

Abstract
Hydrogen sulfide (H2S) has emerged as a crucial biomolecule in physiology and cellular signaling. Key challenges associated with developing new chemical tools for understanding the biological roles of H2S include developing platforms that enable reversible binding of this important biomolecule. Here we report the first synthetic small molecule receptor for hydrosulfide anion, HS−, solely utilizing reversible, hydrogen-bonding interactions in a series of bis(ethynylaniline) derivatives. Binding constants up to 90,300 ± 8700 M−1 were obtained. The fundamental science of reversible sulfide binding—in this case featuring a key CH···S hydrogen bond—will expand the possibility for discovery of sulfide protein targets and molecular recognition agents.
Keywords: anions, supramolecular chemistry, H2S, sulfide, anion binding
Graphical Abstract
Long known for its malodor and toxicity, hydrogen sulfide is the most recently discovered endogenously produced gasotransmitter. Here we report the first synthetic receptor for reversible binding of HS− as characterized spectroscopically in solution and crystallographically.
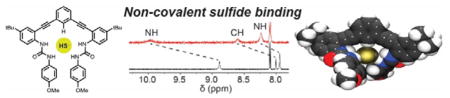
Supramolecular hosts have been developed to selectively bind a variety of anionic species in solution, ranging from inorganic phosphates and phosphorylated biomolecules, to halides, to other anions of environmental and/or biological relevance.[1] These synthetic supramolecular receptors use reversible, mostly non-covalent interactions to select anions based on factors such as their basicity, shape/charge, softness/hardness, position on the Hofmeister series, hydrophobic/solvophobic effects, among others. Notably lacking in the anion binding literature are efforts to target hydrosulfide (HS−), the smallest monoanionic sulfur species, which has recently gained interest as an important biomolecule. We report here the first examples of synthetic receptors that reversibly bind HS− using solely hydrogen bonding interactions. Importantly, a critical CH···S hydrogen bond is key to the strong binding of hydrosulfide, lending support to the hypothesis that appropriately polarized CH hydrogen bond donors[1f, 2] can target softer anions.[1e, 3]
Hydrogen sulfide (H2S) plays diverse roles in the global sulfur cycle and has recently been implicated as an important biologically-relevant signaling molecule.[4] In the last decade, H2S (and its more prevalent HS− conjugate base form under biological conditions) has emerged as the third endogenously produced gasotransmitter, along with CO and NO. H2S is now implicated in diverse (patho)physiological functions in the cardiovascular, immune, gastrointestinal, as well as other systems, making its absence in the supramolecular chemistry of anions even more surprising.[5] In parallel to the biological advances in H2S research, chemical tools for detecting and imaging H2S are rapidly emerging and form a cornerstone of the investigative approaches used to study this critical biomolecule.[6] Despite this importance, current detection methods are plagued by irreversibility, which presents a significant problem in developing chemical tools that provide real-time information on biological processes, suggesting a supramolecular (i.e., reversible) approach to HS− binding would represent an important contribution.
Complicating biological H2S investigations, the pKa of H2S (7.0) ensures that both the neutral (H2S) and monoanionic (HS−) forms are present under physiological conditions, leading to significant and unresolved questions on the specific chemistry and recognition events associated with the individual protonation states. Heightening this dichotomy, HS− was recently identified to be a viable substrate for Cl−/HCO3− anion exchange proteins,[7] and a bacterial ion channel for HS− transport was recently identified (Figure 1a–b).[8] Importantly, the recognition events in sulfide transport in these systems rely on non-covalent, reversible interactions with HS− rather than metal coordination or interaction with the sulfane-sulfur pool. Taken together, these examples suggest that HS−, which has until now been almost entirely overlooked, needs to be included in the complex landscape of biologically-relevant anions, such as Cl−, HCO3−, I−, and NO2−. Despite the emerging importance of sulfide, HS− has only appeared in anion screening sporadically, and we are unaware of any synthetic receptors able to bind H2S or HS− reversibly through well-defined non-covalent interactions.[1b, 9] Systems that could bind H2S or HS− selectively through reversible interactions would not only provide significant insights into potential HS− binding environments in biological contexts, but also provide new strategies for developing reversible and real-time H2S detection methods.
Figure 1.

(a) Protein structure of HSC (PDB:3TDX) showing five individual channels with the bound anion represented as a yellow sphere. (b) Enlargement of the binding pocket showing short contacts to His (2.980 Å), Thr (3.010 Å), Leu (3.725 Å) and Val (3.619 and 4.610 Å). Non-interacting helices are excluded for clarity. (c) Synthetic receptors 1–3.
To approach this challenge, we reasoned that synthetic anion receptors could provide a viable platform to develop reversible HS− binding systems. To optimize selective binding for hydrosulfide, we initially assumed the ideal receptor should feature hydrogen bond donors to target the anionic portion of hydrosulfide and a hydrogen bond acceptor (or suitable pocket of electron density) to accommodate the slightly acidic hydrogen atom. Aligned with these requirements, sulfide has a similar ionic radius to Cl− (S2− = 1.84 Å, Cl− = 1.81 Å) and biological examples reveal that HS− can fill similar roles as Cl−.[10] This similarity has not yet been exploited in the synthetic supramolecular community to target HS−, perhaps because of a prevailing assumption that Cl− and HS− should have quite different binding properties based on their different protonation states, nucleophilicities, hardness/softness, shape, pKb (−8 vs 7, respectively) and resulting hydrogen bond accepting ability.
In this light we reinvestigated the bis(ethynylaniline) anion-binding receptors we have developed for Cl− as a viable platform for non-covalent HS− binding.[11] These modular scaffolds bind anions through tunable urea NH hydrogen bonds, and the central core can be easily modified to incorporate an additional hydrogen bond donating arene (1) or a hydrogen bond accepting pyridine group (2–3).[3] The semi-preorganized binding pocket significantly reduces the entropic penalty for anion encapsulation, while maintaining flexibility to accommodate different anions. The ability to tune the urea hydrogen bond donors as well as the central core binding motif has resulted in a family of receptors that can selectively target a diverse range of analytes.[5a, 12] In addition, recent work has suggested that CH hydrogen bond donors polarized by inductive electron withdrawing groups (e.g., the electronegative sp-hybridized alkyne carbon atoms in 1) should exhibit selectivity for softer anions.[3] Although the place of HS− on the Hofmeister series and HSAB theory tables is not clear, intuition suggests that hydrosulfide should be a softer anion than chloride. Motivated by these challenges, we report here the first examples of synthetic receptors that reversibly bind HS− using solely supramolecular interactions (Figure 1c).
To investigate whether HS− is a suitable guest for hosts 1–3, we titrated NBu4SH[13] into 0.5–1.0 mM solution of each host in 10% DMSO-d6/CD3CN and monitored the titrations by 1H NMR spectroscopy (Figure S1). In each case, we observed that the urea NH resonances shifted significantly downfield upon HS− addition, consistent with anion binding (Figure 2). For example, upon addition of HS− to a solution of 1, the aryl CHa shifted from 7.99 to 9.24 ppm, and the NHb and NHc urea protons shifted downfield from 7.94 and 8.92 to 8.63 and 11.18 ppm, respectively. Highlighting the preference of each receptor 1–3 for HS− rather than H2S, addition of H2S gas to any of the receptors failed to change the UV-Vis or NMR spectra of the hosts. We also confirmed that the observed changes in the NMR spectra upon HS− addition were not due to deprotonation of the urea NH groups. Addition of the strong base 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) produced significantly different UV-Vis and NMR spectra than those observed upon HS− addition (Figure S14). On the basis of the high nucleophilicity of HS−, we also confirmed that the anion did not irreversibly modify the alkyne moieties of the host scaffolds by monitoring the 13C{1H} NMR spectrum of 1 before and after addition of 10 equivalents of HS− (Figure S13). Additionally, titration data of HS− with the host fit to simple 1:1 binding isotherm models. Taken together, these results support the hypothesis that HS− binds within the host pocket and does not covalently modify the host scaffold.
Figure 2.

(a) Scheme showing HS− host-guest equilibrium. (b) Representative UV-Vis difference titration of NBu4SH with 10 μM 2 in CH3CN and fit to a 1:1 binding isotherm (inset). (c) 1H NMR spectra of a titration of 0.988 mM 1 with NBu4SH in 10% DMSO-d6/CD3CN.
To determine whether receptors 1–3 exhibited selectivity for HS− over similar anions, we performed comparison titrations with NBu4Cl under identical conditions. We initially expected that pyridine-based hosts 2 and 3 would exhibit higher binding affinities for HS− because of the hydrogen bond accepting pyridine core; however, titration data established hosts 2 and 3 had significantly lower binding constants for both anions than did phenyl core host 1. This difference suggests that the extra CH hydrogen bond donated from the phenyl core is a key component in establishing the binding magnitude and selectivity. This result was contrary to our initial hypothesis that HS− should also act as a weak hydrogen bond donor to an acceptor on the host receptor (e.g., the pyridine nitrogen of 2 and 3).[14] Despite the lower binding affinities, the pyridine-based hosts 2 and 3 exhibited 6-fold selectivity for HS− over Cl−, whereas host 1 exhibited 2.8-fold selectivity. The higher selectivity could be due to the putative N···HS− hydrogen bond from the pyridine lone pair acting as a hydrogen bond acceptor, which provides an additional stabilizing interaction for HS− and a destabilizing interaction for Cl−. The phenyl core of host 1 donates a hydrogen bond to both anionic guests, resulting in decreased selectivity for hydrosulfide, even if this CH hydrogen bond is an important component to the higher overall binding energy.
To further investigate the difference in anion selectivity, binding constants were also measured by UV-Vis spectroscopy in CH3CN (Table 1). We expected that removal of the DMSO co-solvent would increase the observed binding affinities since acetonitrile is a slightly less competitive solvent (especially as a hydrogen bond acceptor). Addition of NBu4SH to 10 μM solutions of 1, 2, or 3 resulted in attenuation of the 330 nm absorbance with concomitant increase at 360 nm, while proceeding through a well-anchored isosbestic point near 350 nm. As expected, removal of DMSO produced significantly higher binding affinities, with host 1 having a binding constant of 90,300 M−1 and hosts 2 and 3 providing binding constants of ~25,000 M−1. For 1, the selectivity for HS− over Cl− remained similar to the 10% DMSO-d6/CD3CN system, whereas in the case of the pyridine core, a significant increase in selectivity is observed (~18.5:1 HS−:Cl−). The difference between the binding energy of HS− with 1 and 2 is the same in both solvents (ΔΔG = 0.90 kcal mol−1), whereas the Cl− binding energy exhibits a larger solvent dependence (ΔΔG = 1.24 (DMSO/CH3CN) vs. 1.83 (CH3CN) kcal mol−1). For HS−, the ΔΔG is the difference between two stabilizing hydrogen bond motifs and leads to an estimate that a C−H···S hydrogen bond is up to 0.90 kcal mol−1 stronger than an S−H···N hydrogen bond. The ΔΔG of Cl− binding is larger because this represents the difference between a small repulsive N: ···Cl contact and an attractive C−H···Cl hydrogen bond.
Table 1.
HS− and Cl− Binding Parameters in Hosts 1–3.
| Host | Solvent | HS− (log(Ka)) ΔG (kcal mol−1) | Cl− (log(Ka)) ΔG (kcal mol−1) |
|---|---|---|---|
| 1 | 10% DMSO-d6/CD3CN | 3.70 ± 0.07[a] −5.05 |
3.25 ± 0.03[a] −4.43 |
| CH3CN | 4.96 ± 0.04[b] −6.76 |
4.53 ± 0.07[b] −6.18 |
|
| 2 | 10% DMSO−d6/CD3CN | 3.04 ± 0.06[a] −4.15 |
2.34 ± 0.07[a] −3.19 |
| CH3CN | 4.30 ± 0.07[b] −5.86 |
3.19 ± 0.07[b] −4.35 |
|
| 3 | 10% DMSO-d6/CD3CN | 3.12 ± 0.07[a] −4.25 |
2.34 ± 0.02[a] −3.19 |
| CH3CN | 4.45 ± 0.07[b] −6.07 |
3.08 ± 0.06[b] −4.20 |
Fitting NMR spectroscopic data.
Fitting UV-Vis spectroscopic data.
To further establish the reversibility of HS− binding, we treated a solution of 1 in 10% DMSO-d6/CD3CN (Figure 3a) with two equivalents of NBu4SH to form the HS− bound adduct (Figure 3b), after which four equivalents of Zn(OAc)2 were added. Addition of Zn(OAc)2 rapidly resulted in precipitation of ZnS and regenerated the NMR spectrum corresponding to free 1 (Figure 3c). Further addition of five equivalents of NBu4SH regenerated the HS− host-guest complex, confirming reversible binding. Importantly, the 13C{1H} resonances of the alkyne carbons did not shift significantly (Figures 3b, d), confirming that there was no covalent modification of the receptor scaffold.
Figure 3.

(a) Reversibility reaction scheme. (b) 1H NMR spectrum of a 1.0 mM solution of 1 in 10% DMSO-d6/CD3CN. (c) Treatment with 2 equiv. of NBu4SH. (d) Addition of 4 equiv. Zn(OAc)2. Each inset shows the 13C{1H} resonances corresponding to the alkyne region of 1.
Single crystals of [1•HS−][NBu4+] were grown by layering n-hexanes onto an equimolar solution of 1 and NBu4SH in THF in a glovebox. [1•HS−][NBu4+] crystallizes in the space group Pna21 with one molecule of THF per unit cell. Consistent with the solution NMR data, the HS− occupies the binding pocket created by an aryl proton and four urea protons with the NBu4+ cation sitting just above the sulfide – phenyl core plane (Figure S15). The structure shows five hydrogen bonds from the host to the bound sulfide guest. The C–H···S hydrogen bond (3.711 Å) is longer than those formed between the distal bis(urea) protons (3.277, 3.281 Å) (Figure 4a). The average of all five hydrogen bond distances from the host to the guest is 3.56 Å, and all fall within previously defined criteria for hydrogen bonds.[2b, 15] The host conformation in [1•HS−] is remarkably similar to the previously published chloride-bound structure, with an RMS distance between the two structures of only 0.184 Å (Figure S16).[14] These data demonstrate the similar recognition geometries required for Cl− and HS− binding, again highlighting the potential for HS− to be a substrate for classical Cl− binding domains in both native and synthetic systems.
Figure 4.

ORTEP representation showing selected hydrogen bond distances. Atoms are drawn at the 50% probability level. Hydrogens not interacting with the bound HS− are removed for clarity.
In conclusion, we report a series of bis(ethynylaniline) derivatives capable of binding hydrosulfide anion with association constants as high as 90,300 ± 8700 M−1, representing the first reversible binding of the hydrosulfide anion in a synthetic receptor. 1H NMR and UV-Vis spectroscopy both indicate stronger binding of hydrosulfide by the phenyl core receptor 1; however, a greater selectivity for HS− is observed in the pyridine cores (2 and 3). The preference for the phenyl core highlights the unexpected conclusion that a C–H···S contact is favored over an N: ···H–S contact by up to 0.9 kcal mol−1. This difference may be related to the mechanisms that underlie anion binding selectivities beyond shape, size, and charge. Importantly, these results indicate that hydrogen bond polarizability and other aspects of hard/soft acid base theory are relevant to the characterization of anion selective host-guest systems. Additionally, these data suggest that CH hydrogen bond donors are important components in targeting hydrosulfide reversibly. Taken in total, these experiments establish the reversible binding of HS− to synthetic host molecules, and highlight that HS− is an important, and thus far overlooked, biologically-important anion that can be targeted by synthetic molecular architectures. These studies also begin to establish the design rules for targeting hydrosulfide anion using such synthetic receptors. Moreover, we anticipate that the basic science of non-covalent sulfide binding to synthetic targets will help to identify new target proteins for sulfide binding, while also informing on new potential sulfide detection strategies that do not rely on the irreversible covalent modification of sensing platforms.
Supplementary Material
Acknowledgments
This work was supported by the NSF (CHE-1454747 to MDP), Sloan Foundation, and NIH (R01-GM087398 to DWJ/MMH). NIH GM087398 funded early stage intellectual property that was licensed by SupraSensor Technologies/Enterprises, a company co-founded by DWJ/MMH. The NMR facilities at the University of Oregon are supported by the NSF (CHE-1427987).
Footnotes
Supporting information for this article is given via a link at the end of the document.
Contributor Information
Matthew D. Hartle, Department of Chemistry & Biochemistry, Materials Science Institute, and Institute of Molecular Biology University of Oregon Eugene, Oregon 97403-1253, USA.
Ryan J. Hansen, Department of Chemistry & Biochemistry, Materials Science Institute, and Institute of Molecular Biology University of Oregon Eugene, Oregon 97403-1253, USA.
Blakely W. Tresca, Department of Chemistry & Biochemistry, Materials Science Institute, and Institute of Molecular Biology University of Oregon Eugene, Oregon 97403-1253, USA
Samuel S. Prakel, Department of Chemistry & Biochemistry, Materials Science Institute, and Institute of Molecular Biology University of Oregon Eugene, Oregon 97403-1253, USA
Lev N. Zakharov, CAMCOR—Center for Advanced Materials Characterization in Oregon. University of Oregon, Eugene, OR 97403-1443 (USA
Michael M. Haley, Email: haley@uoregon.edu, Department of Chemistry & Biochemistry, Materials Science Institute, and Institute of Molecular Biology University of Oregon Eugene, Oregon 97403-1253, USA
Michael D. Pluth, Email: pluth@uoregon.edu, Department of Chemistry & Biochemistry, Materials Science Institute, and Institute of Molecular Biology University of Oregon Eugene, Oregon 97403-1253, USA
Darren W. Johnson, Email: dwj@uoregon.edu, Department of Chemistry & Biochemistry, Materials Science Institute, and Institute of Molecular Biology University of Oregon Eugene, Oregon 97403-1253, USA
References
- 1.(a) Gale PA, Caltagirone C. Chem Soc Rev. 2015;44:4212–4227. doi: 10.1039/c4cs00179f. [DOI] [PubMed] [Google Scholar]; (b) Lee MH, Kim JS, Sessler JL. Chem Soc Rev. 2015;44:4185–4191. doi: 10.1039/c4cs00280f. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Carnegie RS, Gibb CL, Gibb BC. Angew Chem Int Ed. 2014;53:11498–11500. doi: 10.1002/anie.201405796. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Gibb CL, Oertling EE, Velaga S, Gibb BC. J Phys Chem B. 2015;119:5624–5638. doi: 10.1021/acs.jpcb.5b01708. [DOI] [PubMed] [Google Scholar]; (e) Lisbjerg M, Valkenier H, Jessen BM, Al-Kerdi H, Davis AP, Pittelkow M. J Am Chem Soc. 2015;137:4948–4951. doi: 10.1021/jacs.5b02306. [DOI] [PubMed] [Google Scholar]; (f) Hargrove AE, Nieto S, Zhang T, Sessler JL, Anslyn EV. Chem Rev. 2011;111:6603–6782. doi: 10.1021/cr100242s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Cai J, Sessler JL. Chem Soc Rev. 2014;43:6198–6213. doi: 10.1039/c4cs00115j. [DOI] [PubMed] [Google Scholar]; (b) Hay BP, Bryantsev VS. Chem Commun. 2008;21:2417–2428. doi: 10.1039/b800055g. [DOI] [PubMed] [Google Scholar]; (c) Lee S, Chen CH, Flood AH. Nat Chem. 2013;5:704–710. doi: 10.1038/nchem.1668. [DOI] [PubMed] [Google Scholar]; (d) Hua Y, Flood AH. Chem Soc Rev. 2010;39:1262–1271. doi: 10.1039/b818033b. [DOI] [PubMed] [Google Scholar]; (e) Vyachelsav BS, Hay BP. Org Lett. 2005;7:5031–5034. doi: 10.1021/ol0520119. [DOI] [PubMed] [Google Scholar]; (f) Berryman OB, Johnson DW. Chem Commun. 2009:3143–3153. doi: 10.1039/b823236a. [DOI] [PubMed] [Google Scholar]
- 3.Tresca BW, Hansen RJ, Chau CV, Hay BP, Zakharov LN, Haley MM, Johnson DW. J Am Chem Soc. 2015;137:14959–14967. doi: 10.1021/jacs.5b08767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang R. Physiol Rev. 2012;92:791–896. doi: 10.1152/physrev.00017.2011. [DOI] [PubMed] [Google Scholar]
- 5.(a) Watt MM, Zakharov LN, Haley MM, Johnson DW. Angew Chem Int Ed. 2013;52:10275–10280. doi: 10.1002/anie.201303881. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Singh S, Lin H. Microorganisms. 2015;3:866–889. doi: 10.3390/microorganisms3040866. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Wu D, Si W, Wang M, Lv S, Ji A, Li Y. Nitric Oxide. 2015;50:38–45. doi: 10.1016/j.niox.2015.08.004. [DOI] [PubMed] [Google Scholar]; (d) Yetik-Anacak G, Sorrentino R, Linder AE, Murat N. Br J Pharmacol. 2015;172:1434–1454. doi: 10.1111/bph.12700. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Papapetropoulos A, Whiteman M, Cirino G. Br J Pharmacol. 2015;172:1633–1637. doi: 10.1111/bph.12806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Lin VS, Chen W, Xian M, Chang CJ. Chem Soc Rev. 2015;44:4596–4618. doi: 10.1039/c4cs00298a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yu F, Han X, Chen L. Chem Commun. 2014;50:12234–12249. doi: 10.1039/c4cc03312d. [DOI] [PubMed] [Google Scholar]
- 7.Jennings ML. Am J Physiol Cell Physiol. 2013:C941–C950. doi: 10.1152/ajpcell.00178.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Czyzewski BK, Wang DN. Nature. 2012;483:494–497. doi: 10.1038/nature10881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Sessler JL, Gale P, Cho W-S, Stoddart JF, Rowan SJ, Aida T, Rowan AE. Anion Receptor Chemistry. The Royal Society of Chemistry; 2006. [Google Scholar]; (b) Gale PA, Busschaert N, Haynes CJ, Karagiannidis LE, Kirby IL. Chem Soc Rev. 2014;43:205–241. doi: 10.1039/c3cs60316d. [DOI] [PubMed] [Google Scholar]; (c) Anzenbacher P, Liu YL, Palacios MA, Minami T, Wang Z, Nishiyabu R. Chem Eur J. 2013;19:8497–8506. doi: 10.1002/chem.201204188. [DOI] [PubMed] [Google Scholar]; (d) Palacios MA, Nishiyabu R, Marquez M, Anzenbacher P. J Am Chem Soc. 2007;129:7538–7544. doi: 10.1021/ja0704784. [DOI] [PubMed] [Google Scholar]
- 10.Marcus Y. J Chem Soc Faraday Trans. 1991;87:2995–2999. [Google Scholar]
- 11.(a) Vonnegut CL, Tresca BW, Johnson DW, Haley MM. Chem Asian J. 2015;10:522–535. doi: 10.1002/asia.201403212. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Carroll CN, Naleway JJ, Haley MM, Johnson DW. Chem Soc Rev. 2010;39:3875–3888. doi: 10.1039/b926231h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gavette JV, Mills NS, Zakharov LN, Johnson CA, II, Johnson DW, Haley MM. Angew Chem Int Ed. 2013;52:10460–10464. doi: 10.1002/anie.201302929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hartle MD, Meininger DJ, Zakharov LN, Tonzetich ZJ, Pluth MD. Dalton Trans. 2015;44:19782–19785. doi: 10.1039/c5dt03355a. [DOI] [PubMed] [Google Scholar]
- 14.Tresca BW, Zakharov LN, Carroll CN, Johnson DW, Haley MM. Chem Commun. 2013;49:7240–7242. doi: 10.1039/c3cc44574g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.(a) Desiraju GR, Steiner T. The Weak Hydrogen Bond: in Structural Chemistry and Biology. Oxford University Press; Oxford, New York: 1999. [Google Scholar]; (b) Taylor R, Kennard O. J Am Chem Soc. 1982;104:5063–5070. [Google Scholar]; (c) Wood PA, Allen FH, Pidcock E. CrystEngComm. 2009;11:1563–1571. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.