

Abstract
IMP dehydrogenase (IMPDH) is the rate-limiting enzyme for de novo GMP synthesis. Its activity is correlated with cell growth, and it is the target of a number of proven and experimental drug therapies including mycophenolic acid (MPA). MPA inhibits the enzyme by trapping a covalent nucleotide-enzyme intermediate. Saccharomyces cerevisiae has four IMPDH genes called IMD1—IMD4. IMD2 is transcriptionally regulated and is the only one that enables yeast to grow in the presence of MPA. We show here that de novo synthesis of the IMD2-encoded protein is strongly induced upon MPA treatment. We also monitor the in vivo formation of a covalent nucleotide-enzyme intermediate for Imd2, Imd3, and Imd4 that accumulates in the presence of MPA. Complete formation of the Imd2 intermediate requires drug concentrations manyfold higher than that required to quantitatively trap the Imd3- or Imd4-nucleotide adducts. Purification of the tagged IMD gene products reveals that the family of polypeptides coassemble to form heteromeric IMPDH complexes, suggesting that they form mixed tetramers. These data demonstrate that S. cerevisiae harbor multiple IMPDH enzymes with varying drug sensitivities and offer an assay to monitor the inhibition of IMPDH in living cells. They also suggest that mixed inhibition profiles may result from heteromeric complexes in cell types that contain multiple IMPDH gene products. The mobility shift assay could serve as a tool for the detection of drug-inactivated IMPDH in the cells of patients receiving MPA therapy.
IMP dehydrogenase (IMPDH) is an important enzyme required for the de novo synthesis of guanine nucleotides. It catalyzes the conversion of IMP to XMP with the concomitant reduction of NAD+. Its abundance and activity correlate with the growth of cells, and it has been the target of antiviral, antimicrobial, anti-cancer, and immunosuppressive therapies (1). Yeast has a four-gene family encoding IMPDH-like proteins, of which one, IMD1, is a pseudogene (2). The gene family is essential for growth on rich media but dispensable if guanine is provided exogenously. Either IMD2, IMD3, or IMD4 enables growth in the absence of guanine and therefore supports prototrophy. Only IMD2 provides resistance to the drug mycophenolic acid (MPA), and its expression is tightly regulated by guanine nucleotide levels. It is repressed under guanine-replete conditions and induced when guanine nucleotides are low (3, 4).
MPA is a well characterized inhibitor of IMPDH (5). Across phyla, drug resistance varies by many orders of magnitude. For example, bacterial enzymes are highly resistant, whereas human IMPDH is relatively sensitive (6). Yeast attain resistance in part because IMD2 is transcriptionally induced and highly dependent upon efficient transcription elongation (3, 4, 7). Indeed, many mutations of genes encoding the yeast transcription machinery show sensitivity to MPA due to their inability to up-regulate expression (3). The Imd2 protein, however, also seems intrinsically more MPA-resistant than Imd3 or Imd4, despite having 93% and 84% amino acid identity, respectively (2).
IMPDH enzymes from many organisms are found as tetramers (1, 8). When generated as recombinant proteins for structural and enzymatic characterization, the homotetramer has been the focus of study (9). There are two functional IMPDH genes in the human and mouse genome (10). Specific cell types can display expression of more than one closely related isoform and therefore have the potential to assemble heterotetramers. This fact, and the large gene family in yeast, led us to examine the assembly state of yeast IMPDH. Here, we have studied yeast strains with tandem affinity purification (TAP)-tagged IMD genes. We have developed an assay that can identify the MPA-inhibited species of IMPDH in vivo. We also obtained evidence that the IMPDH family in yeast exists as a heteromeric complex of Imd polypeptides. The rapid accumulation of Imd2 protein in response to drug is confirmed and suggests that a spectrum of tetramers of differing composition can be formed under different conditions in which subunit pool size is altered.
Materials and Methods
Plasmid and Strain Construction. The plasmid pIMD2-TAG-S288C/426 was made by inserting an XhoI and BsrG1 cut PCR product (containing the TAP-tag from chromosomal DNA prepared from strain DY1100 using 5′-TTGGTATGGGTTCAGGCTCT and 5′-ACGGGTCTCGAGTCGGTGTCGATGTAAGTGGA) into pIMD2-S288C (7) cut with the same restriction enzymes. The same XhoI-BsrG1 fragment was removed from pIMD2-TAG-S288C/426 and inserted into pRS316-IMD2 (2) to generate pIMD2TAP-316, a CEN version of this plasmid. The plasmid pIMD2-TAP-C335A was made by site-directed mutagenesis of the TGT codon 335 to GCT (Gene Dynamics, Tigard, OR) of pIMD2TAP-316.
The strains used in this study are shown in Table 1. Strains DY1100 and DY1101 were made by homologous recombination of a TAP-tag-containing PCR product amplified from pBS1479 (CellZome AG, Heidelberg, Germany), as described by Puig et al. (11), into the trp1- strain BY4741-7202 (Research Genetics, Huntsville, AL). The PCR products contained homologous sequences downstream of IMD3 and IMD4 for DY1100 and DY1101, respectively. The upstream primer used to create DY1100 (5′-TGGTGTTCATAACTTACATTCTTACGAAAAGCGTTTACAAAACTCCATGGAAAAGAGAAG) contains 17 nucleotides that anneal to the beginning of the TAP-tag portion of pBS1479 and 43 nucleotides with homology to the final 43 nucleotides of IMD3. The downstream primer, 5′-TTTCTTGATTTCCATACTTAACTGCGAGCAGAATAAATATAAATACGACTCACTATAGGC, anneals to the pBS1479 TAP-tag and contains homology to 43 nucleotides, 18 base pairs downstream of the IMD3 stop. DY1101 was created in a similar manner by using 5′-TGGTGTCCATAATTTGCACTCCTATGAAAA ACGTCTATACA AT TCCATGGAAAGAGAG and 5′-ATTAGGTCTGTTCTTTATTTTAGTTTTGTTGATCTCTATGAAGTACGACTCACTATAGGG. Transformants were tested for correct integration by PCR screening of chromosomal DNA.
Table 1. Yeast strains used in this study.
| Strain | Genotype |
|---|---|
| DY419 | MATα his3Δ1 leu2Δ0 ura3Δ0 MET15 lys2Δ0 Δimd1::HIS3 Δimd2::LEU2 Δimd3::kanMX4 Δimd4::LYS2 [pIMD2-TAG-S288C/426] |
| DY423 | MATα his3Δ1 leu2Δ0 ura3Δ0 MET15 lys2Δ0 Δimd1::HIS3 Δimd2::LEU2 Δimd3::kanMX4 Δimd4::LYS2 [pIMD2TAP-316 (URA3)] |
| DY424 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 [pIMD2TAP-316 (URA3)] |
| DY441 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 IMD2-TAP::HIS3MX6 |
| DY442 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 IMD2-TAP::HIS3MX6 [pRS316 (URA3)] |
| DY1100 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 Δtrp1::kanMX4 IMD3-TAP::TRP1 |
| DY1101 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 Δtrp1::kanMX4 IMD4-TAP::TRP1 |
| DY1103 | MATα his3Δ1 leu2Δ0 ura3Δ0 MET15 lys2Δ0 Δimd1::HIS3 Δimd2::LEU2 IMD3-TAP::TRP1 Δimd4::URA3 |
| DY1107 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 Δtrp1::kanMX4 IMD3::TAP::TRP1 [pRS316 (URA3)] |
| DY1108 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 Δtrp1::kanMX4 IMD4-TAP::TRP1 [pRS316 (URA3)] |
| DY1112 | MATα his3Δ1 leu2Δ0 ura3Δ0 MET15 LYS2 Δimd1::HIS3 Δimd2::LEU2 IMD3::TAP::TRP1 |
| DY2040 | MATα his3Δ1 leu2Δ0 ura3Δ0 MET15 lys2Δ0 Δimd1::HIS3 Δimd2::LEU2 Δimd3::kanMX4 Δimd4::LYS2 [pIMD2-TAP-C335A (URA3)] |
Strains DY1103 and DY1102 were generated by crossing DY887 [deleted for all four IMDs (2)] to DY1100 or DY1101, which contained tagged IMD3 and IMD4, respectively. The resulting diploids were sporulated on guanine-containing medium, and spores were selected that contained either TAP-IMD3 (DY1103) or TAP-IMD4 (DY1102) and no other intact IMDsby selecting for the nutritional marker genes used to delete the respective IMDs. Genotypes were confirmed by PCR on genomic DNA. DY1102 was transformed with pRS316 (12) to generate DY1109.
DY442, DY1107, and DY1108 were obtained by transforming DY441 (YSC1178-7500975, Open Biosystems, Huntsville, AL), DY1100, and DY1101 with pRS316, respectively. DY424 was obtained by transforming BY4741 (Research Genetics, Huntsville, AL) with pIMD2TAP-316.
DY419 and DY423 were generated by transforming DY891 with the plasmids pIMD2-TAG-S288C/426 and pIMD2TAP-316, respectively. DY891 was generated by exchanging the URA3 cassette used to delete IMD4 in DY887 (2) with LYS2. The LYS2 cassette was integrated into the IMD4 locus by homologous recombination by using a PCR product generated from S288C genomic DNA by using the primers 5′-ACCAATTCCATAGCTTTGAAGAAACCTAACAAACATTTTACGATGTATGGGAAGAGCTTTCTAAGTCTGA and 5′-TTATATGCAAAAATAAACTTTTAAATATCTATGGATG CTTACTCAATGCTACAATAAACCAAGATGAAGC. DY1112 was generated by sporulating the diploid generated by mating DY1100 and DY887, selecting spores that were HIS+LEU+TRP+URA- and identifying by PCR an isolate that contained the TAP-IMD3. DY891 was transformed with pIMD2-TAP-C335A to yield DY2040.
MPA Treatment of Living Cells. Cells were grown to saturation in yeast extract/peptone/dextrose (YPD) or synthetic complete medium minus uracil (SC - ura) (13), diluted to an OD600 of 0.1 and grown to an OD600 of 0.5. Untreated aliquots were collected, and the remaining culture was made 15 μg/ml in MPA (Sigma, 10 mg/ml stock in dimethyl sulfoxide). Protein extract was prepared at various times after treatment by vortexing cells with glass beads, and 2 μg of protein was separated on a 7% SDS/polyacrylamide gel, transferred to nitrocellulose, stained with peroxidase-conjugated anti-peroxidase antibody (Sigma) and chemiluminescent reagents (Amersham Pharmacia Biosciences), and visualized by exposure to x-ray film
MPA Treatment of Cell Extracts and Purified Proteins. Cells were grown to OD600 of 0.5, washed with water, and resuspended in 0.2 ml of 10 mM Hepes (pH 7.9), 10 mM KCl, 1.5 mM MgCl2, 0.5 mM DTT, and protease inhibitors (Roche, Mannheim, Germany). A protein extract was prepared by vortexing cells with glass beads. Ten micrograms of extract or 6 μg of purified protein were incubated at 30°C for 30 min with 470 μM MPA, 5 mM NAD+, and 1 mM IMP as indicated. Samples were subjected to electrophoresis and Western blotting.
Purification of TAP-Tagged Proteins. Strains were grown to an OD600 of 0.5–1, washed in water, and frozen at -80°C. Thawed cells were lysed at 4°C in 1 vol of 0.5-mm glass beads and 1 vol of buffer (ref. 11; 10 mM Hepes, pH 7.9/10 mM KCl/1.5 mM MgCl2/0.5 mM DTT/0.5 mM phenylmethylsulfonyl fluoride/2 mM benzamidine/1 μM leupeptin/2 μM pepstatin A/4 μM chymostatin/2.6 μM aprotinin) by using a mechanical bead beater (Biospec Products, Bartlesville, OK). The extract was prepared for chromatography on IgG agarose (Sigma) or IgG Sepharose (Pharmacia) and eluted with tobacco etch virus (TEV) protease as described by Puig et al. (11). Fractions were pooled and dialyzed in 60 mM KCl, 50 mM Tris (pH 7.5), 1 mM DTT, and 10% glycerol.
Enzyme Assays. One microgram (≈16 pmols of monomer) of purified protein was incubated with varying amounts of IMP and NAD+ in 200 μl of 100 mM KCl, 3 mM EDTA, 1 mM DTT, and 50 mM Tris (pH 8). Samples were incubated at 37°C for 30 min, and absorbance at 340 nm was measured. An extinction coefficient of 6.22 mM-1·cm-1 was used for NADH, and activity was plotted by using prism (GraphPad, San Diego).
Results
To study the yeast IMPDH family, we TAP-tagged the chromosomal copies of IMD3 and IMD4 by homologous recombination (11). The TAP-tag enables affinity purification and antibody detection of proteins through the tag's protein A peptides. IMD2 was more refractory to tagging by homologous recombination so we introduced a plasmid with a TAP-tagged version of IMD2 into cells. Each of these TAP-tagged genes rescued the guanine auxotrophy of the imd1Δ imd2Δ imd3Δ imd4Δ quadruple deletant, confirming that the modified proteins are biologically active (data not shown). Plasmid-borne TAP-IMD2 also rescued the MPA sensitivity of the quadruple knockout (data not shown). We did not tag IMD1 because it is likely to be a pseudogene (2).
We have previously shown that IMD2, and to a lesser extent IMD4, mRNA levels are strongly induced during MPA challenge (2). Others have shown that Imd3 protein is also up-regulated by MPA (14). To compare the resting and induced levels of the Imd family of proteins, we treated strains harboring each TAP-tagged Imd with MPA. Total cellular protein was examined by Western blotting to detect the TAP-tag (Fig. 1). A strain with a TAP-tagged chromosomal copy of IMD2 recently became available and was also analyzed (ref. 15; Open Biosystems). As suggested by the relative levels of their mRNAs (2), Imd2 was the least abundant of the three yeast IMPDH-like proteins in the absence of drug (Fig. 1, “0” times). Consistent with previous Northern blotting data, Imd2 was the most strongly induced protein of the three (Fig. 1). A modest drug-induced increase in Imd3 and Imd4 levels was also observed. Interestingly, Imd3 and Imd4 were quantitatively shifted, in a drug-dependent manner, to a lower mobility form that was stable to denaturing gel electrophoresis. A less efficient conversion of Imd2 to a slow mobility form was detected in cells expressing relatively high levels of TAP-Imd2 from a plasmid or endogenous levels from its natural chromosomal location (Fig. 1).
Fig. 1.

Induction and modification of TAP-tagged IMPDH in vivo. Strains DY424 (plasmid IMD2-TAG), DY442 (chromosomal IMD2-TAG), DY1107 (IMD3-TAG), and DY1108 (IMD4-TAG) were grown to an OD600 of 0.5 in synthetic complete medium minus uracil. MPA (470 nM) was added, and protein was harvested at the indicated times. One microgram of total cellular protein was subjected to Western blotting and chemiluminescent detection.
MPA inhibition of IMPDH results from the noncovalent binding of the drug to a naturally occurring, covalent enzymenucleotide intermediate formed between IMP and the active site cysteine's side chain (16). In so doing, MPA traps the inactive enzyme in mid-catalysis before the oxidized nucleotide is released, thereby preventing consummation of the catalytic cycle (16). We tested the idea that the slower mobility form of the enzyme is this intermediate by reconstituting its production in an in vitro cell-free reaction in which we could show dependence of the modification on the substrates IMP and NAD+ as well as MPA. Fig. 2A demonstrates that the shifted form of Imd3 could be produced in a dialyzed extract supplemented with all three small molecules (lane 1 vs. 2). Formation of the slower moving form depended on both substrates (lane 2 vs. lanes 4 and 5). Small amounts of the shifted form were detected in the uninhibited reaction in the presence of substrates (compare lane 3 with lanes 1 and 2). This result may represent small amounts of intermediate that accumulate at steady-state in the natural course of the catalytic cycle. Indeed, release of the oxidized nucleotide is the slow step of the cycle for some IMPDHs (17, 18).
Fig. 2.

Requirements for modification of IMPDH in vitro. Extract from DY1107 (TAP-IMD3), DY424 (TAP-IMD2), or DY2040 (TAP-IMD2C335A) was incubated at 30°C with the indicated substrates ± 47 μM MPA (A) or varying amounts of MPA in the presence of IMP and NAD+ (B). (C) Strain DY424 was incubated in synthetic complete (SC) media containing 15 μg/ml MPA for 120, 150, or 240 min as indicated. An aliquot of cells treated for 120 min was washed twice with half vol of water for 20 min (120 min + 20 min wash), and returned to 30°C in SC media lacking drug for 30 or 120 additional min (120 min + 50 min wash; and 120 min + 140 min wash) and analyzed by Western blotting vs. the TAP-tag.
The nucleotide-enzyme intermediate is formed by attack of the active site cysteine on C2 of the purine ring (19). This cysteine is absolutely required for the activity of mammalian IMPDH (20), and we have shown that substitution of alanine for the cognate cysteine (335) in IMD2 inactivates the protein's ability to provide drug resistance in vivo (7). To provide further evidence that the slower mobility form of the Imds is due to formation of the trapped intermediate, we added MPA to an extract from a strain with a plasmid-borne copy of the Cys335Ala substitution in TAP-Imd2. This Imd2 derivative did not display a shift under conditions that shift the wild-type enzyme (Fig. 2 A, lanes 6 and 7 vs. Fig. 2C). Treatment of living cells also failed to produce the shifted intermediate for the C335A-substituted enzyme (data not shown). We conclude that the slow mobility form of these IMPDHs is a covalent nucleotide-enzyme intermediate and that we can detect MPA inhibition of IMPDHs in vivo and in vitro.
At the commonly used drug concentration used to treat living cells (470 nM), it seemed that Imd3 and Imd4 were quantitatively converted to the slower mobility form in 30 min, whereas Imd2 was only partially converted even after 2 h of treatment (Fig. 1). This result is consistent with the idea that Imd2 is a relatively drug-resistant form of the enzyme. To examine this finding more closely, we titrated MPA into cell-free reactions containing TAP-Imd2 or TAP-Imd3 to compare their susceptibility with the drug (Fig. 2B). TAP-Imd3 was quantitatively trapped in the covalent nucleotide-bound state at <5 μM MPA. TAP-Imd2 required ≈500 μM for complete conversion. Similarly, halfmaximal conversion was achieved with <500 nM MPA for TAP-Imd3 vs. 50 μM for TAP-Imd2. We conclude that Imd2 is greater than two orders of magnitude more resistant to MPA than Imd3.
To test the reversibility of the inhibition, we treated cells expressing TAP-Imd2 with MPA for 120, 150, or 240 min to generate the shifted species (Fig. 2C). Cells were washed in water and resuspended in fresh medium lacking drug. As quickly as 20 min after drug removal, the shifted species had disappeared (Fig. 2C). These data suggest that the two forms of the enzyme inhibited in vivo are interconvertible, as would be expected from reversible drug binding, although it remains possible that the inhibited form of the enzyme is degraded and that uninhibited enzyme was regenerated by new synthesis. This result would have to be true in our cell-free conditions, however, because similar results were obtained with in vitro-modified enzyme upon dilution of the MPA after inhibition (data not shown).
Imd2, -3, and -4 were independently purified by means of their TAP-tag and were resolved by denaturing gel electrophoresis. Each preparation possessed two predominant polypeptides as visualized by staining denaturing gels with Coomassie blue (Fig. 3, +all IMDs lanes). Mass spectrometry of tryptic peptides derived from each revealed that both were Imd polypeptides (data not shown). Because IMPDH enzymes from numerous organisms are known to form tetramers (8, 9, 21, 22), we considered the possibility that TAP-tagged Imds were coisolating the untagged isoforms in a tetrameric complex. In other words, TAP-Imd2 had copurified untagged Imd3 and/or Imd4, and, similarly, TAP-Imd3 was isolated with tightly bound Imd2 and/or Imd4. To test this idea, we established strains with TAP-IMD2 or TAP-IMD3 as the sole IMD and purified the respective TAP-complexes (Fig. 3, +no other IMDs lanes). Indeed, only the larger, TAP-tagged protein was isolated from these cells. We conclude that yeast can generate a complex family of IMPDH with subunit contributions from more than one of the three gene products.
Fig. 3.

TAP-tagged Imds copurify with untagged Imds. TAP-tagged proteins were isolated on an IgG-containing matrix from strains DY441, DY1100, DY1101, DY419, and DY1103 (left to right), resolved by polyacrylamide gel electrophoresis, and stained with Coomassie blue. The polypeptide at ≈100 kDa in some samples is a contaminant derived from the affinity matrix.
Having the purified forms of the TAP-Imds enabled us to test whether the enzyme alone was able to generate the shifted intermediate in the absence of additional proteins. Purified TAP-Imd2 and TAP-Imd3 isolated from strains lacking any other Imds were incubated with MPA, IMP, and NAD+ and analyzed by direct staining after polyacrylamide gel electrophoresis. TAP-Imd2 and TAP-Imd3 were able to generate the shifted species, and again Imd3 did so more completely than Imd2 at 470 μM MPA (Fig. 4). We also tested tagged-Imd3 purified from cells containing untagged IMD4 as the only other IMD. Both polypeptides were efficiently shifted to slower mobility forms in the presence of MPA, further confirming the identity of both bands as active Imd subunits. These data provide strong evidence that the MPA-inhibited form of all IMPDHs in yeast can be identified and that heteromeric IMPDH complexes form naturally.
Fig. 4.

Modification of purified yeast IMPDH in vitro. Affinity-purified TAP-Imd2 and TAP-Imd3 were purified from DY423, DY1103, and DY1112 (Left, Center, and Right, respectively) and incubated alone or in complete reactions with IMP, NAD+, and MPA. Two micrograms of each protein were separated by polyacrylamide gel electrophoresis and stained with Coomassie blue.
Formation of the covalent nucleotide intermediate represent a partial reaction in which IMP is oxidized but the product, XMP, is not released. IMPDH activity has not yet been demonstrated for purified yeast IMPDH although activity has been detected in crude extracts (23). We assayed TAP-Imd3 (Fig. 3, lane 2) for IMPDH activity by the conventional measurement of NADH production. We obtained catalytic activity that was linear with time (up to 2 h) and enzyme concentration (up to 130 pmol per reaction) (data not shown). The enzyme displayed saturable kinetics toward NAD+ (Fig. 5A) and IMP (Fig. 5B). The 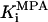 of IMPDH enzymes from different organisms correlates directly with the magnitude of the Km for NAD+ (6). The apparent Km for NAD+ of 314 ± 61 μM measured here (Fig. 5A) was between that measured for the MPA-sensitive human type II enzyme (21 μM; ref. 24) and the relatively MPA-resistant E. coli enzyme (2 mM; ref. 25).
of IMPDH enzymes from different organisms correlates directly with the magnitude of the Km for NAD+ (6). The apparent Km for NAD+ of 314 ± 61 μM measured here (Fig. 5A) was between that measured for the MPA-sensitive human type II enzyme (21 μM; ref. 24) and the relatively MPA-resistant E. coli enzyme (2 mM; ref. 25).
Fig. 5.

Kinetics of TAP-Imd3 activity and MPA inhibition. TAP-Imd3 purified from DY1100 was assayed for IMPDH activity at 1 mM IMP while varying NAD+ (A) or at 1.8 mM NAD+ while varying IMP (B). (C) TAP-Imd2 (from DY423) and TAP-Imd3 (DY1103) were purified and assayed for enzyme activity at 2 mM NAD+ and 0.5 mM IMP in the presence of increasing amounts of MPA. Activity was set at 100% for the samples without drug (n = 3 for Imd3 and n = 7 for Imd2). The drug-containing reactions (n = 3 for Imd3 and n = 7 for all Imd2 points except 25 μM, which was n = 4) were expressed as a percentage of this control and averaged. Error bars represent the SEM. Under these conditions, the absolute specific activity of Imd2 was approximately one-fourth that of Imd3 in the absence of drug.
To directly test whether Imd2 is more MPA-resistant than Imd3, we assayed both enzymes purified from strains lacking any other Imds (Fig. 3, right two lanes) in the presence of increasing concentrations of drug (Fig. 5C). Consistent with this idea, Imd2 displayed an EC50 value approximately two orders of magnitude greater than Imd3.
Discussion
The enzymology of IMPDH and the mechanism of its inhibition by MPA are well understood. Much of our biochemical knowledge has been derived from studies using recombinant and therefore homotetrameric IMPDH. Here, we have exploited molecular genetics to tag the Saccharomyces cerevisiae IMD gene products, biochemically isolate them in vitro, and genetically isolate them in vivo to investigate the complex IMPDH family in this organism. We confirmed that de novo synthesis of Imd2 protein is strongly induced after drug challenge, which follows from the known transcriptional induction of its promoter by MPA (3, 4). Our data also support the previous suggestion that the ability of IMD2 to provide yeast with MPA-resistant growth is due to intrinsic features of the Imd2 protein in addition to its enhanced production after drug treatment (2).
We provide strong evidence that the MPA-inhibited form of each of the family members can be detected in living cells. The basis of the discrimination between inhibited and uninhibited enzyme is due to a drug-dependent and substrate-dependent change in migration of the protein upon gel electrophoresis that requires its enzymatic activity. It seems likely that the shift results from a conformational difference of the modified and unmodified form as well as the chemical difference of a covalently attached nucleotide, because the magnitude of the mobility change (corresponding to ≈8,000 Da) is greater than that expected based upon the additional mass of the added nucleotide. The modification is acid- and thiol-stable. We were able to show that Imd3 and Imd4, which cannot support growth in the presence of drug, were quantitatively modified. In contrast, Imd2, which enables growth in the presence of drug, is less readily inhibited in vivo and in vitro, confirming our earlier hypothesis that its activity in vivo is intrinsically more resistant to MPA than that of Imd3 or Imd4 (2). This finding was borne out in a direct enzymatic assay. It has been suggested that part of the differential MPA sensitivity of phylogenetically distinct IMPDH is due to the differing lifetime of the covalent intermediates formed by the different enzymes (17, 18). Our data are consistent with this idea and provide a physical measure of the drug-inhibited enzyme. By using kinetic measurements, 30% of the Trichomonas foetus enzyme, and the majority of the human type II enzyme, populate the covalently modified state during the course of an in vitro reaction (17, 18). Under our conditions, we observed only a small fraction of yeast enzyme (Imd3) that accumulated as this intermediate (Fig. 2 A, lane 3). This finding does not seem to be due to a large fraction of inactive enzyme in the mixture because the entire pool of protein can ultimately be converted to the modified form and formation of the intermediate requires enzyme activity. The rate-limiting step in the reactions catalyzed by Imd2, -3, and -4 and mixed heteromeric complexes formed therefrom remain to be determined. Nevertheless, the assay described here provides a way to measure accumulation of this intermediate.
The differences in MPA sensitivity seen for these yeast isoforms mirrors that seen for IMPDH across the phylogenetic spectrum. In general, microbial IMPDHs are relatively resistant to MPA (e.g., T. foetus Ki ≈ 9 μM; ref. 18), whereas mammalian enzymes are far more sensitive (e.g., human IMPDH-II Ki ≈ 10–20 nM; refs. 9 and 24). The two human IMPDH gene products differ by ≈5-fold in MPA sensitivity (9). This heterogeneity has potential clinical significance because these two isoforms coexist in the same cell type and both seem MPA-inducible (5, 26–28). Extension of this assay to patient material should facilitate the detection and measurement of the end point of MPA therapy: inhibition of IMPDH.
S. cerevisiae is somewhat unusual in that it possesses genes encoding three IMPDHs, representing at least two classes of MPA susceptibility. A combination of yeast genetics and biochemistry has enabled us to show that the different Imd gene products coassemble into heteromeric complexes. Study of subunit mixing and the enzymatic activity of heterotetramers has received relatively little attention. Our findings raise the interesting problem of the identity and stoichiometry of yeast IM-PDH multimers as well as the potential for dynamic changes in that stoichiometry. For example, after MPA treatment, tetramers formed from two Imd3 and two Imd4 polypeptides might be replaced by Imd2-enriched mixed heterotetramers. Due to the diversity of the IMPDH genes in yeast, these cells display a rich potential to form many different forms of the IMPDH tetramer and potentially form a complex set of IMPDH with varying drug sensitivities. The findings reported here suggest that significant heterogeneity may also exist in human cells and that there may be a dynamic shift in subunit composition of the tetramer upon drug challenge during mycophenolate-based therapies.
Acknowledgments
We thank Drs. D. Edmondson, L. Hedstrom, R. Kahn, and J. Pohl for advice and helpful discussions. We gratefully acknowledge CellZome AG for plasmid pBS1479. This work was supported by National Institutes of Health Grant GM46331.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: IMPDH, IMP dehydrogenase; MPA, mycophenolic acid; TAP, tandem affinity purification.
References
- 1.Hedstrom, L. (1999) Curr. Med. Chem. 6, 545-560. [PubMed] [Google Scholar]
- 2.Hyle, J. W., Shaw, R. J. & Reines, D. (2003) J. Biol. Chem. 278, 28470-28478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shaw, R. J. & Reines, D. (2000) Mol. Cell. Biol. 20, 7427-7437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Escobar-Henriques, M. & Daignan-Fornier, B. (2001) J. Biol. Chem. 276, 1523-1530. [DOI] [PubMed] [Google Scholar]
- 5.Allison, A. C. & Eugui, E. M. (2000) Immunopharmacology 47, 85-118. [DOI] [PubMed] [Google Scholar]
- 6.Zhang, R., Evans, G., Rotella, F., Westbrook, E., Huberman, E., Joachimiak, A. & Collart, F. R. (1999) Curr. Med. Chem. 6, 537-543. [PubMed] [Google Scholar]
- 7.Shaw, R. J., Wilson, J. L., Smith, K. T. & Reines, D. (2001) J. Biol. Chem. 276, 32905-32916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sintchak, M. D., Fleming, M. A., Futer, O., Raybuck, S. A., Chambers, S. P., Caron, P. R., Murcko, M. A. & Wilson, K. P. (1996) Cell 85, 921-930. [DOI] [PubMed] [Google Scholar]
- 9.Carr, S. F., Papp, E., Wu, J. C. & Natsumeda, Y. (1993) J. Biol. Chem. 268, 27286-27290. [PubMed] [Google Scholar]
- 10.Zimmermann, A. G., Gu, J. J., Laliberte, J. & Mitchell, B. S. (1998) Prog. Nucleic Acid Res. Mol. Biol. 61, 181-209. [DOI] [PubMed] [Google Scholar]
- 11.Puig, O., Caspary, F., Rigaut, G., Rutz, B., Bouveret, E., Bragado-Nilsson, E., Wilm, M. & Seraphin, B. (2001) Methods 24, 218-229. [DOI] [PubMed] [Google Scholar]
- 12.Sikorski, R. S. & Hieter, P. (1989) Genetics 122, 19-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sherman, F. (1991) Methods Enzymol. 194, 3-20. [DOI] [PubMed] [Google Scholar]
- 14.Escobar-Henriques, M., Balguerie, A., Monribot, C., Boucherie, H. & Daignan-Fornier, B. (2001) J. Biol. Chem. 276, 46237-46242. [DOI] [PubMed] [Google Scholar]
- 15.Ghaemmaghami, S., Huh, W. K., Bower, K., Howson, R. W., Belle, A., Dephoure, N., O'Shea, E. K. & Weissman, J. S. (2003) Nature 425, 737-741. [DOI] [PubMed] [Google Scholar]
- 16.Link, J. O. & Straub, K. (1996) J. Am. Chem. Soc. 118, 2091-2092. [Google Scholar]
- 17.Digits, J. & Hedstrom, L. (1999) Biochemistry 38, 2295-2306. [DOI] [PubMed] [Google Scholar]
- 18.Digits, J. & Hedstrom, L. (1999) Biochemistry 38, 15388-15397. [DOI] [PubMed] [Google Scholar]
- 19.Huete-Perez, J. A., Wum, J. C., Whitby, F. G. & Wang, C. C. (1995) Biochemistry 34, 13889-13894. [DOI] [PubMed] [Google Scholar]
- 20.Futer, O., Sintchak, M. D., Caron, P. R., Nimmesgern, E., DeCenzo, M. T., Livingston, D. J. & Raybuck, S. A. (2002) Biochim. Biophys. Acta 1594, 27-39. [DOI] [PubMed] [Google Scholar]
- 21.Gilbert, H., Lowe, C. & Drabble, W. (1979) Biochem. J. 183, 481-494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yamada, U. & Natsumeda, Y & Weber, G. (1988) Biochemistry 27, 2193-2196. [DOI] [PubMed] [Google Scholar]
- 23.Exinger, F. & Lacroute, F. (1992) Curr. Genet. 22, 9-11. [DOI] [PubMed] [Google Scholar]
- 24.Farazi, T., Leichman, J., Harris, T., Cahoon, M. & Hedstrom, L. (1997) J. Biol. Chem. 272, 961-965. [DOI] [PubMed] [Google Scholar]
- 25.Kerr, K. M. & Hedstrom, L. (1997) Biochemistry 36, 13365-13373. [DOI] [PubMed] [Google Scholar]
- 26.Glesne, D. A., Collart, F. R. & Huberman, E. (1991) Mol. Cell. Biol. 11, 5417-5425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zimmermann, A. G., Wright, K. L., Ting, J. P. & Mitchell, B. S. (1997) J. Biol. Chem. 272, 22913-22923. [DOI] [PubMed] [Google Scholar]
- 28.Gu, J. J., Spychala, J. & Mitchell, B. S. (1997) J. Biol. Chem. 272, 4458-4466. [DOI] [PubMed] [Google Scholar]