 1352458515614093. doi: 10.1177/1352458515614093. [DOI] [PubMed] [Google Scholar]
- 18.Kim SH, Kim W, Li XF, Jung IJ, Kim HJ. Does interferon beta treatment exacerbate neuromyelitis optica spectrum disorder? Mult Scler. 2012;18:1480–3. doi: 10.1177/1352458512439439. [DOI] [PubMed] [Google Scholar]
- 19.Kim SH, Kim SM, Vincent A, Ahn SW, Hong YH, Park KS, et al. Clinical characteristics, prognosis, and seropositivity to the anti-aquaporin-4 antibody in Korean patients with longitudinally extensive transverse myelitis. J Neurol. 2010;257:920–5. doi: 10.1007/s00415-009-5438-2. [DOI] [PubMed] [Google Scholar]