

Abstract
Oil palm is the world’s leading source of vegetable oil and fat. Dura, Pisifera and Tenera are three forms of oil palm. The genome sequence of Pisifera is available whereas the Dura form has not been sequenced yet. We sequenced the genome of one elite Dura palm, and re-sequenced 17 palm genomes. The assemble genome sequence of the elite Dura tree contained 10,971 scaffolds and was 1.701 Gb in length, covering 94.49% of the oil palm genome. 36,105 genes were predicted. Re-sequencing of 17 additional palm trees identified 18.1 million SNPs. We found high genetic variation among palms from different geographical regions, but lower variation among Southeast Asian Dura and Pisifera palms. We mapped 10,000 SNPs on the linkage map of oil palm. In addition, high linkage disequilibrium (LD) was detected in the oil palms used in breeding populations of Southeast Asia, suggesting that LD mapping is likely to be practical in this important oil crop. Our data provide a valuable resource for accelerating genetic improvement and studying the mechanism underlying phenotypic variations of important oil palm traits.
Keywords: palm, genome, sequencing, SNP, breeding
1. Introduction
Oil palm (Elaeis guineensis Jacquin) is a monoecious species in the family Arecaceae.1 It originates from West Africa and is the world’s leading source of vegetable oil and fat. Oil palm does not produce offshoots, and its propagation is by sowing the seeds. Commercial plantation of oil palm commenced on the West African coast and in Southeast Asia. Plantations of oil palm help to provide cooking oil and fuel to many developing countries. There are three major and different forms/varieties in the species E. guineensis: Dura, Pisifera and Tenera.1 The majority of oil palm trees planted for oil production in Asia are the offspring of four Dura trees brought from Africa to Asia in 1848 and a few Pisifera trees imported to Asia from Africa.1 The hybrid Tenera form, generated by crossing the Dura (♀) and Pisifera (♂) forms, shows a yield advantage (hybrid vigor) of 25% over the pure Dura form; hence, it is preferred for plantations. It is generally believed that the SHELL gene is the major gene controlling oil yield.2 Conventional breeding and changes in plantation management in the past 60 years have improved quality traits and also elevated the crude palm oil (CPO) yield from 2.0 to 4.1 tons/hectare/year.3 As the estimated potential CPO yield of the crop may be as high as 18 tons of oil per hectare/year,4 there is substantial potential to increase CPO yield. Besides E. guineensis, another palm species, Elaeis oleifera, has been used to produce oil in South and Central America.1 E. oleifera diverged from E. guineensis as long as 51 Myr ago.5 However, its oil yield is much lower than that of E. guineensis while its oil quality is better.1 A natural E. oleifera × E. guineensis hybrid was found in Costa Rica. Palms with a short trunk and short leaves, known as Compact, have been developed by repeated backcrossing of this palm with E. guineensis.6 The improved Compact palm is expected to be commercially exploited in Southeast Asia in the future due to its high oil quality and short trunk.
Genetic variation is the basis of genetic improvement. The draft genomes of Pisifera form of the species E. guineensis, and another species E. oleifera oil palm are available.5 The genome size of oil palm is 1.8 Gb.5 The genome sequence of the Dura form in the species E. guineensis has not been reported yet. Considerable efforts have been made to understand the genetic variation in oil palms.7–9 However, information about genome-wide genetic variation is still lacking. The purpose of this study was to obtain a whole genome sequence of an elite Dura palm and genome-wide overview of genetic variation in oil palms including Compact palms. We sequenced the genome of an elite palm Dura from a breeding population and re-sequenced the genomes of 17 palm trees from Southeast Asia, Africa and Central America. The draft genome sequence of Dura is 1.701 Gb, containing 36,015 genes. Re-sequencing additional 17 palm trees identified 18.1 million SNPs. We have mapped over 10,000 SNPs to the linkage map of oil palm. Our data provide a valuable resource to accelerate oil palm genetic improvement and to investigate mechanisms underlying phenotypic variations of important traits (e.g. oil yield and quality, disease resistance) in oil palms.
2. Materials and methods
Methods and any associated references are available in Supplementary Materials. Here we only presented the major methods.
2.1. Genome sequencing, assembly and quality assessment
High-quality genomic DNA was extracted from leaf samples of a Dura elite palm D1 (Supplementary Table S1), from a breeding program of Wilmar International Ltd. Plantation, using plant DNA extraction kits (Qiagen, Hilden, Germany). The extracted DNA was treated with RNase A and proteinase K to remove RNA and protein contamination, respectively, and the DNA further precipitated with ethanol. Illumina short-insert paired-end (size: 300 bp) and long-insert mate-pair (3–5, 10 and 20 kb) libraries were prepared following the manufacturer’s instructions. The template DNA fragments were sequenced using the Illumina HighSeq 2500 and Miseq. For Roche 454 sequencing, library construction and sequencing were conducted following the recommendations of Roche.
We developed a comprehensive pipeline (Supplementary Fig. S1) and used it to assemble the Dura oil palm genome (Details see Supplementary Materials). The Pisifera palm genome5 was used as a reference genome for the assembly. Our pipeline included five components: (i) de novo assembly, (ii) mis-assembly scaffold identification and correction, (iii) alignment to reference genome, (iv) repeat scaffold identification, and (v) solution of overlapping scaffolds.
We evaluated the quality of the genome assembly using three methods, including expressed sequence tag (EST) coverage, completeness of genome and coverage of markers in linkage maps. We checked EST coverage. A total of 41,695 ESTs which were collected from oil palm leaf and mesocarp tissues were used to assess the gene coverage of this draft oil palm genome. ESTs were aligned to the genome by BLAT. Only ESTs with alignments of identity≥0.9 were retained. We analysed the completeness of the draft genome of Dura palm. A computational method CEGMA,10 which defined a set of much conserved protein families that occur in a wide range of eukaryotes, was adopted to check the completeness of our draft genome. The completeness of each genome could be measured by the number of conserved proteins defined by the program. The existing marker dataset from oil palm was used to evaluate the quality of our draft genome. In our study, the accuracy of each scaffold and mapping of the scaffolds into real chromosomes were determined by linkage maps. This was done by examining the number of known markers which could be found in our draft genome. We used 256 SSR markers mapped by Billotte et al.11 and 454 SSR markers mapped by ourselves12 to investigate the completeness of the draft genome.
2.2. Genome annotation and identification of gene synteny and whole genome duplication
We annotated repeats, gene and ncRNA using a number of software (details see ‘Materials and Methods’ section). We attempted to identify gene synteny and whole genome duplication. To generate a pair-wise alignment of gene models between oil palm and grape, oil palm and soybean, and oil palm and date palm all predicted gene models were aligned to the respective reference gene models using the software MUMmer.13 The criterion used was that the number of genes in one synteny block should be more than 5.
2.3. Re-sequencing the genomes of 17 additional oil palm trees
After assembling the draft genome for the elite Dura palm tree D1, we also collected samples from oil palm trees from the major plantation area (i.e. Southeast Asia), its place of origin (i.e. Africa) and Central America for re-sequencing. From the collection of oil palms across the world, we selected 13 trees from three forms (i.e. Dura, Pisifera and Tenera) of the species E. guineensis, which included the elite Dura tree D1 used in generating the draft genome (see details in Supplementary Table S1). In addition, we also selected five trees of the Compact palm for re-sequencing, which is a hybrid between the two species E. guineensis, and E. oleifera and expected to be exploited in Southeast Asia in the future. We re-sequenced the genomes of 17 trees using paired-end sequencing using the Illumina HighSeq 2500 as described earlier. The sequencing depth of each whole genome was around 5–8-fold (Supplementary Table S1).
2.4. Detection of SNPs and genetic variations in 18 palm trees
For the 18 accessions of oil palm trees we sequenced, SNPs were called in three steps: (i). All reads were aligned to our assembled draft genome using Bowtie14 with the default setting. (ii). The SNPs were then called with SAMtools.15 (iii) And then four thresholds (i.e. Samtool_pileup SNP score ≥30, Read sequence depth for this allele ≥10, The minimum distance for adjacent SNPs ≥10 bp and Only one polymorphism detected at each SNP location) were used to post-filter unreliable SNPs:
To explore the various information of SNPs in different groups: Dura, Pisifera, Tenera and Compact, we compared the following information for different groups. (i) Location information for each SNP: intergenic region, UTR, intron, exon, CDS, downstream (length: 5 Kb), upstream (length 5 K). (ii) Coding feature: NON_Synonymous_coding (SNP causing a codon change that produces a different amino acid), Synonymous_coding (SNP causing a codon change that produces the same amino acid). (iii) Codon level: Codon_change (one or many codons are changed), Codon_Insert (one or many codons are inserted), Codon_deletion (one or many codons are deleted), Exon_deleted (a deletion that removes the whole exon), Start_Lost (the start codon is mutated into a non-start codon), Synonymous_start (the start codon is mutated into another start codon), Synonymous_stop (the stop codon is mutated into another stop codon), Stop_lost (the stop codon is mutated into a non-stop codon).
To analyse the genetic relationship between different oil palm trees, two methods were used to construct the phylogenic trees. For the first method, SNPs were first used to calculate the genetic distances between different accessions/strains of oil palm.16 Next, a Neighbour-Joining method was used to construct the phylogenetic tree on the basis of the distance matrix, calculated by the software PHYLIP (http://evolution.genetics.washington.edu/phylip.html). Finally, MEGA417 was used to visualize the phylogenetic tree. For the second method, we used TreeMix18 to infer the population evolution history.
We performed a Principal Component Analysis (PCA) following the procedure as reported.19 The eigenvector decomposition of the genotype data was performed by the eigen function in R. The whole program was provided by the SNPrelate package in R.20 After excluding SNPs from individuals that had missing data, the remaining SNPs were used to construct the population structure using the program STRUCTURE.21 The length of the burn-in period and the number of MCMC reps after burn in were set to default value. The number of populations was set from 3 to 7. We estimated FST, πbetween, and πwithin to examine population differentiation using vcftools.15
2.5. Detection of LD and identification of selection signatures
We explored haplotype patterns in different oil palm strains using the SNP dataset. Correlation coefficient (r2) of alleles was calculated to measure the LD level in three groups: Dura, Pisifera and Tenera, using Haploview.22 The same parameters as soybean23 were set: −maxdistance 1000 –dprime –minMAF 0.1 –hwcutoff 0.01. The average r2 value was calculated for each length of distance and LD decay figures were drawn by R for different groups of oil palm.
We used the rehh24 package in R to identify selection signatures on the oil palm genome. The integrated haplotype Score (iHS) is a measure of the amount of extended haplotype homozygosity at a given SNP along the ancestral allele relative to the derived allele.25 The RSB score measured the difference between two different populations based on iHS scores.26
3. Results
3.1. Sequencing and assembly of the Dura palm genome
We sequenced the genome of one Dura tree using Illumina HighSeq 2500, Miseq and Roche 454, and obtained 211 Gb raw data. After removing low-quality data, we assembled the genome using 171 Gb high quality sequences (Supplementary Table S2). The assembled draft genome was 1.701 Gb, containing 10,971 scaffolds with an N50 size of 0.76 Mb. The longest scaffold was 22.37 Mb (Table 1). The draft genome sequence covered 94.49% of the oil palm genome based on that the genome size of oil palm is 1.8 Gb.5
Table 1.
Statistics of the genome sequence assembly of a Dura oil palm
| Total length of raw sequencing reads | 211.99 Gb |
| Total length of clean sequencing reads | 171.33 Gb |
| Number of scaffolds | 10,971 |
| N50 | 761,236 bp |
| Longest scaffold length) | 23.37 Mb |
| GC content | 36.80% |
| Assembled genome size | 1,700.81 Mb |
| Genome coverage | 94.49% |
| Number of genes | 36,105 |
| Number of protein-coding genes | 27,229 |
| Number of R genes | 566 |
| Average length of genes | 3,573 |
| Average number of exon per gene | 3.7 |
We evaluated the accuracy of the genome assembly by BLASTing the assembled genomic DNA sequence of Dura against the draft genome of Pisifera.5 85.6% of the Dura genome sequences are identical to the Pisifera genome sequences, suggesting that most of our assembled sequences are consistent with the Pisifera genome.
We checked the completeness of the draft Dura genome by using three independent approaches. First, we used the public EST database27 to examine the gene coverage of this draft genome. The draft genome had a high coverage (∼80%) of protein coding genome regions (Supplementary Table S3). Second, we found that of the 248 highly conserved proteins defined in CEGMA,10 around 87% of them can be found in our draft genome. Third, using DNA markers mapped on published linkage maps of oil palm,11,12 710 SSR markers (Supplementary Fig. S2) were aligned to the draft genome using BWA28 with only one mismatch. About 98% of the total number of available markers could be successfully aligned to our draft genome. Taken together, our data suggest that the draft Dura genome has a relatively high completeness. The missing portions of the Dura palm genome may be highly repetitive and thus difficult to assemble using our sequence dataset and current assembly method.
3.2. Annotation of the Dura genome
De novo and homology-based gene prediction were carried out using repeat masked scaffolds. These two gene sets were then merged to form a comprehensive and non-redundant reference gene set by MAKER2.29 A total of 36,015 gene models were predicted. 75.8% of them predicted protein-coding genes showing significant sequence similarity to known genes deposited in public databases (e.g. NCBI non-redundant database30). The GC content of the protein coding genes was 39.00% whereas that for the assembled draft genome was 36.80%. The average gene length was 3,573 bp (Supplementary Table S4).
The species which has the highest gene sequence homology with our Dura sample is grape (Vitis vinifera)31 (Supplementary Fig. S3), a eudicotyledonous crop, followed by another monocotyledonous crop, Oryza sativa.32 This kind of high amino acid sequence similarity with a phylogenetically unrelated plant (the oil palm is monocotyledonous while the grapevine is eudicotyledonous) has also been observed in the date palm33 and in proteins encoded by oil palm ESTs.34 We do not know the exact reasons for this relationship. Sequencing more plant species may give a clear answer about this relationship.
In the assembled genome, repetitive sequences accounted for 14.37% (Supplementary Table S5) whereas unassembled repeats and gaps accounted for around 27.81%. Thus, repeat sequences accounted for 42.18% of the draft genome, which is similar to that of the Pisifera genome.5 Among the repetitive sequences, retroelements were most abundant (7.96%), followed by low complexity repeats (3.84%), simple repeats (1.00%) and tandem repeats (0.69%) (Supplementary Table S5). Satellites were least abundant (0.001%).
In addition, we identified genes for non-coding RNAs in the Dura palm draft genome (Supplementary Tables S6 and S7). A total of 636 genes for tRNAs and 1,182 genes for rRNAs were detected. The number of tRNA genes is similar to the 699 tRNA genes for Arabidopsis35 and 606 for sorghum.36 It is interesting that one tRNA selenocysteine gene was detected in the oil palm genome. This gene was previously only found in maize,37 sorghum36 and bamboo,38 but absent in Oryza sativa,32 Arabidopsis thaliana35 and date palm.33 The specific function of this tRNA needs further investigation. We also identified genes for 199 known miRNA families (Supplementary Table S7).
3.3. Diversity and divergence between palms from Southeast Asia and Africa
Re-sequencing of 17 additional palm trees identified 18.1 million SNPs (Table 2). The heterozygosity rate in oil palms was 1.01% (i.e. one SNPs in 99.2 bp), which is higher than that in soybean (0.57%)23 and in date palm (0.47%).33 The genetically improved Dura and Pisifera palms used in producing the hybrid Tenera in Southeast Asia showed the lowest number of SNPs and genetic diversity (Table 2) as compared with the Tenera palms collected in Africa. The total distribution of genome wide diversity was lower for Dura and Pisifera compared with Tenera (Fig. 1). On the other hand, the Dura and Pisifera trees from Southeast Asia contained 1,419 and 341 form-specific SNPs respectively (Fig. 2). The genomic diversity in Compact palms from Central America was lower than that in Tenera from Africa (Table 2 and Fig. 1).
Table 2.
Number of SNPs in the whole genomes of different groups of oil palm and Ka/Ks ratio in coding genes
| Number of SNPs | Non-synoymous SNPs | Synoymous SNPs | Ka/Ks | Θπ (10−3) | UTR | Intron | Intergenic | |
|---|---|---|---|---|---|---|---|---|
| Dura | 5,964,480 | 55,288 | 39,848 | 1.387 | 2.32 | 11,301 | 411,488 | 4,405,899 |
| Pisifera | 8,224,740 | 70,040 | 52,324 | 1.339 | 3.06 | 17,710 | 627,176 | 5,962,105 |
| Tenera | 12,156,312 | 105,541 | 78,414 | 1.346 | 3.62 | 25,047 | 893,848 | 8,859,415 |
| Compact | 8,329,603 | 84,542 | 61,586 | 1.373 | 2.72 | 18,644 | 635,842 | 5,972,771 |
| all | 18,138,943 | 160,290 | 114,450 | 1.401 | 3.54 | 33,907 | 1,218,992 | 13,369,315 |
Θπ, genome diversity; UTR, untranslated region; Non-syn, non-synonymous mutation; and Syn, synonymous mutation.
Figure 1.
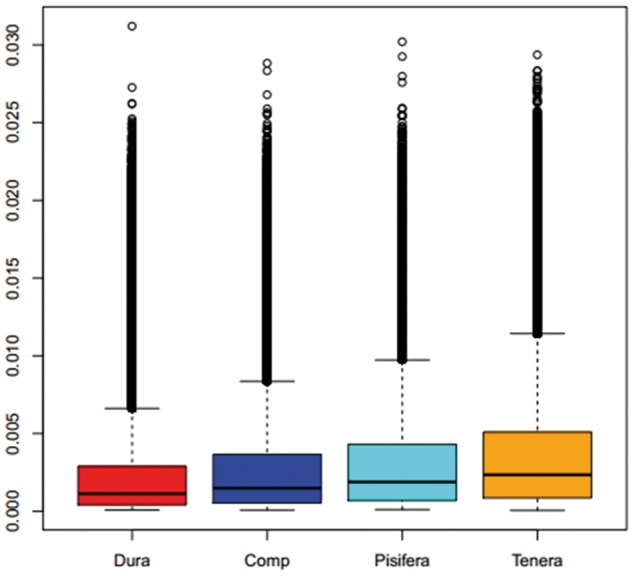
Box plot for the genome diversity (parameter θπ) for different oil palm groups: Dura, Tenera, Pisifera and Compact (Comp).
Figure 2.
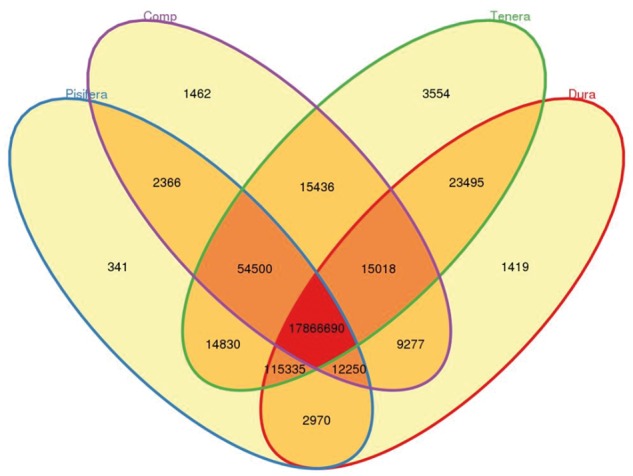
Venn graph for SNP number between Dura, Pisifera, Tenera and Compact palms.
To examine the genetic relationships among the 18 palm trees from Southeast Asia, Africa and Central America, we constructed a NJ-phylogenetic tree (Fig. 3A) using the identified SNPs. The phylogenetic tree showed that four Dura trees (D1, D2, D3, and D4) from Southeast Asia and two hybrid Tenera trees (T1 and T2) from Africa formed a subclade, whereas the five Compact palms (C1, C2, C3, C4, and C5) from Central America formed another subclade. The three Pisifera trees (P1, P2, and P3) from Southeast Asia and four hybrid Tenera (T3, T4, T5, and T6) from Africa constituted other subclades. A PCA gave similar results (Fig. 3B). Using the software STRUCTURE,39 with k between 3 and 7, subpopulations were detected among trees from Southeast Asia and Africa (Supplementary Fig. S4). As the average value of Ln likelihood was the highest when K= 4, we presented the results of the analysis at K = 4 (Fig. 3C). We found that several trees showed evidence of admixture, especially the Tenera group (Fig. 3A–C), which is generally in agreement with the fact that Tenera is the hybrid generated by crossing Dura and Pisifera trees. These data indicate that there is substantial genetic differentiation among palms used for breeding and plantations in Southeast Asia, Africa and Central America, and that the Compact palms are quite different from Dura, Pisifera and Tenera, which may be because the Compact palm is the hybrid between two palm species (i.e. E. guineensis and E. oleifera) of palm.6,40
Figure 3.

Analysis of the phylogenetic relationship, population structure and LD decay in oil palm trees. (A) A NJ-phylogenetic tree for 18 different oil palm trees (for details about the trees, see Supplementary Table S1). (B) PCA plots for 18 different oil palm trees. (C) Cluster for 18 different oil palm trees by STRUCTURE with K = 4 (C). (D) LD distribution by different pairwise distance. x-axis: pairwise distance and y-axis: LD(r2).
3.4. High LD in the oil palm genome
To obtain an overview of the genome-wide LD patterns in oil palm, we used the software SAMtools15 for LD analysis. In general, Dura, Pisifera, Tenera and Compact palms showed very high LD (Fig. 1D). The average distance over which LD decayed to 50% of its maximum value in oil palm was > 200 kb, which is substantially longer than the distance reported in any other crop (e.g. 75–150 kb in soybean,41 < 1 Kb in maize and rice,32,37 3–4 kb in A. thaliana35). Unlike humans and livestock, agronomic plant species usually show very short LD.35,37,41 Therefore, oil palm may be a good object to study effects of long LD on genome evolution and population structure. In addition, the results of LD analysis showed that the LD patterns in palms from different locations were different. The Dura palms displayed the most LD, followed by Pisifera, Tenera and Compact palms (Fig. 1D). The very high LD in oil palms, especially among trees from Southeast Asia, implies that LD mapping is likely to be practical in oil palms. Therefore, only a small set of SNPs covering the whole genome would be required to conduct genome-wide association studies (GWAS) for important traits for marker-assisted selection (MAS). By using the published linkage maps12 and in-silico mapping,42 over 10,000 detected SNPs (Supplementary Fig. S1, Supplementary Materials) were mapped to 16 linkage groups, supplying a useful resource for quantitative trait loci (QTL) mapping and GWAS for traits.
3.5. Signatures of selection, and positive selection on R genes of oil palm
Analysis of Fst revealed that some regions of the genome showed much higher values of Fst than most other regions (Supplementary Fig. S5), suggesting that these regions may contain genes under positive selection during the breeding of palms in different regions. Similarly, we also explored the iHS score across the whole genome (Supplementary Fig. S6) and detected conserved regions in the genomes of palms from different geographical locations, suggesting that these regions may be essential for maintaining the basic biological functions of palms. By comparing the iHS score of Dura with Tenera (Supplementary Fig. S7), and Pisifera with Tenera (Supplementary Fig. S8), across the whole draft genome, some interesting gene candidates were found to be under positive selection, such as the gene for asparagine synthase-like protein in chromosome 5, which has been reported to be related to the height of oil palms.12 Other interesting gene candidates, such as fatty acid desaturase and disease-resistance genes (R genes), were also under positive selection. Special attention should be paid to these genes in future breeding efforts.
The genes encoding proteins accounted for around 7% of the oil palm genome. However, only about 2% of the total SNPs were found to be present in these regions. The remaining (∼98%) SNPs were found in non-coding regions (Table 1 and Supplementary Fig. S9). The average Ka/Ks ratio in oil palms was 1.4 (Table 1), which is among the highest out of all plants reported so far (1.31–1.61 in soybean,23 1.2 in rice32 and 0.83 in A. thaliana35). We further analysed gene functional categories using genes whose Ka/Ks ratios were significantly different from the average ratio of all genes in the palm genome. We found that gene families with essential functions (e.g. mRNA translation and maintenance of protein location in the nucleus) tended to have substantially lower substitution ratios (X2 test, P < .01) (Supplementary Fig. S10), which is in agreement with the finding in soybean23 and bacteria.43. In contrast, gene families that function in regulatory processes, such as fatty acid metabolic processes, R genes, and steroid biosynthetic processes, had higher ratios (Supplementary Fig. S11), similar to previous findings in Arabidopsis35 and soybean.23
A total of 566 R genes (Supplementary Table S8) were identified from the Dura genome, substantially less than the number of 1,085 in the genome of rice, whose genome size is only 25% of oil palm.32 The average Ka/Ks ratio of R genes was 1.7, much higher than that (1.4) of all genes in the palm genome, suggesting a strong positive selection of R genes in palms. Based on conserved encoded protein domains, these R genes were distributed into 10 different groups (Supplementary Table S8). We found that the CNL, TNL and FLS groups had a higher Ka/Ks ratio than the average ratio of all R genes. Among the 50 genes with the highest Ka/Ks ratio in the sequenced oil palm genomes, 17 were putative R genes and these had an average Ka/Ks ratio of 3.
4. Discussion
We sequenced, assembled and annotated the genome of one elite palm Dura. Evaluation by three independent methods (i.e. EST coverage, genome completeness and linkage map) demonstrated the accuracy and completeness of our draft genome for the Dura tree. Previously published genomes of the Pisifera palms and E. oleifera5 have provided important genetic resources for oil palm breeding. Here, we present the first draft genome of the Dura palm. In comparison to the published draft genome of a Pisifera tree,43 the draft genome of our Dura tree covers more of the palm genome. The draft genome sequence of the Dura tree and its annotations, in combination with the draft genomes of Pisifera and E. oleifera palms, should facilitate further research on oil palm biology. We note, however, that there are still over 10 thousand scaffolds for the draft Dura genome. Filling in the gaps and connecting the scaffolds to make a well-assembled genome is an important task for the future.
Genetic variation is the basis of selective breeding for genetic improvement, and is also the safeguard from future challenges (e.g. emerging diseases and climate change). By sequencing 18 palm trees from Southeast Asia, Africa and Central America, we identified over 18 million SNPs, and revealed a low genetic diversity among Dura palms in Southeast Asia. Similarly, in comparison to the Tenera palms from Africa, the Pisifera palms from Southeast Asia contained a lower number of SNPs and genome diversity. These results indicate that the elite Dura and Pisifera palms used in commercial production/breeding in Southeast Asia contain only a part of the genetic variation in oil palms. To broaden the genetic diversity of oil palm in Southeast Asia, it is essential to import germplasm from Africa. Meanwhile, we found that the elite Dura and Pisifera palms used in producing the hybrid Tenera for large scale plantations in Southeast Asia contained a number of form-specific SNPs. It is well known that the average oil yield of oil palms in Southeast Asia is much higher than that in Africa due to the extensive selective breeding for oil yield in Southeast Asia.1 Therefore, these form-specific alleles in the elite Dura and Pisifera palms in Southeast Asia may be useful in improving the production performances of oil palms in Africa. Taken together, our data suggest that exchange of oil palm germplasm could benefit both Southeast Asia and Africa for future improvements of this important oil crop. The Compact palm, a hybrid between E. oleifera and E. guineensis, has the potential to become a new form of palm for plantation in Southeast Asia due to its high oil quality and lower trunk, which makes the harvest easier.1 This study showed that genetic diversity in the Compact palms was lower than that in the Tenera palms.1 Therefore, to ensure sustainability of plantation of the Compact palm, more individuals should be imported from Central America. Genotyping of palms with the identified SNPs could help to identify source materials for import and exchange.
Although the average CPO yield of oil palm has already reached 4.1 tons/ha/year, there is still huge room for increase to its estimated maximum yield (18 tons/ha/year).4 The availability of the sequence data and the selection of over 10,000 SNPs covering the 16 linkage groups of the whole Dura palm genome, will facilitate mapping of QTL and GWAS for important traits, thus accelerating genetic improvement in oil palms. The very high LD in oil palms suggests that in oil palms compared with other agronomic crops, MAS could be a better choice whereas positional cloning of genes encoding important traits could be more challenging.
Diseases are a major factor in determining yield in oil palm plantations. Diagnosis and prevention or cure of diseases, as well as selection of palms resistant to diseases, have proven to be difficult. To combat diseases, DNA markers could be used to search for disease resistance genes within the E. guineensis genome. We identified 566 R genes in oil palms. The number of R genes in oil palms is less than that in other plants,32,36,37 but they are more variable. We postulate that the extreme diversity and rapid evolution of R genes may compensate for the relative low number of R genes in palm trees in comparison to other plants, and provide a higher capacity to protect them against challenges from diverse pathogens. Further detailed functional characterization of R genes may render them useful in reducing the impact of pathogens on oil palm production.
Accession codes
Short-read genomic sequence data from this project have been deposited in the DNA Data Bank of Japan under the accession no. DRA002154. All assembly results and annotation results can be downloaded from http://chuanh.tll.org.sg/oilpalm/.
Conflict of interest
None declared.
Supplementary data
Supplementary data are available at www.dnaresearch.oxfordjournals.org.
Funding
This research was supported by Wilmar International Ltd. and Temasek Life Sciences Laboratory. J.J. was partially supported by a Singapore Ministry of Education Tier-2 grant, MOE2009-T2-2-004. We also thank Temasek Life Sciences Laboratory central facilities for technical assistance.
Supplementary Material
References
- 1.Corley R., Tinker P. 2015, The Oil Palm. Blackwell: Oxford. [Google Scholar]
- 2.Singh R., Low E.-T. L., Ooi L. C.-L., et al. 2013, The oil palm SHELL gene controls oil yield and encodes a homologue of SEEDSTICK. Nature, 500, 340–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sambanthamurthi R., Singh R., Kadir A. P. G., Abdullah M. O., Kushairi A. 2009, Opportunities for the oil palm via breeding and biotechnology In: Shri Mohan J., Priyadarshan P. M. (eds.), Breeding Plantation Tree Crops: Tropical Species, Springer, pp. 377–421. [Google Scholar]
- 4.Corley R. 1983, Potential productivity of tropical perennial crops. Exp. Agric., 19, 217–37. [Google Scholar]
- 5.Singh R., Ong-Abdullah M., Low E. T., et al. 2013, Oil palm genome sequence reveals divergence of interfertile species in old and new worlds. Nature, 500, 335–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Escobar R., Alvarado A. 2004, Strategies in production of oil palm compact seeds and clones. Exp. Agric., 27, 13–26. [Google Scholar]
- 7.Cochard B., Adon B., Rekima S., et al. 2009, Geographic and genetic structure of African oil palm diversity suggests new approaches to breeding. Tree Genet. Genom., 5, 493–504. [Google Scholar]
- 8.Hayati A., Wickneswari R., Maizura I., Rajanaidu N. 2004, Genetic diversity of oil palm (Elaeis guineensis Jacq.) germplasm collections from Africa: implications for improvement and conservation of genetic resources. Theor. Appl. Genet., 108, 1274–1284. [DOI] [PubMed] [Google Scholar]
- 9.Purba A., Noyer J., Baudouin L., Perrier X., Hamon S., Lagoda P. 2000, A new aspect of genetic diversity of Indonesian oil palm (Elaeis guineensis Jacq.) revealed by isoenzyme and AFLP markers and its consequences for breeding. Theor. Appl. Genet., 101, 956–961. [Google Scholar]
- 10.Parra G., Bradnam K., Korf I. 2007, CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics, 23, 1061–1067. [DOI] [PubMed] [Google Scholar]
- 11.Billotte N., Marseillac N., Risterucci A. M., et al. 2005, Microsatellite-based high density linkage map in oil palm (Elaeis guineensis Jacq.). Theor. Appl. Genet., 110, 754–765. [DOI] [PubMed] [Google Scholar]
- 12.May Lee, Jun Hong Xia, Zhongwei Zou, et al. 2014, A consensus linkage map of oil palm and a major QTL for stem height. Sci. Rep., 5, 8232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kurtz S., Phillippy A., Delcher A. L., et al. 2004, Versatile and open software for comparing large genomes. Genome Biol., 5, R12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Langmead B., Salzberg S. L. 2012, Fast gapped-read alignment with Bowtie 2. Nat. Methods, 9, 357–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li H., Handsaker B., Wysoker A., et al. 2009, The sequence alignment/map format and SAMtools. Bioinformatics, 25, 2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu X., Liu X., Ge S., et al. 2012, Resequencing 50 accessions of cultivated and wild rice yields markers for identifying agronomically important genes. Nat. Biotechnol., 30, 105–11. [DOI] [PubMed] [Google Scholar]
- 17.Kumar S., Nei M., Dudley J., Tamura K. 2008, MEGA: a biologist-centric software for evolutionary analysis of DNA and protein sequences. Brief. Bioinform., 9, 299–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pickrell J. K., Pritchard J. K. 2012, Inference of population splits and mixtures from genome-wide allele frequency data. Plos Genet., 8, e1002967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thirunavukkarasu N., Hossain F., Shiriga K., et al. 2013, Unraveling the genetic architecture of subtropical maize (Zea mays L.) lines to assess their utility in breeding programs. BMC Genomics, 14, 877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zheng X., Levine D., Shen J., Gogarten S. M., Laurie C., Weir B. S. 2012, A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics, 28, 3326–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pritchard J. K., Stephens M., Donnelly P. 2000, Inference of population structure using multilocus genotype data. Genetics, 155, 945–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barrett J. C., Fry B., Maller J., Daly M. J. 2005, Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics, 21, 263–5. [DOI] [PubMed] [Google Scholar]
- 23.Lam H. M., Xu X., Liu X., et al. 2010, Resequencing of 31 wild and cultivated soybean genomes identifies patterns of genetic diversity and selection. Nat. Genet., 42, 1053–9. [DOI] [PubMed] [Google Scholar]
- 24.Gautier M., Vitalis R. 2012, rehh: an R package to detect footprints of selection in genome-wide SNP data from haplotype structure. Bioinformatics, 28, 1176–7. [DOI] [PubMed] [Google Scholar]
- 25.Voight B. F., Kudaravalli S., Wen X., Pritchard J. K. 2006, A map of recent positive selection in the human genome. Plos Biol., 4, e72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tang K., Thornton K. R., Stoneking M. 2007, A new approach for using genome scans to detect recent positive selection in the human genome. Plos Biol., 5, e171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bourgis F., Kilaru A., Cao X., et al. 2011, Comparative transcriptome and metabolite analysis of oil palm and date palm mesocarp that differ dramatically in carbon partitioning. Proc. Natl. Acad. Sci. USA, 108, 12527–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li H., Durbin R. 2009, Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics, 25, 1754–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Holt C., Yandell M. 2011, MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics, 12, 491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pruitt K. D., Tatusova T., Maglott D. R. 2007, NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res., 35, D61–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jaillon O., Aury J. M., Noel B., et al. 2007, The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature, 449, 463–7. [DOI] [PubMed] [Google Scholar]
- 32.Goff S. A., Ricke D., Lan T. H., et al. 2002, A draft sequence of the rice genome (Oryza sativa L. ssp. japonica). Science, 296, 92–100. [DOI] [PubMed] [Google Scholar]
- 33.Al-Dous E. K., George B., Al-Mahmoud M. E., et al. 2011, De novo genome sequencing and comparative genomics of date palm (Phoenix dactylifera). Nat. Biotechnol., 29, 521–7. [DOI] [PubMed] [Google Scholar]
- 34.Jouannic S., Argout X., Lechauve F., et al. 2005, Analysis of expressed sequence tags from oil palm (Elaeis guineensis). FEBS Lett., 579, 2709–14. [DOI] [PubMed] [Google Scholar]
- 35.Initiative A. G. 2000, Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature, 408, 796. [DOI] [PubMed] [Google Scholar]
- 36.Paterson A. H., Bowers J. E., Bruggmann R., et al. 2009, The Sorghum bicolor genome and the diversification of grasses. Nature, 457, 551–6. [DOI] [PubMed] [Google Scholar]
- 37.Schnable P. S., Ware D., Fulton R. S., et al. 2009, The B73 maize genome: complexity, diversity, and dynamics. Science, 326, 1112–5. [DOI] [PubMed] [Google Scholar]
- 38.Peng Z., Lu Y., Li L., et al. 2013, The draft genome of the fast-growing non-timber forest species moso bamboo (Phyllostachys heterocycla). Nat. Genet., 45, 456–61 [DOI] [PubMed] [Google Scholar]
- 39.Pritchard J. K., Wen W., Falush D. 2003, Documentation for STRUCTURE software: version 2 http://pritchardlab.stanford.edu/structure_software/release_versions/v2.3.4/structure_doc.pdf.
- 40.Alvarado A., Escobar R., Peralta F., Chinchilla C. 2006, Compact seeds and clones and their potential for high density planting. International Seminar on Yield Potential in the Oil Palm, The International Society for Oil Palm Breeders (ISOPB), Phuket, Thailand, pp. 27–8.
- 41.Schmutz J., Cannon S. B., Schlueter J., et al. 2010, Genome sequence of the palaeopolyploid soybean. Nature, 463, 178–83. [DOI] [PubMed] [Google Scholar]
- 42.Xia J. H., Wan Z. Y., Ng Z. L., et al. 2014, Genome-wide discovery and in silico mapping of gene-associated SNPs in Nile tilapia. Aquaculture, 432, 67–73. [Google Scholar]
- 43.Jordan I. K., Rogozin I. B., Wolf Y. I., Koonin E. V. 2002, Essential genes are more evolutionarily conserved than are nonessential genes in bacteria. Genome Res., 12, 962–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.