

Abstract
The x-ray structure of lactose permease of Escherichia coli (LacY) exhibits a single sugar-binding site at the apex of a hydrophilic cavity open to the cytoplasm, and it has been postulated that the binding site has alternating access to either side of the membrane during turnover. Here, the affinity of LacY for ligand in right-side-out or inside-out membrane vesicles is measured in the absence or presence of an H+ electrochemical gradient (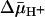 ) by utilizing ligand protection against alkylation. Right-side-out or inside-out membrane vesicles containing LacY with a single cysteine residue at position 148 exhibit KD values for lactose or β-d-galactopyranosyl 1-thio-β-d-galactopyranoside of ≈1.0 mM or 40 μM, respectively, and no systematic change is observed in the presence of
) by utilizing ligand protection against alkylation. Right-side-out or inside-out membrane vesicles containing LacY with a single cysteine residue at position 148 exhibit KD values for lactose or β-d-galactopyranosyl 1-thio-β-d-galactopyranoside of ≈1.0 mM or 40 μM, respectively, and no systematic change is observed in the presence of 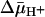 under conditions in which there is little or no accumulation of ligand. The results are consistent with a mechanism in which the major effect of
under conditions in which there is little or no accumulation of ligand. The results are consistent with a mechanism in which the major effect of 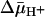 on sugar accumulation is caused by an increased rate of deprotonation on the inner face of the membrane, leading to an increase in the rate of return of the unloaded symporter to the outer face of the membrane.
on sugar accumulation is caused by an increased rate of deprotonation on the inner face of the membrane, leading to an increase in the rate of return of the unloaded symporter to the outer face of the membrane.
Keywords: membranes, bioenergetics, transport, H+ symport, membrane protein structure
The lactose permease of Escherichia coli (LacY), a particularly well studied representative of the major facilitator super-family (MFS) (1), is solely responsible for all translocation reactions catalyzed by the galactoside transport system (2). Similar to many members of the MFS, LacY couples free energy released from downhill translocation of H+ in response to an H+ electrochemical gradient (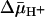 ) to drive the energetically uphill stoichiometric accumulation of α- or β-d-galactopyranosides. Although it has been argued that accumulation of sugar is likely caused by a
) to drive the energetically uphill stoichiometric accumulation of α- or β-d-galactopyranosides. Although it has been argued that accumulation of sugar is likely caused by a 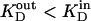 -induced change in the affinity of LacY for substrate on either side of the membrane (
-induced change in the affinity of LacY for substrate on either side of the membrane (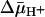 ) (3–5), evidence for this notion is weak (6).
) (3–5), evidence for this notion is weak (6).
Recently, the x-ray structure of LacY mutant C154G in an inward-facing conformation with bound ligand β-d-galactopyranosyl 1-thio-β-d-galactopyranoside (TDG) was solved at a resolution of 3.5 Å (7), confirming many conclusions derived from biochemical and biophysical studies carried out over the past 20 years (reviewed in ref. 2). The molecule is a monomer composed of N- and C-terminal domains, each with six transmembrane helices, symmetrically positioned within the molecule, similar to the crystal structure of GlpT (8), which was reported simultaneously, and the helix-packing model suggested for OxlT (9). A large internal hydrophilic cavity is exposed to the cytoplasm, and a single molecule of ligand is bound at the pseudo twofold axis of symmetry at the apex of the hydrophilic cavity and in the approximate middle of the molecule (7) (Fig. 1A).
Fig. 1.

Postulated structural changes between inward- and outward-facing LacY conformations with bound TDG. (A) Inward-facing conformation viewed parallel to the membrane. (B) A possible model for the outward-facing conformation based on chemical modification and cross-linking experiments, viewed parallel to the membrane. The model was obtained as described in ref. 7.
As shown in the x-ray structure (7), with the exception of Glu-269 (helix VIII), all side chains involved in specificity (i.e., interactions with the galactopyranosyl moiety) are located in the N-terminal domain. A primary interaction is found between the irreplaceable residue Arg-144 (helix V) and the O3 and O4 atoms of the galactopyranosyl ring through a bidentate H bond, as suggested (10–13). Another essential residue, Glu-126 (helix IV), is in close proximity to Arg-144 and likely interacts with the O4, O5, or O6 atoms of TDG through water molecules. Although a proposed salt bridge between Arg-144 and Glu-126 (14, 15) is not observed in the structure, such an interaction may form in the absence of ligand or in another conformation. There is also a hydrophobic interaction between the bottom of the galactopyranosyl ring and the indole ring of Trp-151 (helix V), as proposed (16). Recent fluorescence studies support the contention that Trp-151 is located in the hydrophilic cavity, and phosphorescence experiments demonstrate hydrophobic stacking between the galactopyranosyl and indole rings (17). The binding site in the N-terminal domain bears a striking similarity to that of many other sugar-binding proteins (18, 19). Glu-269 in helix VIII in the C-terminal domain, another irreplaceable residue, forms a salt bridge with Arg-144 as well as an H bond with Trp-151. More recent studies with N-bromosuccinimide (J. Vázquez-Ibar, L.G., A. Weinglass, G. Verner, R. Gordillo, and H.R.K., unpublished data), a tryptophan reagent, provide support for the presence of an H bond between Glu-269 and Trp-151. It has also been suggested that the charge pair between Glu-269 and Arg-144 is necessary to maintain the H bond between Trp-151 and Glu-269 in such a manner as to orient Trp-151 correctly for a proper ligand binding. Although the Trp-151–Glu-269 H bond seems to be important for binding affinity, it is not required for turnover, because mutant W151F exhibits very good lactose transport despite decreased affinity (16). Furthermore, evidence has been presented indicating that Glu-269 plays an important role in ligand binding (13, 20) as well as H+ translocation (21, 22). Finally, excimer fluorescence with pyrene-labeled E269C/H322C LacY (23) and Mn(II) binding by native His-322/E269H LacY (24, 25) indicates that Gly-269 may come into close approximation with His-322 in at least one conformer of LacY. Therefore, it is conceivable that interplay between the two most important residues in the N- and C-terminal domains of LacY (Arg-144 and Glu-269) plays a key role in coupling substrate binding and H+ translocation.
Clearly, an alternative, outward-facing conformation open to the periplasmic side is absolutely required for substrate transport across the membrane. A simulation of the outward-facing conformation has been constructed on the basis of structural flexibility, ligand-induced increases in the reactivity of certain Cys-replacement mutants in the periplasmic region of LacY with N-ethylmaleimide (NEM), and a discrepancy between distances in the crystal structure and distances approximated from thiol cross-linking across the hydrophilic cavity facing the cytoplasm (7) (Fig. 1B). Based on these considerations as a whole, it was postulated that LacY contains a single binding site with alternating access to either side of the membrane during turnover (7) (Fig. 1).
We report here that LacY exhibits comparable binding affinities in right-side-out (RSO) or inside-out (ISO) membrane vesicles for lactose or TDG in the absence or presence of 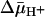 . The observations are consistent with a transport mechanism in which the primary effect of
. The observations are consistent with a transport mechanism in which the primary effect of 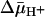 is kinetic and does not involve a significant change in the affinity of the binding site.
is kinetic and does not involve a significant change in the affinity of the binding site.
Materials and Methods
Materials. N-[14C]ethylmaleimide was purchased from DuPont/NEN. Immobilized monomeric avidin was from Pierce, and all unlabeled sugars were obtained from Sigma. All other materials were reagent-grade and obtained from commercial sources.
Construction of Plasmids. Cloning of cassette lacY was as described in ref. 26. Construction of plasmid pKR35/single-Cys-148 lacY containing a C-terminal biotin acceptor domain has also been described (4, 26).
Growth of Cells. E. coli T184 [lacI+ O+ Z-Y- (A) rpsL, met-, thr-, recA, hsdM, hsdR/F′, laclq O+ ZD118 (Y+A+)] (27) containing given plasmid was grown in Luria–Bertani broth with 100 mg/liter ampicillin. Overnight cultures were diluted 10-fold and allowed to grow for 2 h at 37°C before induction with 1 mM isopropyl 1-thio-β-d-galactopyranoside. After additional growth for 2 h at 37°C, cells were harvested by centrifugation.
Preparation of RSO or ISO Membrane Vesicles. RSO membrane vesicles were prepared by osmotic lysis as described in refs. 28 and 29. ISO membrane vesicles were also prepared as described in ref. 30 and washed three times with 50 mM potassium phosphate (pH 7.5)/5 mM MgSO4 and resuspended with the same buffer at a protein concentration of ≈20 mg/ml, frozen in liquid N2, and stored at -80°C until use.
[14C]NEM Labeling. The KD for TDG was determined in situ by alkylation of single-Cys-148 LacY with 0.5 mM [14C]NEM [40 mCi/mmol (1 Ci = 37 GBq)] in the absence or presence of given concentrations of lactose or TDG as described in ref. 31. The procedure for labeling was modified in the following manner. Reactions were carried out on ice, initiated by addition of membrane vesicles, and terminated at 5 min. When the effect of 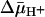 was tested, the vesicles were incubated on ice with d-lactate under oxygen or ATP, as indicated, for 5 min before starting the reaction. In addition, the concentration of membrane protein applied to use was decreased to 10 mg/ml to obtain a membrane potential of at least -80 mV, as measured by accumulation of [3H]tetraphenylphosphonium in the presence of 20 mM lithium d-lactate under oxygen with RSO vesicles at 0°C (32). With ISO vesicles, quenching of bis-(1,3-dibutylbarbituric acid)pentamethine oxonol yielded a membrane potential of approximately +90 to +100 mV in the presence of 10 mM Mg(II)ATP as described (33).
was tested, the vesicles were incubated on ice with d-lactate under oxygen or ATP, as indicated, for 5 min before starting the reaction. In addition, the concentration of membrane protein applied to use was decreased to 10 mg/ml to obtain a membrane potential of at least -80 mV, as measured by accumulation of [3H]tetraphenylphosphonium in the presence of 20 mM lithium d-lactate under oxygen with RSO vesicles at 0°C (32). With ISO vesicles, quenching of bis-(1,3-dibutylbarbituric acid)pentamethine oxonol yielded a membrane potential of approximately +90 to +100 mV in the presence of 10 mM Mg(II)ATP as described (33).
KD values were determined by using the origin computer program (Microcal Software, Northampton, MA) with nonlinear least-squares curve fitting to the following user-defined equation: Y = (1 - P1)/(1 + X/P2) + P1, where P1 is the residual labeling and P2 is the KD. In general, the average KD values given are derived from two to four independent experiments, and the variation was no more than ±10%.
Results
Ligand Protection Against Alkylation of Cys-148 with [14C]NEM. As shown by ligand protection against alkylation with [14C]NEM, single-Cys-148 LacY binds ligand in RSO vesicles with high affinity (4, 12). Here, binding affinity for two substrates in both RSO and ISO vesicles in the absence or presence of 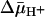 was measured by using the same methodology. Because
was measured by using the same methodology. Because 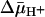 drives transport, it is important to minimize accumulation of the ligand to avoid underestimating KD in RSO vesicles in particular. Therefore, several aspects of the assay were modified. Labeling was carried out on ice for 5 min, at which time the rate of NEM labeling is linear (Fig. 2), and the level of substrate accumulation is essentially nil (34). Furthermore, reactions were started by addition of vesicles without preincubating with sugar. RSO vesicles exhibit a KD of ≈1 mM for lactose (Fig. 3A) and ≈40 μM for TDG (Fig. 4A), values similar to those obtained at room temperature (12, 16). Moreover, under the same conditions, KD values of ≈1 mM for lactose and 49 μM for TDG are obtained for ISO vesicles (Figs. 3B and 4B, respectively). Therefore, it is clear that there is no significant difference within experimental error in the KD of LacY for ligand from either side of the membrane in the absence of
drives transport, it is important to minimize accumulation of the ligand to avoid underestimating KD in RSO vesicles in particular. Therefore, several aspects of the assay were modified. Labeling was carried out on ice for 5 min, at which time the rate of NEM labeling is linear (Fig. 2), and the level of substrate accumulation is essentially nil (34). Furthermore, reactions were started by addition of vesicles without preincubating with sugar. RSO vesicles exhibit a KD of ≈1 mM for lactose (Fig. 3A) and ≈40 μM for TDG (Fig. 4A), values similar to those obtained at room temperature (12, 16). Moreover, under the same conditions, KD values of ≈1 mM for lactose and 49 μM for TDG are obtained for ISO vesicles (Figs. 3B and 4B, respectively). Therefore, it is clear that there is no significant difference within experimental error in the KD of LacY for ligand from either side of the membrane in the absence of 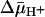 .
.
Fig. 2.

Rate of NEM labeling. Ice-cold RSO vesicles containing single-Cys-148 LacY at 10 mg of protein per ml were added to a solution containing [14C]NEM (40 mCi/mmol, 0.5 mM final concentration). Reactions were carried out on ice and quenched with 10 mM dithiothreitol at the given time. Biotinylated LacY was solubilized in dodecyl β-d-maltopyranoside and purified by affinity chromatography on monomeric avidin Sepharose. Samples were subjected to SDS/12% polyacrylamide gel electrophoresis, and 14C-labeled protein was quantitated with a Typhoon 9410 PhosphorImager (Molecular Dynamics). A.U., arbitrary units.
Fig. 3.

Lactose protection against [14C]NEM labeling of single-Cys-148 LacY. (A and B) Ice-cold RSO or ISO vesicles containing single-Cys-148 mutant at 10 mg of protein per ml were added to a solution containing [14C]NEM (40 mCi/mmol, 0.5 mM final concentration) and given concentrations of lactose and incubated on ice for 5 min. Reactions were quenched with 10 mM dithiothreitol, and biotinylated LacY was purified and analyzed as described for Fig. 2. Labeling in the presence of a given concentration of lactose is expressed as percent labeling observed in the absence of ligand. (C and D) Effect of 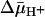 on the lactose protection against Cys-148 alkylation by NEM. Ice-cold RSO or ISO membrane vesicles were added with 20 mM lithium d-lactate under oxygen or 10 mM Mg(II)ATP, respectively, and incubated on ice for 5 min before addition to a solution containing [14C]NEM and given concentrations of lactose as described above (final protein concentration, 10 mg/ml). KD values were calculated as described in Materials and Methods.
on the lactose protection against Cys-148 alkylation by NEM. Ice-cold RSO or ISO membrane vesicles were added with 20 mM lithium d-lactate under oxygen or 10 mM Mg(II)ATP, respectively, and incubated on ice for 5 min before addition to a solution containing [14C]NEM and given concentrations of lactose as described above (final protein concentration, 10 mg/ml). KD values were calculated as described in Materials and Methods.
Fig. 4.

TDG protection against [14C]NEM labeling of single-Cys-148 LacY in RSO or ISO vesicles in the absence and presence of 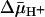 . Experiments were carried out as described for Fig. 3 with given concentrations of TDG. KD values were calculated as described in Materials and Methods.
. Experiments were carried out as described for Fig. 3 with given concentrations of TDG. KD values were calculated as described in Materials and Methods.
Effect of 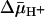 on Binding. With RSO membrane vesicles at a protein concentration of 10 mg/ml, a membrane potential (ΔΨ, interior negative; Fig. 5A)of -80 to -100 mV is measured in the presence of 20 mM d-lactate under oxygen after a 5-min incubation on ice, and the ΔΨ is maintained for at least 5 min, as judged by [3H]tetraphenylphosphonium accumulation (data not shown; see ref. 35). Under these conditions, measurement of ligand binding with RSO vesicles containing single-Cys-148 LacY exhibits KD values of ≈0.9 mM for lactose and 35 μM for TDG (Figs. 3C and 4C, respectively), values within experimental error of those obtained in the absence of
on Binding. With RSO membrane vesicles at a protein concentration of 10 mg/ml, a membrane potential (ΔΨ, interior negative; Fig. 5A)of -80 to -100 mV is measured in the presence of 20 mM d-lactate under oxygen after a 5-min incubation on ice, and the ΔΨ is maintained for at least 5 min, as judged by [3H]tetraphenylphosphonium accumulation (data not shown; see ref. 35). Under these conditions, measurement of ligand binding with RSO vesicles containing single-Cys-148 LacY exhibits KD values of ≈0.9 mM for lactose and 35 μM for TDG (Figs. 3C and 4C, respectively), values within experimental error of those obtained in the absence of 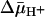 . In contrast, it was reported that generation of
. In contrast, it was reported that generation of 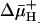 causes a very dramatic decrease in Km for both lactose and TDG (36).
causes a very dramatic decrease in Km for both lactose and TDG (36).
Fig. 5.

Effect of 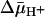 of opposite polarities on substrate translocation in RSO or ISO vesicles. (A)
of opposite polarities on substrate translocation in RSO or ISO vesicles. (A) 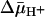 with RSO vesicles generated by addition of d-lactate (ΔΨ, interior negative); substrate is accumulated. (B)
with RSO vesicles generated by addition of d-lactate (ΔΨ, interior negative); substrate is accumulated. (B) 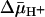 with ISO vesicles generated by addition of 10 mM Mg(II)ATP or 20 mM lithium d-lactate under oxygen (interior positive and/or acid), which is opposite to that of RSO vesicles; substrate effluxes from the vesicles.
with ISO vesicles generated by addition of 10 mM Mg(II)ATP or 20 mM lithium d-lactate under oxygen (interior positive and/or acid), which is opposite to that of RSO vesicles; substrate effluxes from the vesicles.
With ISO vesicles, 10 mM Mg(II)ATP was added to generate a 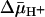 of opposite polarity to that of RSO vesicles (interior positive and/or acid; Fig. 5B). Under these conditions, the internal concentration of ligand should be less than the external concentration, because the polarity of the
of opposite polarity to that of RSO vesicles (interior positive and/or acid; Fig. 5B). Under these conditions, the internal concentration of ligand should be less than the external concentration, because the polarity of the 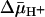 causes substrate to efflux from the vesicles (37). KD values of ≈2.2 mM for lactose and ≈56 μM for TDG are observed (Figs. 3D and 4D, respectively). Therefore, the results indicate that there is little or no effect of
causes substrate to efflux from the vesicles (37). KD values of ≈2.2 mM for lactose and ≈56 μM for TDG are observed (Figs. 3D and 4D, respectively). Therefore, the results indicate that there is little or no effect of 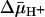 on binding affinity from either side of the membrane.
on binding affinity from either side of the membrane.
Discussion
Recently, the x-ray structures of two members of the MFS, the LacY (7) and the Pi/glycerol-3-phosphate antiporter (GlpT) (8) have been solved. Physiologically, LacY utilizes free energy released from the downhill translocation of H+ to drive galactoside accumulation (symport), whereas GlpT utilizes free energy released from the downhill translocation of Pi from inside the cell to drive glycerol-3-phosphate accumulation (antiport). Remarkably, despite little similarity in primary sequence, both proteins exhibit similar structures. Thus, both transporters are comprised of two bundles of six transmembrane helices symmetrically disposed within the membrane, and the packing of the helices is almost identical (7, 8, 38, 39). In LacY, the bound ligand is located at the pseudo twofold axis of symmetry between the two six-helix bundles in the approximate middle of the molecule. Although the GlpT structure is devoid of substrate, there are two Arg residues likely involved in Pi and glycerol-3-phosphate binding that are also located in the middle of the molecule. Moreover, both proteins exhibit a large hydrophobic cavity open to the cytoplasm, with the binding site at the apex of the cavity, and in both instances, it has been postulated that each transporter has a single binding site with alternating accessibility to either side of the membrane during turnover. However, it is not clear whether 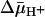 changes binding affinity, particularly with LacY, although it has been shown that ΔΨ and ΔpH have quantitatively the same kinetic (36) and thermodynamic (32, 40) effects on lactose transport.
changes binding affinity, particularly with LacY, although it has been shown that ΔΨ and ΔpH have quantitatively the same kinetic (36) and thermodynamic (32, 40) effects on lactose transport.
Technically, it is difficult to obtain a true KD value for a transport protein, because the ligands used are translocated across the membrane and may accumulate in RSO vesicles in the presence of 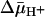 , thereby leading to underestimation of KD. Thus, the experiments presented here were carried out on ice, which decreases substrate accumulation drastically (34). Under these experimental conditions, in the absence of
, thereby leading to underestimation of KD. Thus, the experiments presented here were carried out on ice, which decreases substrate accumulation drastically (34). Under these experimental conditions, in the absence of 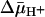 , both ice-cold RSO and ISO vesicles likely equilibrate with the external medium. In the presence of
, both ice-cold RSO and ISO vesicles likely equilibrate with the external medium. In the presence of 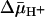 , RSO vesicles still may be able to accumulate lactose or TDG 2- to 3-fold even though the reactions were carried out on ice for only 5 min. Therefore, the measured KD values for RSO vesicles in the presence of
, RSO vesicles still may be able to accumulate lactose or TDG 2- to 3-fold even though the reactions were carried out on ice for only 5 min. Therefore, the measured KD values for RSO vesicles in the presence of 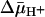 may be underestimated by 2- to 3-fold. However, this is unlikely, because ISO vesicles in the presence of ATP generate a
may be underestimated by 2- to 3-fold. However, this is unlikely, because ISO vesicles in the presence of ATP generate a 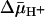 of opposite polarity (interior positive and/or acid) (41), which causes a decrease in the intravesicular concentration of ligand relative to the concentration in the medium (Fig. 5B). Remarkably, the results presented here with two ligands of LacY demonstrate that the KD manifested by ISO vesicles exhibits a <2-fold change in the absence or presence of
of opposite polarity (interior positive and/or acid) (41), which causes a decrease in the intravesicular concentration of ligand relative to the concentration in the medium (Fig. 5B). Remarkably, the results presented here with two ligands of LacY demonstrate that the KD manifested by ISO vesicles exhibits a <2-fold change in the absence or presence of 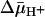 . Moreover, the KD values observed with RSO or ISO vesicles in the absence or presence of
. Moreover, the KD values observed with RSO or ISO vesicles in the absence or presence of 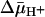 are similar within experimental error. The results provide a strong indication that
are similar within experimental error. The results provide a strong indication that 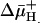 has little or no effect on binding affinity, a conclusion that raises a number of interesting considerations regarding the mechanism by which
has little or no effect on binding affinity, a conclusion that raises a number of interesting considerations regarding the mechanism by which 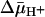 drives accumulation.
drives accumulation.
In the presence of 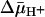 (interior negative and/or alkaline), wild-type LacY can accumulate lactose against an ≈100-fold concentration gradient, and single-Cys-148 LacY accumulates the disaccharide against an ≈20-fold gradient. Without a significant decrease in binding affinity on the inside of the membrane, how does
(interior negative and/or alkaline), wild-type LacY can accumulate lactose against an ≈100-fold concentration gradient, and single-Cys-148 LacY accumulates the disaccharide against an ≈20-fold gradient. Without a significant decrease in binding affinity on the inside of the membrane, how does 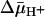 drive lactose accumulation against a concentration gradient? Based on the effect of 2H2O on various translocation reactions, it has been postulated (2, 42) that the rate-limiting step for turnover in the absence of
drive lactose accumulation against a concentration gradient? Based on the effect of 2H2O on various translocation reactions, it has been postulated (2, 42) that the rate-limiting step for turnover in the absence of 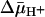 is deprotonation, which precedes return of the unloaded protein to the outer surface of the membrane. It is also noteworthy that the primary kinetic effect of
is deprotonation, which precedes return of the unloaded protein to the outer surface of the membrane. It is also noteworthy that the primary kinetic effect of 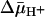 on both lactose and TDG transport is a dramatic decrease in Km (36). Because the most energetically stable form of LacY seems to be the inward-facing conformation with Glu-325 protonated (7), it seems reasonable to suggest that
on both lactose and TDG transport is a dramatic decrease in Km (36). Because the most energetically stable form of LacY seems to be the inward-facing conformation with Glu-325 protonated (7), it seems reasonable to suggest that 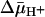 enhances the rate of deprotonation on the inner surface of the membrane and thereby allows unloaded LacY to return to the outward-facing conformation more rapidly. Thus, the major contribution of
enhances the rate of deprotonation on the inner surface of the membrane and thereby allows unloaded LacY to return to the outward-facing conformation more rapidly. Thus, the major contribution of 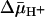 on active transport by LacY seems to be kinetic, with little or no change in affinity for sugar.
on active transport by LacY seems to be kinetic, with little or no change in affinity for sugar.
Acknowledgments
We thank Miklós Sahin-Tóth for initiating this project, Shushi Nagamori for technical advice with preparation of the ISO vesicles, and Ernest Wright for critical and insightful comments and suggestions. This work was supported in part by National Institutes of Health Grant DK51131:09 (to H.R.K.).
Abbreviations: LacY, lactose permease; MFS, major facilitator superfamily; TDG, β-d-galactopyranosyl 1-thio-β-d-galactopyransoside; NEM, N-ethylmaleimide; RSO, right-side out; ISO, inside out; 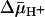 , H+ electrochemical gradient.
, H+ electrochemical gradient.
References
- 1.Saier, M. H., Jr. (2000) Mol. Microbiol. 35, 699-710. [DOI] [PubMed] [Google Scholar]
- 2.Kaback, H. R., Sahin-Tóth, M. & Weinglass, A. B. (2001) Nat. Rev. Mol. Cell Biol. 2, 610-620. [DOI] [PubMed] [Google Scholar]
- 3.Schuldiner, S. & Kaback, H. R. (1977) Biochim. Biophys. Acta 472, 399-418. [DOI] [PubMed] [Google Scholar]
- 4.Frillingos, S. & Kaback, H. R. (1996) Biochemistry 35, 3950-3956. [DOI] [PubMed] [Google Scholar]
- 5.Lolkema, J. S., Carrasco, N. & Kaback, H. R. (1991) Biochemistry 30, 1284-1290. [DOI] [PubMed] [Google Scholar]
- 6.Overath, P., Teather, R. M., Simoni, R. D., Aichele, G. & Wilhelm, U. (1979) Biochemistry 18, 1-11. [DOI] [PubMed] [Google Scholar]
- 7.Abramson, J., Smirnova, I., Kasho, V., Verner, G., Kaback, H. R. & Iwata, S. (2003) Science 301, 610-615. [DOI] [PubMed] [Google Scholar]
- 8.Huang, Y., Lemieux, M. J., Song, J., Auer, M. & Wang, D. N. (2003) Science 301, 616-620. [DOI] [PubMed] [Google Scholar]
- 9.Hirai, T., Heymann, J. A., Maloney, P. C. & Subramaniam, S. (2003) J. Bacteriol. 185, 1712-1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Frillingos, S., Gonzalez, A. & Kaback, H. R. (1997) Biochemistry 36, 14284-14290. [DOI] [PubMed] [Google Scholar]
- 11.Venkatesan, P., Hu, Y. & Kaback, H. R. (2000) Biochemistry 39, 10656-10661. [DOI] [PubMed] [Google Scholar]
- 12.Sahin-Tóth, M., le Coutre, J., Kharabi, D., le Maire, G., Lee, J. C. & Kaback, H. R. (1999) Biochemistry 38, 813-819. [DOI] [PubMed] [Google Scholar]
- 13.Sahin-Tóth, M., Karlin, A. & Kaback, H. R. (2000) Proc. Natl. Acad. Sci. USA 97, 10729-10732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wolin, C. D. & Kaback, H. R. (2000) Biochemistry 39, 6130-6135. [DOI] [PubMed] [Google Scholar]
- 15.Zhao, M., Zen, K. C., Hubbell, W. L. & Kaback, H. R. (1999) Biochemistry 38, 7407-7412. [DOI] [PubMed] [Google Scholar]
- 16.Guan, L., Hu, Y. & Kaback, H. R. (2003) Biochemistry 42, 1377-1382. [DOI] [PubMed] [Google Scholar]
- 17.Vazquez-Ibar, J. L., Guan, L., Svrakic, M. & Kaback, H. R. (2003) Proc. Natl. Acad. Sci. USA 100, 12706-12711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Merritt, E. A., Sarfaty, S., Feil, I. K. & Hol, W. G. (1997) Structure (London) 5, 1485-1499. [DOI] [PubMed] [Google Scholar]
- 19.Duan, X. & Quiocho, F. A. (2002) Biochemistry 41, 706-712. [DOI] [PubMed] [Google Scholar]
- 20.Weinglass, A. B., Sondej, M. & Kaback, H. R. (2002) J. Mol. Biol. 315, 561-571. [DOI] [PubMed] [Google Scholar]
- 21.Ujwal, M. L., Sahin-Tóth, M., Persson, B. & Kaback, H. R. (1994) Mol. Membr. Biol. 11, 9-16. [DOI] [PubMed] [Google Scholar]
- 22.Franco, P. J. & Brooker, R. J. (1994) J. Biol. Chem. 269, 7379-7386. [PubMed] [Google Scholar]
- 23.Jung, K., Jung, H., Wu, J., Privé, G. G. & Kaback, H. R. (1993) Biochemistry 32, 12273-12278. [DOI] [PubMed] [Google Scholar]
- 24.Jung, K., Voss, J., He, M., Hubbell, W. L. & Kaback, H. R. (1995) Biochemistry 34, 6272-6277. [DOI] [PubMed] [Google Scholar]
- 25.He, M., Voss, J., Hubbell, W. L. & Kaback, H. R. (1997) Biochemistry 36, 13682-13687. [DOI] [PubMed] [Google Scholar]
- 26.van Iwaarden, P. R., Driessen, A. J., Menick, D. R., Kaback, H. R. & Konings, W. N. (1991) J. Biol. Chem. 266, 15688-15692. [PubMed] [Google Scholar]
- 27.Teather, R. M., Bramhall, J., Riede, I., Wright, J. K., Furst, M., Aichele, G., Wilhelm, U. & Overath, P. (1980) Eur. J. Biochem. 108, 223-231. [DOI] [PubMed] [Google Scholar]
- 28.Kaback, H. R. (1971) Methods Enzymol. 22, 99-120. [Google Scholar]
- 29.Short, S. A., Kaback, H. R. & Kohn, L. D. (1974) Proc. Natl. Acad. Sci. USA 71, 1461-1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nagamori, S., Vazquez-Ibar, J. L., Weinglass, A. B. & Kaback, H. R. (2003) J. Biol. Chem. 278, 14820-14826. [DOI] [PubMed] [Google Scholar]
- 31.Sahin-Tóth, M., Gunawan, P., Lawrence, M. C., Toyokuni, T. & Kaback, H. R. (2002) Biochemistry 41, 13039-13045. [DOI] [PubMed] [Google Scholar]
- 32.Ramos, S., Schuldiner, S. & Kaback, H. R. (1976) Proc. Natl. Acad. Sci. USA 73, 1892-1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Matsushita, K., Ohnishi, T. & Kaback, H. R. (1987) Biochemistry 26, 7732-7737. [DOI] [PubMed] [Google Scholar]
- 34.Kaback, H. R. & Barnes, E. M., Jr. (1971) J. Biol. Chem. 246, 5523-5531. [PubMed] [Google Scholar]
- 35.Felle, H., Porter, J. S., Slayman, C. L. & Kaback, H. R. (1980) Biochemistry 19, 3585-3590. [DOI] [PubMed] [Google Scholar]
- 36.Robertson, D. E., Kaczorowski, G. J., Garcia, M. L. & Kaback, H. R. (1980) Biochemistry 19, 5692-5702. [DOI] [PubMed] [Google Scholar]
- 37.Kaczorowski, G. J., Robertson, D. E. & Kaback, H. R. (1979) Biochemistry 18, 3697-3704. [DOI] [PubMed] [Google Scholar]
- 38.Abramson, J., Kaback, H. R. & Iwata, S. (2004) Curr. Opin. Struct. Biol., in press. [DOI] [PubMed]
- 39.Abramson, J., Iwata, S. & Kaback, H. R. (2004) Mol. Membr. Biol., in press. [DOI] [PubMed]
- 40.Ramos, S. & Kaback, H. R. (1977) Biochemistry 16, 854-859. [DOI] [PubMed] [Google Scholar]
- 41.Reenstra, W. W., Patel, L., Rottenberg, H. & Kaback, H. R. (1980) Biochemistry 19, 1-9. [DOI] [PubMed] [Google Scholar]
- 42.Viitanen, P., Garcia, M. L., Foster, D. L., Kaczorowski, G. J. & Kaback, H. R. (1983) Biochemistry 22, 2531-2536. [DOI] [PubMed] [Google Scholar]