

Abstract
Chenopodium quinoa Willd. (quinoa) originated from the Andean region of South America, and is a pseudocereal crop of the Amaranthaceae family. Quinoa is emerging as an important crop with the potential to contribute to food security worldwide and is considered to be an optimal food source for astronauts, due to its outstanding nutritional profile and ability to tolerate stressful environments. Furthermore, plant pathologists use quinoa as a representative diagnostic host to identify virus species. However, molecular analysis of quinoa is limited by its genetic heterogeneity due to outcrossing and its genome complexity derived from allotetraploidy. To overcome these obstacles, we established the inbred and standard quinoa accession Kd that enables rigorous molecular analysis, and presented the draft genome sequence of Kd, using an optimized combination of high-throughput next generation sequencing on the Illumina Hiseq 2500 and PacBio RS II sequencers. The de novo genome assembly contained 25 k scaffolds consisting of 1 Gbp with N50 length of 86 kbp. Based on these data, we constructed the free-access Quinoa Genome DataBase (QGDB). Thus, these findings provide insights into the mechanisms underlying agronomically important traits of quinoa and the effect of allotetraploidy on genome evolution.
Keywords: Chenopodium quinoa, draft genome, NGS, inbred accession
1. Introduction
Quinoa (Chenopodium quinoa Willd.) is an annual herbaceous plant that originated from the Andean region of South America, and is a pseudocereal crop of the Amaranthaceae family, which also includes sugar beet (Beta vulgaris L. ssp. vulgaris) and spinach (Spinacia oleracea L.).1,2 The major area of quinoa cultivation ranges from Columbia to central Chile,3,4 and includes altitudes from sea level up to 4,000 m above sea level5 and annual rainfalls of 80 mm to 2,000 mm.1,2,6 Quinoa is therefore well adapted to grow under adverse climatic and soil conditions7 and displays high tolerance to drought,8,9 soil salinity,10,11 and frost.12 Furthermore, quinoa is an exceptional nutritional source of a wide spectrum of minerals (e.g. Ca, Fe, Mg, P, and Zn), vitamins (e.g. A, B1, B2, B9, C, and E), dietary fiber, linolenate, natural antioxidants (e.g. polyphenols), and high-quality protein, containing high levels of essential amino acids, particularly methionine and lysine.13–16 Being gluten-free, quinoa is suitable for consumption by individuals who are allergic or intolerant to wheat.14 Owing to the outstanding nutritional value of quinoa seeds and the great adaptability of quinoa plants to adverse environments, quinoa is considered by the Food and Agriculture Organization of the United Nations (FAO) to be an important crop with the potential to contribute to food security worldwide.17 Moreover, the National Aeronautics and Space Administration, USA (NASA) deems quinoa as an optimal food source for astronauts on long-term space missions in isolated conditions.18
Quinoa has been cultivated in the Andes for several thousand years.2 Although quinoa cultivation was forbidden during the Spanish Conquest of South America in the sixteenth century, quinoa is cultivated in over 50 countries today.2,19 Indeed, several thousand quinoa accessions are stored in germplasm banks.2 Although quinoa is considered to be a predominantly autogamous (i.e. self-pollinated) species, multiple reports indicate that quinoa accessions are genetically heterogeneous due to outcrossing based on carrying two kinds of flowers on the same plant.2,20 Nevertheless, no inbred quinoa accessions have been reported to date, and this is problematic because molecular genetics and biology studies of quinoa rely on the development of an inbred quinoa line.
Quinoa is an allotetraploid species (2n = 4x = 36 with a genome size of 1,448 Mbp)21,22 that consists of two distinct genomes, A and B.23 Genetic mapping of quinoa has been conducted using amplified fragment length polymorphism (AFLP) markers,1 simple sequence repeat (SSR) markers,1 and array-platform markers.24 However, the most recent quinoa map contains just 511 single nucleotide polymorphism (SNP) sites and does not span the entire quinoa genome.24 To obtain a sufficient number of molecular markers to cover the entire genome, the draft genome sequence must be established. This would serve as a reference to identify not only SNPs/SSRs, but also next-generation sequencing (NGS)-based markers, such as genotyping-by-sequencing (GBS) markers.25
Illumina and Pacific Biosciences (PacBio) have developed powerful NGS techniques to sequence the genomes of all living organisms. The Illumina HiSeq 2500 sequencer generates a high number of short reads (<250 bp) with a high quality of base calls. In contrast, the PacBio RS II sequencer produces long reads (average read length, 7 kbp), though its throughput and quality of base calls are lower.26 Recently, long reads have been shown to fill gaps within and between scaffolds assembled by short reads.27 Prompted by the finding that different types of sequencers have been successfully combined for the de novo assembly of genome scaffolds of a heterozygous plant, Primula veris,28 we used two distinct types of sequencers to produce the draft genome sequence of the tetraploid species quinoa.
In this study, we established an inbred and standard quinoa accession, Kd, suitable for molecular analyses, and provided the draft genome sequence of the quinoa accession using the Illumina HiSeq 2500 and PacBio RS II sequencers. Based on these data, we constructed the free-access Quinoa Genome DataBase (QGDB; http://quinoa.kazusa.or.jp), which provides annotations of in silico predicted genes. Furthermore, we utilized comparative genomics and experimental approaches to identify genes in quinoa that are involved in abiotic and biotic stress responses.
2. Materials and methods
2.1. Plant materials and growth conditions
Quinoa (Chenopodium quinoa Willd.) seeds had been propagated in a temperature-controlled plant growth room at the Laboratory of Plant Pathology, Graduate School of Agriculture, Kyoto University in the absence of the other quinoa accessions for over 20 years.29 Then at the Japan International Research Center for Agricultural Sciences (JIRCAS), to establish an inbred quinoa accession, quinoa seeds have been propagated from a single plant derived from the seeds propagated in Kyoto University. To prevent cross-pollination, all of the inflorescences of these plants grown in JIRCAS were covered with non-woven pollination bags (Rizo, Tsukuba, Japan). The quinoa seeds were sown in a peat moss mix (Jiffy Mix, Sakata Seeds, Yokohama, Japan) in a cell tray and were grown in a growth chamber at 27 °C with a short-day photoperiod (11 h light/13 h darkness). After 14 days, the seedlings were transferred to a standard potting mix (Tsuchitaro, Sumitomo Forestry, Tokyo, Japan) in 20-L plant pots and were grown under ambient light in a temperature-controlled phytotron in JIRCAS with a temperature of 25 ± 5 °C and relative humidity of 55 ± 25%. For NGS analyses, a single plant was selected, and the progeny seeds were harvested.
2.2. Evaluation of salt-tolerance in quinoa and Arabidopsis
Quinoa seeds (Kd) were sown in the peat moss mix in a cell tray and were grown in a growth chamber at 27 °C with a short-day photoperiod (11 h light/13 h darkness). At 14 days after sowing, the seedlings were transferred to a standard potting mixture (Professional-baido, Daio Chemical, Tokyo, Japan) in 0.16-L plant pots and were further grown under the same conditions. Arabidopsis thaliana (CS60000) seeds were sown on MS agar plates and then were stored at 4 °C in the darkness for 3 days. Then, the seedlings were grown in a growth cabinet (Biotron; NK systems, Japan) at 22 °C with a long-day photoperiod (16 h light/8 h darkness). At 14 days after sowing, the seedlings were transferred to the standard potting mix in 0.16-L plant pots and were grown in a growth chamber at 27 °C with a short-day photoperiod (11 h light/13 h darkness). At 21 days after sowing, the quinoa and the Arabidopsis seedlings were treated with 0 mM or 300 mM NaCl. The survival rates were measured at 22 days after the salt treatments. To maintain the concentration (300 mM NaCl corresponds to 2.90 S/m) and the default volume of the salt solution, the electrical conductivities of the salt solutions were measured using an electrical conductivity meter (Laqua DS-71: HORIBA, Kyoto, Japan) every 2 days.
2.3. Inoculation of quinoa plants
For virus inoculation, quinoa plants were grown in potting mix (Tsuchitaro, Sumitomo Forestry, Tokyo, Japan) in a plant growth room at 25 °C with 16 h of illumination per day. Capped full-length RNA transcripts of two bromoviruses, Brome mosaic virus (BMV) and Cowpea chlorotic mottle virus (CCMV), were synthesized in vitro using T7 RNA polymerase (Takara Bio, Kusatsu, Japan), as described previously.30,31 A mixture of transcripts of viral RNAs 1, 2, and 3 was inoculated mechanically with Carborundum onto the four youngest fully expanded leaves of 5-week-old quinoa plants. The inoculated plants were kept in the growth room and observed for symptom expression.
2.4. Extraction and purification of nuclear DNA
Approximately 80 g of quinoa leaves was harvested from 52-day-old single plants. Cell lysis and the isolation of nuclei were performed using CelLytic PN Isolation/Extraction Kit (Sigma-Aldrich) according to the manufacturer’s instructions with some modifications. Twenty grams of leaves were homogenized in liquid nitrogen, and then the tissue powder was suspended in 800 ml Nuclei Isolation Buffer (Sigma-Aldrich). Homogenization and suspension were repeated four times. The suspension was passed through a Filter Mesh 100 (Sigma-Aldrich), and the supernatant was centrifuged at 1,300 g for 10 min at 4 °C. The precipitate was resuspended in 15 ml of Wash Buffer (Sigma-Aldrich), followed by centrifugation at 2,200 g for 3 min at 4 °C. The wash step was repeated three times. The washed precipitate was resuspended in 10 ml of AP1 Buffer (Qiagen), 40 μl of RNase A solution (Qiagen) was added, and the solution was incubated at 65 °C for 18 min. After centrifugation at 21,500 g for 10 min at 4 °C, the supernatant was transferred to a 50-ml tube. Then, 3.3 ml of Buffer P3 (Qiagen) was added to the tube, which was centrifuged at 21,500 g for 10 min at 4 °C. An equal volume of 2-propanol was added to the supernatant. After gentle mixing, the supernatant was centrifuged at 17,200 g for 3 min at 4 °C. The precipitate was rinsed twice with 70% ethanol. Finally, the resulting dried, pure nuclear DNA was dissolved in 10 ml of Buffer AE (Qiagen).
2.5. DNA sequencing
Library construction and sequencing were performed at Takara Bio. For sequencing using the Illumina HiSeq platform, a paired-end (PE) library with insert sizes of 185 bp and two mate-pair (MP) libraries with expected insert sizes of 2,700–3,500 bp and of 9,000–11,000 bp (Supplementary Table S1) were constructed from nuclear DNA according to the manufacturer’s protocol (Illumina Inc., CA, USA). These libraries were sequenced using a HiSeq 2500 sequencer. For sequencing by PacBio RS II platform, libraries with expected insert sizes (25,300–28,700 bp) (Supplementary Table S1) were constructed according to the manufacture’s protocol (Pacific Biosciences of California, Inc., CA, USA). The libraries were sequenced on 40 single-molecule real-time (SMRT) cells of PacBio RS II.
2.6. Amplification and sequencing of RDR1 genomic fragments
Genomic fragments of CqRDR1A and CqRDR1B from quinoa (Kd), and CpRDR1 of C. pallidicaule were amplified using PrimeSTAR GXL DNA Polymerase (Takara Bio). For amplification of CqRDR1B, two overlapping genomic fragments, named the 5′-half and 3′-half, were separately amplified. The amplified genomic fragments were purified using the Wizard SV Gel and PCR Clean-Up System (Promega). Sequence data were obtained using the ABI PRISM 3130x1 Genetic Analyzer (Life Technologies). To verify the segregation pattern of CqRDR1A and CqRDR1B genes in the F1 progeny of Kd, a co-dominant PCR marker, rdr1-MF2/MR2, was designed. Genomic DNA from 20 self-pollinated F1 progeny was used as PCR templates, and PCR products were analysed on a 4% agarose gel stained with ethidium bromide. Primers used are listed in Supplementary Table S2.
2.7. Extraction of total RNA and DNase I treatment
For RNA Sequencing (RNA-Seq) analysis, total RNA was extracted from various quinoa organs (Supplementary Table S3), using a phenol-SDS method,32 with minor modifications. Quinoa organs were homogenized with liquid nitrogen, and the tissue powders were suspended in TE-saturated-phenol/RNA extraction buffer (0.2 M Tris, 0.2 M LiCl, 5 mM EDTA, and 1% SDS) solution (1/1; vol/vol), mixed well, and centrifuged at 21,500 g for 5 min at 4 °C. The upper aqueous layer was transferred to phenol-chloroform-isoamyl alcohol solution followed, vortexed, and then centrifuged at 21,500 g for 5 min at 4 °C. The total RNA in the upper aqueous phase was precipitated by adding 1/100 volume of acetic acid and an equal volume of 2-propanol. After incubation at −30 °C for 20 min, the suspension was centrifuged at 21,500 g for 15 min at 4 °C. The precipitate was dissolved in nuclease-free water and centrifuged to remove insoluble matter. A quarter volume of 10 M LiCl was added to the supernatant in tubes. The samples were vortexed and incubated at 4 °C for at least 1 h. After centrifuging at 21,500 g for 15 min at 4 °C, the precipitate was rinsed with 2 M LiCl solution and then dissolved in nuclease-free water. The total RNA solution was subjected to phenol-chloroform extraction followed by ethanol precipitation, and then the precipitates were rinsed with 70% ethanol. The dried total RNA was dissolved in nuclease-free water, and the quality was checked using a Bioanalyzer (model 2100; Agilent). Forty micrograms of total RNA was treated with RQ1 DNase I (Promega), according to the manufacturer’s instructions. After DNase I treatment, the total RNA was purified using an RNeasy Plant Mini Kit (Qiagen). First-strand cDNA was synthesized from 1.0 µg total RNA using the PrimeScript High Fidelity RT-PCR Kit (Takara Bio) with oligo-dT (20) primer, according to the manufacturer’s instructions.
2.8. RNA sequencing
Library preparation, sequencing, and assembly were performed at Eurofins Genomics (Ebersberg, Germany). The normalization library was prepared according to Shimizu et al.33 The normalized library was sequenced using an Illumina MiSeq with a 2× 150-bp read module. Low-quality reads and adapter sequences were trimmed using Trimmomatic v0.32.34
2.9. Estimation of genome size
For genome size estimation, PE reads with a k-mer size of 17 were used, as reported previously.35 The k-mer distribution was investigated using Jellyfish 2.1.3.36 The genome size and coverage (i.e. the number of base pairs sequenced as a multiple of the number of base pairs present in the genome) were estimated using the peak at 97 on the k-mer frequency distribution curve (Supplementary Fig. S1) as described previously.35
2.10. Genome assembly
The genome was assembled at Takara Bio. The adaptor sequences of Illumina reads were trimmed using cutadapt v1.2.1.37 The trimmed Illumina reads were assembled using ALLPATHS-LG v52488,38 with the setting HAPLOIDIFY= TRUE. PacBio reads of longer than 1,000 bp were used for gap-closing and further scaffolding of the assemblies obtained from the Illumina reads by PBJelly2 v14.1.14 under default settings.27 Sequences homologous to bacterial, fungal, and human (hg19) genome sequences, vector sequences from UniVec (http://www.ncbi.nlm.nih.gov/tools/vecscreen/univec/), chloroplast (Accession number: NC_000932.1) and mitochondrial (Accession number: NC_001284.2) genome sequences from A. thaliana, and the PhiX sequence used for background controlling in Illumina sequencing by BLASTN39 searches with an E-value cutoff of 1E-10 and length coverage of ≥10%, were excluded as probable contamination. Finally, scaffolds longer than 300 bp were selected and designated Cqu_r1.0. Repetitive sequences in Cqu_r1.0 were detected using RepeatScout 1.0.5.40 and RepeatMasker 4.0.3 (http://www.repeatmasker.org) as described previously.35
2.11. Gene prediction and annotation
To analyse the relationship between the transcriptome and the quinoa genome sequences, we mapped a de novo assembly of transcriptome sequences against Cqu_r1.0. De novo assembly was conducted using the software tools Velvet v1.2.10 and Oases v0.2.08.41,42 A multi-kmer approach was applied. In this approach, separate kmers are first assembled (kmers 59, 69, 79, 89) and then the set of assemblies is merged into a single ‘merged’ assembly (kmer 29). The assembled transcripts were clustered based on sequence identity (≥99%) using the software CD-HIT-EST v4.6.43 These transcripts were mapped against the genome sequence (Cqu_r1.0) using GMAP v2016-05-01 software44 with the threshold option of ≥95% identity and ≥80% coverage.
RNA-Seq reads were mapped onto the draft genome sequence (Cqu_r1.0) with TopHat 2.0.12.45 The bam file obtained was used to generate the training set for the gene prediction of BRAKER1 pipeline.46 Using the training set, the genes were predicted by Augustus 3.0.3.47 The RNA-Seq reads were mapped onto the predicted genes, and splicing variants were excluded by RSEM 1.2.15.48 The predicted genes in the quinoa genome together with those in S. oleracea (Spinach-1.0, 21,702 genes), B. vulgaris (RefBeet-1.1, 27,421 genes), Amaranthus hypochondriacus (AhG2s, 30,564 genes), and A. thaliana genomes (TAIR10, 35,386 genes) were clustered using the CD-HIT program43 with the parameters c = 0.4 and aL = 0.4.
The predicted genes were subjected to similarity searches against the NCBI NR database (ftp://ftp.ncbi.nlm.nih.gov/blast/db/FASTA/nr.gz) and amino acid sequences of A. thaliana from TAIR10 (https://www.arabidopsis.org) using BLASTX with an E-value cutoff of 1E-10. The top hit was used to assign the product name. BLAST searches against UniProt (TrEMBL + Swiss-Prot) with an E-value cut-off of 1E-20 were also carried out. A domain search against InterPro (http://www.ebi.ac.uk/interpro/) was conducted using InterProScan49 with an E-value cutoff of 1.0. Finally, genes were classified based on the NCBI euKaryotic clusters of Orthologous Groups (KOG) database50 by performing BLAST searches with an E-value cutoff of 1E-4. In addition, the genes were mapped onto the KEGG reference pathways by BLAST searches against the KEGG GENES database (http://www.genome.jp/kegg/genes.html) with an E-value cut-off of 1E-4, length of coverage of 25%, and identity of 50%.
Genes related to transposable elements (TEs) were inferred based on a BLAST search against the NCBI NR database and conserved domains were identified based on a search against InterPro and GyDB 2.051 using hmmsearch in HMMER 3.052 with an E-value cutoff of 1.0. Transfer RNA genes (tRNAs) were predicted using tRNAscan-SE v.1.23.53 Ribosomal RNA genes (rRNAs) were predicted in BLASTN searches with an E-value cutoff of 1E-10 using A. thaliana 5.8S and 25S rRNAs (Accession number: X52320.1) and 18S rRNA (Accession number: X16077.1) as queries.
2.12. Estimation of the pair-wise nucleotide divergence at synonymous site (Ks) between putative homoeologous genes
CD-HIT was used to identify putative homoeologous genes. Using the amino acid data set containing 62,512 sequences annotated by BLASTP searches against the NCBI NR database, gene clusters containing two genes (i.e. putative homoeologous genes) were surveyed by the CD-HIT program with the parameters c = 0.9 and aS = 0.5. Paired genes on the same scaffold were not used for further analyses, as they might be tandemly duplicated genes. Alignments of two protein sequences were conducted by BLASTP, and subsequent codon alignment was conducted by PAL2NAL.54 Finally, Ks values were estimated by PAML55 and the frequency of Ks (0 < Ks < 0.5) was plotted.
2.13. Phylogenetic analyses
Alignments of sequences were carried out using CLUSTALW2.56 The evolutionary history was inferred using the neighbor-joining method,57 and the evolutionary distances were computed using the JTT matrix-based method.58
3. Results and discussion
3.1. A standard quinoa accession Kd for molecular genetic analyses
3.1.1. Kd is an appropriate quinoa accession for genome sequencing
Although quinoa is essentially self-pollinating, this species does exhibit genetic heterogeneity due to outcrossing.2,20 Quinoa is a gynomonoecious species that has both hermaphrodite and female flowers on the same plant, so that quinoa plants can accept pollen from the other individuals. Heterozygous alleles at a microsatellite locus were observed in 32% of quinoa accessions.20 Thus, reducing the genome complexity by repeated self-pollination is a crucial step for sequencing the genome of the allotetraploid species quinoa.
A quinoa line has been propagated in an air-conditioned plant growth room without outcrossing for over 20 years.29 We multiplied seeds derived from this line with special non-woven pollination bags, in which inflorescences had been enclosed during flowering stage to prevent cross-pollination, producing inbred accession Kyoto-d (Kd) seeds. The inbred accession Kd (Fig. 1A–H) is more stable in terms of phenotypic uniformity, which has been evaluated based on the extent of variation in size, colour, and morphological characteristics of seeds, seedlings, and flowers in every generation obtained thus far, than any of the more than 150 accessions collected from research institutes (Fig. 1A; more detailed data will be published elsewhere), indicating that Kd is a suitable standard accession for molecular genetics and biology analyses. Kd seeds are approximately 2 mm in diameter (Fig. 1D). Like other quinoa lines (for example, Bonales-Alatorre et al.59), epidermal bladder cells (salt bladders) are present on the surface of leaves (Fig. 1E and F). Guard cells are also observed on both abaxial and adaxial surface of leaves (Fig. 1G and H).
Figure 1.

Morphological characteristics of quinoa (Kd) plants. (A) 14 day-old quinoa (Kd) seedlings grown in soil. Scale bar = 1 cm. (B) 32 day-old quinoa (Kd) plant grown in soil. Scale bar = 5 cm. (C) A main panicle. Scale bar = 5 cm. (D) Dried mature quinoa (Kd) seeds. Scale bar = 1 cm. (E) Epidermal bladder cells (salt bladders) on abaxial surface of a young quinoa leaf (the leaf blade length: 15 mm). Scale bar = 0.5 mm. (F) Epidermal bladder cells (salt bladders) on abaxial surface of a fully expanded quinoa leaf. Scale bar = 0.5 mm. (G) Epidermal bladder and guard cells on abaxial surface of a fully expanded quinoa leaf were observed using a colour laser three-dimensional profile microscope (Keyence, Osaka, Japan), which shows epidermal bladder and stomatal aperture with no pretreatments. White arrows indicate epidermal bladder cells. Scale bar = 50 µm. (H) Enlarged view of guard cells on abaxial surface of a fully expanded quinoa leaf was provided using the microscope as described in (G). Scale bar = 50 µm.
3.1.2. Kd responds well to abiotic and biotic stresses
Quinoa plants are able to grow under a wide spectrum of harsh environments60 and are facultative C3 halophytes.11,59,61 Indeed, all of the Kd plants survived for at least 22 days after treatment with 300 mM NaCl, whereas no A. thaliana plants (CS60000, the inbred line used for whole-genome sequencing) did (Fig. 2A–D), indicating that Kd also displays enhanced tolerance to high salinity in comparison with the C3 glycophyte A. thaliana. These data are consistent with previous findings for quinoa plants subjected to salt tolerance tests,11,59 suggesting that Kd is appropriate for molecular analyses to identify the mechanisms underlying enhanced tolerance of quinoa to abiotic stresses.
Figure 2.

Quinoa (Kd) plants exhibited higher salt tolerance and characteristic symptoms of virus infections. (A–D) Comparative analysis of salinity tolerance in quinoa (Kd) versus Arabidopsis plants grown in soil. 3 week-old quinoa (A, C) and Arabidopsis (B, D) plants were treated with 0 mM NaCl (A, B) or 300 mM NaCl (C, D) for 3 weeks and photographed at 6 weeks after germination. Scale bar = 8 cm. (E) Chlorotic or necrotic local lesions induced on the inoculated quinoa leaves mechanically inoculated with Brome mosaic virus (BMV) or Cowpea chlorotic mottle virus (CCMV) are viewed at 7 days post-inoculation, respectively. Mock, mock inoculation. Scale bar = 2 cm. (F) Systemic or nonsystemic infection of quinoa plants inoculated with BMV or CCMV viewed at 21 days post-inoculation, respectively. Scale bar = 8 cm.
Quinoa and the tobacco species Nicotiana benthamiana are widely used as diagnostic hosts to identify virus species.62 One well-studied and monocot-adapted bromovirus, BMV, induced chlorotic local lesions that developed into large chlorotic blotches on the inoculated Kd leaves, and then continued to spread systemically, resulting in severe symptoms, including leaf distortions and dwarfing (Fig. 2E and F). In contrast, a closely related and dicot-adapted bromovirus, CCMV, caused just small necrotic local lesions on the inoculated Kd leaves. Interestingly, CCMV was arrested within the lesions and did not spread systemically (Fig. 2E and F). These findings are in accordance with previous reports in the other accessions.63,64 Thus, Kd responds to abiotic and biotic stresses in a manner that is representative of quinoa species, and is therefore suitable for molecular analyses of its great adaptability and susceptibility to a wide range of viruses.
3.2. Genome size estimation
In de novo genome assemblies, most scaffolds are in fragmented, and thus it is often difficult to determine the genome size accurately from the total assembly length. As the first step of our quinoa genome project, we estimated the genome size using the frequency distribution curve of 17-mer obtained from Illumina short reads, as successfully reported in previous studies.35,65 The k-mer frequency distribution curve (k-mer =17) using paired-ends (PEs) with a 185-bp insert size is shown in Supplementary Figure S1. The highest peak, at a multiplicity of 97, was expected to be a ‘homo-peak’, that contained 17-mers from a homozygous region of the genome.66 Based on the highest peak, the genome size of quinoa was estimated to be 1.5 Gbp, in good agreement with that of 1,448 Mbp from a cytometry analysis.21 It is noteworthy that no peaks around half of the multiplicity of 97 were detected. A previous study indicated that a ‘hetero-peak’, i.e. a peak at half the multiplicity of the homo-peak, is detected in heterozygous species.66 A simulation also indicated that the greater the degree of heterozygosity, the greater the height of the hetero-peak.66 The finding that no distinct hetero-peaks were present in our k-mer analysis implies that the repeated self-pollination of Kd has resulted in a highly homogenized genetic background. The smaller peak at around the doubled multiplicity of the homo-peak (i.e. at around ∼200) is of interest, because it may contain k-mers related to allotetraploidy. However, the k-mer curve distribution for the ‘diploid’ genome is known to show a smaller peak at the doubled multiplicity of the homo-peak.67 Thus, the smaller peak at around ∼200 in our k-mer analysis may relate to genomic sequences caused by both allotetraploidy and other simple duplications.
3.3. Genome assembly of quinoa
We obtained a large amount of quinoa DNA sequence data using Illumina HiSeq 2500 and PacBio RS II platforms (Supplementary Table S1). In total, 290.8 Gbp and 45.8 Gbp of raw data were generated by the HiSeq and PacBio RS II platforms, respectively. The average size of raw reads from PacBio RS II was 10.1 kbp. These Illumina and PacBio sequence data corresponded to 196× and 31× coverage of the quinoa genome, respectively. First, de novo genome assembly was performed using the short reads generated from the HiSeq platform using ALLPATHS-LG. We obtained 110,092 contigs, with a total length of 830.0 Mbp and an N50 size of 14,505 bp (Supplementary Table S4). These contigs were grouped into 36,423 scaffolds, with a total length of 946.6 Mbp and an N50 size of 53,276 bp. Then, using PacBio long reads and PBJelly2 software, gap-closing and further scaffolding of the assembly generated from HiSeq reads were performed. This strategy reduced the gap lengths in the scaffolds generated using short reads, improving the genome assembly.28 After processing with PBJelly2 software, we obtained 24,847 scaffolds, with a total length of 1.1 Gbp and an N50 length of 86,941 bp. The number of observed gaps (number of ‘N’ and ‘Others’) resulted in a 76% reduction of the number observed using only HiSeq data, and the N50 length was 1.6-fold greater than in the assembly before an application of PBJelly2 (Supplementary Table S4). After trimming of two scaffolds that exhibited signs of contamination (identified in a BLAST search), 24,845 scaffolds were designated as the draft genome sequence, Cqu_r1.0 (Table 1). The scaffolds were named ‘Cqu_sc’ followed by a five-digit identifier and the sequence version (e.g. Cqu_sc00001.1). The total length of Cqu_r1.0 was 1.1 Gbp, and the N50 length was 86,941 bp. As with the results of the genome assembly of the heterozygous plant P. veris,28 the length of the assembly for the tetraploid species quinoa was improved by including PacBio long reads. However, the genome coverage rates of the assembly were low both in quinoa (73%) and P. veris (63%). Recently, near-complete genome assembly of a diploid species, Vigna angularis (azuki bean), was achieved using 27.6 Gbp of the PacBio long reads, which corresponds to 51× coverage of the genome size.68 The authors constructed 2,529 scaffolds (N50 = 3.0 Mbp), covering 97.1% of the V. angularis genome. In our analysis of quinoa, we attained a genome coverage of 30×. Given the complexity of the quinoa genome resulting from ploidy and large genome size, a much greater coverage than 51× of the genome size is needed to use the method reported by Sakai et al.68 Attaining a greater genome coverage is our next objective for refining our quinoa assembly.
Table 1.
Statistics of the draft genome sequences (Cqu_r1.0)
| Number of sequences | 24,845 |
| Cumulative length of sequences (bases) | 1,087,413,657 |
| Average length of sequences per contig (bases) | 43,768 |
| Max length of sequences (bases) | 641,516 |
| Min length of sequences (bases) | 332 |
| N50 length (bases) | 86,941 |
| Number of undetermined bases | 28,385,628 |
| GC% (GC/ATGC) | 36.9 |
3.4. Gene prediction and annotation
Transcriptome sequencing of the normalized cDNA library, which was derived from 12 different tissues of quinoa, produced 14.5 million reads, corresponding to 4.3 Gbp after quality trimming (Supplementary Table S5). We obtained a de novo transcriptome assembly containing 87,877 transcripts (≥100 bp). Of these transcripts, we were able to successfully map 71,744 (81.6%) onto Cqu_r1.0, which is higher than the mapping efficiency reported for barley69 (74.9%). Our genome assembly covers much of the transcriptome assembly, suggesting that it represents a suitable framework for gene prediction of C. quinoa. Using these RNA-Seq reads as the training set to predict genes for Augustus 3.0.3, 226,647 CDSs (Cqu_r1.0_cds) consisting of 190.5 Mbp, were obtained (Supplementary Table S6).
Genes related to transposable elements (TEs) were inferred according to BLAST searches against the NCBI NR database (Supplementary Table S7). The total length of known repeats was 132.7 Mbp (12.2% of Cqu_r1.0) and Class I LTR elements were frequently found (7.2% of Cqu_r1.0). In this analysis, we identified unique repeats that had not previously been sequenced, and these had a total length of 535.5 Mbp and accounted for 49.2% of Cqu_r1.0. Genes annotated as transposons were tagged ‘TE’ in the sequence name.
Based on BLAST searches against the NCBI NR database, the genes were divided into four classes. The first class contains genes that include both a start and stop codon; the second contains genes that include either a start codon or a stop codon, or that lack both a start and stop codon; the third contains genes that include a stop codon in the coding region; and the fourth contains short sequences (encoding < 50 amino acids). The tags based on this classification system and the numbers of genes in each tag are listed in Supplementary Table S8. We found that 150,029 genes in the first and second classes were not related to TEs, and we successfully annotated 62,512 of these 150,029 genes by BLAST searches against the UniProtKB database. Thus, the number of annotated genes (62,512 genes) is roughly twice that predicted in most other plant diploid genomes.70 This appears to be reasonable, since quinoa is a tetraploid species. However, given that a large number of genes (87,517 genes) were not annotated by the BLAST searches against the UniProtKB database, the number of protein-coding genes of quinoa predicted in this study might still be an underestimation. In addition to the protein-coding genes, we identified 2,592 genes for tRNAs, and the number of genes for each tRNA is summarized in Supplementary Table S9. The draft genome sequence (Cqu_r1.0), predicted gene sequences, deduced amino acid sequences, annotations derived from BLAST searches against the NCBI NR and TAIR10 databases, and domains identified in the search against InterPro were included in the free-access Quinoa Genome DataBase (QGDB; http://quinoa.kazusa.or.jp). In addition, local BLAST searches and keyword searches for gene names and their annotations were also implemented in the QGDB.
An amino acid sequence similarity search was performed for the predicted proteins encoded by the subset of 62,512 genes using the CD-HIT program with the parameters c = 0.9 and aS = 0.5, and 46,695 of sequence homology clusters were identified. Of these, 9,768 contained two genes; i.e. 19,536 of 62,512 predicted genes were paired. Of the 9,768 pairs, 131 were located on the same scaffold, and might be tandemly duplicated genes. The average of Ks in the remaining 9,637 clusters was 0.08, and the distribution of Ks values peaked at ∼0.035 (Supplementary Fig. S2). Considering that the Ks values of these duplicated genes are similar to the previously estimated Ks average between two homoeologous SOS1 genes (Ks = 0.077),71 most of these duplicated genes in a single cluster can be considered as putative homoeologs derived from two genomes. A major challenge in the de novo assembly of polyploids is to differentiate the homoeologous genomes and to assign the genes/scaffolds to each subgenome. In hexaploid wheat, transcriptome assemblies in ancestral diploid species were used to classify the genes predicted from a de novo genome assembly into the A, B, and D genomes.72 This was not possible in quinoa because the donor diploid species of the B-genome has yet to be identified,23 and no genome or transcriptome data of the A-genome species were available. Nevertheless, the fact that we could identify several putative homeologous genes suggests that we were successful in capturing the two subgenomes at least to a certain extent. Genome or transcriptome sequencing of C. standleyanum or other A-genome species, such as C. pallidicaule, will facilitate efforts to assign the predicted genes/scaffolds to the A- or B-genome in quinoa.
3.5. Comparative analysis of the quinoa and other Amaranthaceae species gene sequences
The entire data set of 226,647 genes was mapped onto the KEGG metabolic pathway and classified into the categories under ‘1. Metabolism’ and the numbers of mapped genes in each pathway are summarized in Supplementary Table S10. Briefly, 1,703 genes from quinoa were mapped onto 133 of the 158 categories of metabolic pathway in the KEGG database, whereas 1,748 genes of S. oleracea, 1,753 genes of B. vulgaris, and 1,009 genes of A. hypochondriacus were mapped onto 133, 135, and 124 pathways, respectively. Twenty-six pathways were only associated with genes in the quinoa genome. However, genes with similarity to the quinoa genes on these 26 pathways were identified in S. oleracea, B. vulgaris, or A. hypochondriacus by subsequent BLASTP searches against the NR database (Supplementary Table S11). These would be false positive by the mapping analyses for KEGG metabolic pathways under the parameters with relatively low thresholds (E-value cut-off of 1E-4, length of coverage of 25%, and identity of 50%).
The entire data set of 226,647 genes was also annotated by conducting similarity searches using the CD-HIT program (-c:0.4, -aL:0.4; Fig. 3). First, the predicted genes in the quinoa genome were clustered together with 30,564 genes in A. hypochondriacus, 27,421 in B. vulgaris, 21,702 in S. oleracea, and 35,386 in A. thaliana. The 3,393 clusters were common among five species, and 9,534, 4,543, 6,114, 5,805, and 9,996 genes belonged to the common clusters in C. quinoa, A. hypochondriacus, B. vulgaris, S. oleracea, and A. thaliana, respectively. Quinoa has 565 clusters of genes in a section shared only with A. thaliana, which is much less than the 1,224 genes present in sections shared with S. oleracea, 1,498 with B. vulgaris, and 3,215 with A. hypochondriacus in Amaranthaceae (Fig. 3). Similar results were obtained in the comparative analysis of the filtered dataset of 62,512 genes of quinoa annotated by performing BLASTP searches against the NR database in NCBI (Supplementary Fig. S3). In the Amaranthaceae family, the genera Chenopodium and Spinacia belong to Chenopoideae, the genus Beta to Betoideae, and the genus Amaranthus to Amaranthoideae.73 The genes, which were selected from a common section (central part in Fig. 3) on condition of a single copy being present in A. hypochondriacus, B. vulgaris, S. oleracea, and A. thaliana and of two copies being present in quinoa, were concatenated. Multiple alignment was carried out for the concatenated genes by ClustalW2,56 and the dendrogram was constructed by the neighbour-joining method (Supplementary Fig. S4). Quinoa and S. oleracea were phylogenetically closely related as expected, confirming that genes obtained from the QGDB were appropriate derivatives of those in the three other Amaranthaceae species. These basically single copy genes might be useful in phylogenetic analyses of core eudicots, as previously proposed for orthologous gene sets in P. veris.28
Figure 3.
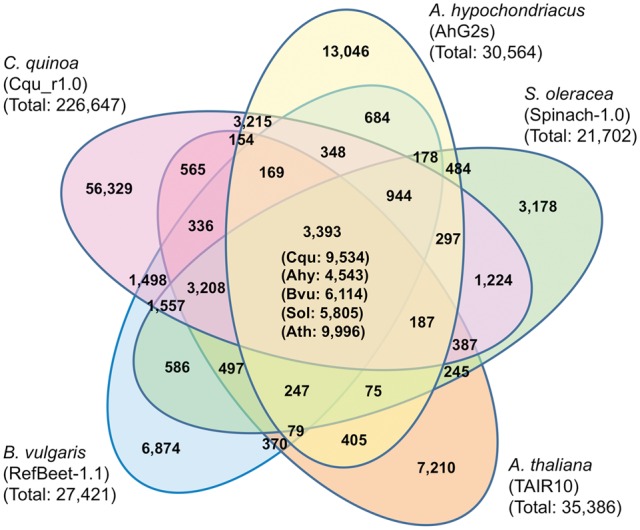
Cluster analysis of the predicted gene sequences. Predicted genes in Chenopodeum quinoa, Amaranthus hypochondriacus, Beta vulgaris, Spinacia oleracea, and Arabidopsis thaliana were clustered into gene families. The number in each section represents the number of clusters, and the numbers in parentheses in the central section represent the numbers of genes included from each species. The number below the species shows the total number of genes used as input for the CD-HIT (-c: 0.4, -aL: 0.4).
We also detected several expanded genes in quinoa from the results of CD-HIT analysis. Ten lysine histidine transporter 2 (LHT2) like genes, which are related to amino acid uptake, were detected in the quinoa genome,74,75 but 2, 3, and 1 genes were found in S. oleracea, B. vulgaris, and A. hypochondriacus, respectively (Supplementary Table S12). Some cytochrome P450 genes were also expanded in the quinoa genome. Two of these were annotated as cytochrome P450 71A1 and 76AD1, which play a role in the polyphenol- and betalain-biosynthesis pathway, respectively.76,77 Expansion and subsequent functional differentiation of these genes might be related to quinoa having a high protein content and abundant secondary metabolites.
3.6. Genome designed for adaptation to hostile environments
3.6.1. ABA signalling genes in quinoa
Quinoa can withstand a variety of abiotic stresses,61,78 including high salinity (Fig. 2A–D). Transporter genes implicated in salt tolerance in quinoa, such as CqSOS1A and CqSOS1B, which are homologs of Arabidopsis SOS1 involved in plasma membrane-localized Na+/H+ transport, have been analysed so far.71 Meanwhile, the phytohormone abscisic acid (ABA) is known to regulate abiotic stress tolerance.79 Phylogenetic analyses revealed that gene families involved in ABA signalling were substantially expanded in the quinoa genome compared with other Amaranthaceae plant species. For example, the PYR/PYL/RCAR gene family, which encodes a bona fide ABA receptor, plays key roles in ABA signalling. Neighbour-joining analyses of AtPYLs and PYLs from Amaranthaceae species revealed three phylogenetic groups (Supplementary Fig. S5). In each group, we identified subgroups consisting of genes of Amaranthaceae species, including three in group I, two in group II, and three in group III (Supplementary Fig. S5). Six of these subgroups contained two phylogenetically similar genes from C. quinoa and one from each of the other Amaranthaceae species included in the analysis. These pairs of C. quinoa genes are putative homoeologs, thought to have arisen from allotetraploidization. The gene family encoding SnRK2 protein kinases includes core positive regulators of ABA signalling, and similar phylogenetic results were obtained: four of the six subgroups identified contained two genes from C. quinoa and one from each of the other Amaranthaceae species examined (Supplementary Fig. S6). The Ks values between putative homoeologs found in PYLs and SnRK2 ranged from 0 to 0.088 and from 0 to 0.076, respectively (Supplementary Table S13). These findings are roughly consistent with the distribution of Ks values estimated from putative homoeologous gene sets obtained by CD-HIT analysis (Supplementary Fig. S2). Thus, our sequence results provide an important basis for studies aimed at determining the roles and precise numbers of candidate genes and pathways that regulate tolerance to abiotic stress in allotetraploid quinoa.
3.6.2. RDR1 homoeologs in quinoa
Quinoa and N. benthamiana have been widely used as diagnostic hosts to identify virus species based on their own characteristic symptoms.62 However, N. benthamiana is usually systemically infected with viruses, whereas quinoa is either locally or systemically infected, as shown in Figure 2E and F. Although mutations in Rdr1, which is involved in antiviral defence, have provided intriguing clues as to the mechanism by which a wide range of viruses can amplify well in N. benthamiana,80,81 it remains unknown why a broad range of viruses can infect quinoa. We therefore analysed the RDR1 genes in quinoa.
We identified two RDR1 orthologs in quinoa derived from a predicted gene (Cqu_c21957.1_g001.1) based on QGDB and a truncated gene on a scaffold (Cqu_c12210.1), and named these CqRDR1A and CqRDR1B, respectively. RT-PCR and Sanger sequencing revealed that CqRDR1A carries a complete ORF encoding 1,122 amino acids, and that CqRDR1B harbours a 2-bp deletion in the protein-coding region that induces a nonsense mutation (Supplementary Fig. S7), leading to the absence of a large portion of the RNA-dependent RNA polymerase (RdRP) domain that includes the active site (DxDGD).82 Along with the observation that the RdRP domains are well conserved in angiosperms (Fig. 4), these findings clearly indicate that RdRP function is impaired in CqRDR1B.
Figure 4.

RDR1 of Amaranthaceae and other plant species. Amino acid sequences of Amaranthus hypochondriacus, Arabidopsis thaliana, and Oryza sativa were obtained from Phytozome 11,84 sequences of Beta vulgaris and Spinacia oleracea were from the Beta vulgaris Resource,85 and sequences of Nicotiana benthamiana (AAS78669.1) and N. tabacum were from NCBI. The nucleotide sequence of N. benthamiana (WA) was from Nicotiana benthamiana Genome and Transcriptome86 and translated to the putative amino acid sequence by EMBOSS Transeq.87 (A) Unrooted neighbor-joining tree based on amino acid sequences. The bootstrap values (500 replicates) not less than 50 are shown next to the branches. The scale bar corresponds to 0.05 substitutions per site. The root was assumed as the midpoint of the tree. (B) Schematic view of RDR1. The RNA-dependent RNA polymerase (RdRP) domain88 is indicated by a rounded rectangle and the active site of RDR182 is indicated by an arrowhead. The scale bar corresponds to 1,200 amino acids.
Our phylogenetic analysis revealed that CqRDR1 duplication occurred after the speciation between Chenopodium and Spinacia (Fig. 4). Based on a co-dominant marker that distinguishes between two CqRDR1 genes, we confirmed that all 20 tested Kd progeny retained both genes at different loci (Supplementary Fig. S8). Comparison of RDR1 sequences between quinoa and C. pallidicaule indicated that CqRDR1A was derived from the Chenopodium A-genome (Supplementary Fig. S9). In addition, no PCR products were amplified from four C. pallidicaule lines using a B-genome-specific primer pair (data not shown), suggesting that CqRDR1A and CqRDR1B are homoeologs. Although further analyses will be required to reveal the mechanism supporting the susceptibility to a wide variety of viruses, these findings provide insight into the mechanism underlying the response to virus infections in quinoa.
4. Conclusion and future perspectives
The year 2013 was declared the International Year of Quinoa by FAO to heighten public awareness of the nutritional benefits of this durable plant, as part of a sustainable food production effort aimed at food security and nutrition.17 However, molecular analysis of quinoa is limited by its genetic heterogeneity and its genome complexity, due to allotetraploidy. To overcome the former limitation, we established the inbred accession Kd that possesses standard quinoa properties in response to abiotic and biotic stresses. This provides a useful experimental standard material for basic and applied studies of quinoa, which have recently focused on its exceptional nutritional value, tolerance to unfavourable environments, and susceptibility to a broad range of viruses.
The allotetraploid nature of quinoa makes it difficult to construct a genome assembly using high-throughput sequencing. To overcome this limitation, we employed a combination of Illumina HiSeq 2500 and PacBio RS II NGSs, which produce short (approximately 200 bp) and long (mean length of 10 kbp) reads, respectively. The long reads were used for gap-closing and for further scaffolding of the assembly obtained from the Illumina short reads to improve the genome assembly, and resulted in a 76% reduction in gaps and a 1.6-fold increase in the N50 length. In our trial usage of the database QGDB, ABA signalling genes and RDR1 were detected, and gene expansion probably caused by tetraploidization was observed in all cases. It should be noted that we were also able to identify putative homoeologous genes in both the clustering and Ks distribution analyses. Thus, although our assembly does not cover the entire genome and still contains large gaps, these findings have clearly demonstrated that the genome database created from the assembly is a useful basis for identifying agronomically important genes. Nevertheless, the length of scaffolds of our assembly could be increased by applying new technologies, such as high-throughput optical mapping,83 which provides long-range genomic information through constructing restriction maps. In addition, a high-density linkage map generated using NGS-based GBS markers with Kd-based recombinant inbred lines (RILs) will also be required to anchor the improved scaffolds to each chromosome to generate pseudomolecules of the 18 chromosomes in quinoa.
5. Data availability
The Illumina and PacBio reads used in this study are available from DDBJ/EMBL/Genbank under the accession numbers listed in Supplementary Table S1 and S5. The scaffold sequences are available under the accession numbers BDCQ01000001-BDCQ01024845 (24,845 entries). The draft genome sequence Cqu_r1.0, CDS and protein sequences, and annotation file (gff file) are also available from the free-access Quinoa Genome DataBase (QGDB; http://quinoa.kazusa.or.jp). DNA sequences of RDR1s are deposited in the DDBJ/EMBL/Genbank database under the accession numbers LC146405, LC146407, and LC149497.
Supplementary Material
Acknowledgements
We thank S. Tabata (Kazusa) and A. Nakagawa (Ishikawa Pref. Univ.) for valuable suggestions, K. Amano, E. Ohgawara and E. Kishi (JIRCAS) for their excellent technical support, M. Fujita (RIKEN) for helpful comments and skilful graphics assistance, and K. L. Farquharson for language-editing support of the manuscript. We are deeply grateful to I. Furusawa (prof. emeritus of Kyoto Univ.), T. Okuno (Ryukoku Univ.; prof. emeritus of Kyoto Univ.), H. Nagano (Kyoto Univ.), and the other past members at the Lab. of Plant Pathology, Kyoto Univ., involved in the quinoa growth for more than 20 years. We apologize to all authors whose work could not be cited here due to space constraints.
Conflict of interest
None declared.
Supplementary data
Supplementary data are available at www.dnaresearch.oxfordjournals.org.
Funding
This research was supported by cooperative research funds from Actree Co. Our work at JIRCAS was partly supported by the Ministry of Agriculture, Forestry and Fisheries (MAFF) of Japan.
References
- 1.Maughan P.J., Bonifacio A., Jellen E.N., et al. 2004, A genetic linkage map of quinoa (Chenopodium quinoa) based on AFLP, RAPD, and SSR markers. Theor. Appl. Genet., 109, 1188–95. [DOI] [PubMed] [Google Scholar]
- 2.Zurita-Silva A., Fuentes F., Zamora P., Jacobsen S.-E., Schwember A.R. 2014, Breeding quinoa (Chenopodium quinoa Willd.): potential and perspectives. Mol. Breed., 34, 13–30. [Google Scholar]
- 3.Risi J., Galwey N.W. 1989, The pattern of genetic diversity in the Andean grain crop quinoa (Chenopodium quinoa Willd.). I. Associations between characteristics. Euphytica, 41, 147–62. [Google Scholar]
- 4.Fuentes F., Bhargava A. 2011, Morphological analysis of quinoa germplasm grown under lowland desert conditions. J. Agron. Crop Sci., 197, 124–34. [Google Scholar]
- 5.González J.A., Bruno M., Valoy M., Prado F.E. 2011, Genotypic variation of gas exchange parameters and leaf stable carbon and nitrogen isotopes in ten quinoa cultivars grown under drought. J. Agron. Crop Sci., 197, 81–93. [Google Scholar]
- 6.Martínez E.A., Veas E., Jorquera C., San Martín R., Jara P. 2009, Re-introduction of quínoa into arid Chile: cultivation of two lowland races under extremely low irrigation. J. Agron. Crop Sci., 195, 1–10. [Google Scholar]
- 7.Garcia M., Raes D., Jacobsen S.-E. 2003, Evapotranspiration analysis and irrigation requirements of quinoa (Chenopodium quinoa) in the Bolivian highlands. Agric. Water Manage., 60, 119–34. [Google Scholar]
- 8.Vacher J.J. 1998, Responses of two main Andean crops, quinoa (Chenopodium quinoa Willd) and papa amarga (Solanum juzepczukii Buk.) to drought on the Bolivian Altiplano: significance of local adaptation. Agric. Ecosyst. Environ., 68, 99–108. [Google Scholar]
- 9.Razzaghi F., Ahmadi S.H., Adolf V.I., Jensen C.R., Jacobsen S.E., Andersen M.N. 2011, Water relations and transpiration of quinoa (Chenopodium quinoa Willd.) under salinity and soil drying. J. Agron. Crop Sci., 197, 348–60. [Google Scholar]
- 10.Jacobsen S.E., Mujica A. 2003, Quinoa: an alternative crop for saline soils. J. Exp. Bot., 54, i25. [Google Scholar]
- 11.Hariadi Y., Marandon K., Tian Y., Jacobsen S.E., Shabala S. 2011, Ionic and osmotic relations in quinoa (Chenopodium quinoa Willd.) plants grown at various salinity levels. J. Exp. Bot., 62, 185–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jacobsen S.E., Monteros C., Christiansen J.L., Bravo L.A., Corcuera L.J., Mujica A. 2005, Plant responses of quinoa (Chenopodium quinoa Willd.) to frost at various phenological stages. Eur. J. Agron., 22, 131–9. [Google Scholar]
- 13.Vega-Galvez A., Miranda M., Vergara J., Uribe E., Puente L., Martinez E.A. 2010, Nutrition facts and functional potential of quinoa (Chenopodium quinoa willd.), an ancient Andean grain: a review. J. Sci. Food Agric., 90, 2541–7. [DOI] [PubMed] [Google Scholar]
- 14.Maradini Filho A.M., Pirozi M.R., Da Silva Borges J.T., Pinheiro Sant'Ana H.M., Paes Chaves J.B., Dos Reis Coimbra J.S. 2015, Quinoa: nutritional, functional and antinutritional aspects. Crit. Rev. Food Sci. Nutr., doi: 10.1080/10408398.2014.1001811. [DOI] [PubMed] [Google Scholar]
- 15.Nowak V., Du J., Charrondiere U.R. 2016, Assessment of the nutritional composition of quinoa (Chenopodium quinoa Willd.). Food Chem., 193, 47–54. [DOI] [PubMed] [Google Scholar]
- 16.Mota C., Santos M., Mauro R., et al. 2016, Protein content and amino acids profile of pseudocereals. Food Chem., 193, 55–61. [DOI] [PubMed] [Google Scholar]
- 17.Bazile D., Bertero D., Nieto C. 2015, State of the art report on quinoa around the world in 2013. Food and Agriculture Organization of the United Nations (FAO) & Centre de Coopération Internationale en Recherche Agronomique pour le Développement (CIRAD): Rome. [Google Scholar]
- 18.Schlick G., Bubenheim D.L. 1993, Quinoa: an emerging “new” crop with potential for celss. NASA Tech. Pap., 3422, 1–9. [Google Scholar]
- 19.González J.A., Eisa S.S.S., Hussin S.A.E.S., Prado F.E. 2015, In: K.S. Murphy, J. Matanguihan. (eds), Quinoa: improvement and sustainable production. John Wiley & Sons, Inc, New Jersey, pp. 1–18. [Google Scholar]
- 20.Christensen S.A., Pratt D.B., Pratt C., et al. 2007, Assessment of genetic diversity in the USDA and CIP-FAO international nursery collections of quinoa (Chenopodium quinoa Willd.) using microsatellite markers. Plant Genet. Resour., 5, 82–95. [Google Scholar]
- 21.Palomino G., Hernández L.T., de la Cruz Torres E. 2008, Nuclear genome size and chromosome analysis in Chenopodium quinoa and C. berlandieri subsp. nuttalliae. Euphytica, 164, 221–30. [Google Scholar]
- 22.Yangquanwei Z., Neethirajan S., Karunakaran C. 2013, Cytogenetic analysis of quinoa chromosomes using nanoscale imaging and spectroscopy techniques. Nanoscale Res Lett, 8, 463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Walsh B.M., Adhikary D., Maughan P.J., Emshwiller E., Jellen E.N. 2015, Chenopodium polyploidy inferences from Salt Overly Sensitive 1 (SOS1) data. Am. J. Bot., 102, 533–43. [DOI] [PubMed] [Google Scholar]
- 24.Maughan P.J., Smith S.M., Rojas-Beltrán J.A., et al. 2012, Single nucleotide polymorphism identification, characterization, and linkage mapping in quinoa. Plant Genome, 5, 114–25. [Google Scholar]
- 25.Elshire R.J., Glaubitz J.C., Sun Q., et al. 2011, A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS One, 6, e19379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carneiro A.R., Ramos R.T., Barbosa H.P., et al. 2012, Quality of prokaryote genome assembly: indispensable issues of factors affecting prokaryote genome assembly quality. Gene, 505, 365–7. [DOI] [PubMed] [Google Scholar]
- 27.English A.C., Richards S., Han Y., et al. 2012, Mind the gap: upgrading genomes with Pacific Biosciences RS long-read sequencing technology. PLoS One, 7, e47768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nowak M.D., Russo G., Schlapbach R., Huu C.N., Lenhard M., Conti E. 2015, The draft genome of Primula veris yields insights into the molecular basis of heterostyly. Genome Biol., 16, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nagano H., Okuno T., Mise K., Furusawa I. 1997, Deletion of the C-terminal 33 amino acids of cucumber mosaic virus movement protein enables a chimeric brome mosaic virus to move from cell to cell. J. Virol., 71, 2270–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kroner P., Ahlquist P. 1992, In: S.J. Gurr, M.J. McPherson, D.J. Bowles. (eds), Molecular plant pathology: a practical approach, vol. I IRL Press at Oxford University Press, Oxford, 23–34. [Google Scholar]
- 31.Mise K., Allison R.F., Janda M., Ahlquist P. 1993, Bromovirus movement protein genes play a crucial role in host specificity. J. Virol., 67, 2815–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reddy K.J., Gilman M. 1995, In: F.M. Ausubel, R. Brent, R.E. Kingston, et al. (eds), Current Protocols in Molecular Biology. John Wiley & Sons Inc, New Jersey. Chapter 4, Section 4.3. [Google Scholar]
- 33.Shimizu M., Iwano S., Uno Y., et al. 2014, Qualitative de novo analysis of full length cDNA and quantitative analysis of gene expression for common marmoset (Callithrix jacchus) transcriptomes using parallel long-read technology and short-read sequencing. PLoS One, 9, e100936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bolger A.M., Lohse M., Usadel B. 2014, Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics, 30, 2114–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hirakawa H., Shirasawa K., Kosugi S., et al. 2014, Dissection of the octoploid strawberry genome by deep sequencing of the genomes of Fragaria species. DNA Res., 21, 169–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marçais G., Kingsford C. 2011, A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics, 27, 764–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Martin M. 2011, Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J., 17, 10–2. [Google Scholar]
- 38.Gnerre S., Maccallum I., Przybylski D., et al. 2011, High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc. Natl Acad. Sci. U. S. A., 108, 1513–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J. 1990, Basic local alignment search tool. J. Mol. Biol., 215, 403–10. [DOI] [PubMed] [Google Scholar]
- 40.Price A.L., Jones N.C., Pevzner P.A. 2005, De novo identification of repeat families in large genomes. Bioinformatics, 21 Suppl 1, i351–8. [DOI] [PubMed] [Google Scholar]
- 41.Zerbino D.R., Birney E. 2008, Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res., 18, 821–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schulz M.H., Zerbino D.R., Vingron M., Birney E. 2012, Oases: robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics, 28, 1086–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fu L., Niu B., Zhu Z., Wu S., Li W. 2012, CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics, 28, 3150–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu T.D., Watanabe C.K. 2005, GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics, 21, 1859–75. [DOI] [PubMed] [Google Scholar]
- 45.Trapnell C., Pachter L., Salzberg S.L. 2009, TopHat: discovering splice junctions with RNA-Seq. Bioinformatics, 25, 1105–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hoff K.J., Lange S., Lomsadze A., Borodovsky M., Stanke M. 2016, BRAKER1: unsupervised RNA-seq-based genome annotation with GeneMark-ET and AUGUSTUS. Bioinformatics, 32, 767–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stanke M., Waack S. 2003, Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics, 19 Suppl 2, ii215–25. [DOI] [PubMed] [Google Scholar]
- 48.Li B., Dewey C.N. 2011, RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics, 12, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Quevillon E., Silventoinen V., Pillai S., et al. 2005, InterProScan: protein domains identifier. Nucleic Acids Res., 33, W116–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tatusov R.L., Fedorova N.D., Jackson J.D., et al. 2003, The COG database: an updated version includes eukaryotes. BMC Bioinformatics, 4, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Llorens C., Futami R., Covelli L., et al. 2011, The Gypsy Database (GyDB) of mobile genetic elements: release 2.0. Nucleic Acids Res., 39, D70–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Eddy S.R. 2009, A new generation of homology search tools based on probabilistic inference. Genome Inform, 23, 205–11. [PubMed] [Google Scholar]
- 53.Lowe T.M., Eddy S.R. 1997, tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res., 25, 955–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Suyama M., Torrents D., Bork P. 2006, PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res., 34, W609–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang Z. 2007, PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol., 24, 1586–91. [DOI] [PubMed] [Google Scholar]
- 56.Larkin M.A., Blackshields G., Brown N.P., et al. 2007, Clustal W and Clustal X version 2.0. Bioinformatics, 23, 2947–8. [DOI] [PubMed] [Google Scholar]
- 57.Saitou N., Nei M. 1987, The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol., 4, 406–25. [DOI] [PubMed] [Google Scholar]
- 58.Jones D.T., Taylor W.R., Thornton J.M. 1992, The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci., 8, 275–82. [DOI] [PubMed] [Google Scholar]
- 59.Bonales-Alatorre E., Shabala S., Chen Z.H., Pottosin I. 2013, Reduced tonoplast fast-activating and slow-activating channel activity is essential for conferring salinity tolerance in a facultative halophyte, quinoa. Plant Physiol., 162, 940–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sun Y., Liu F., Bendevis M., Shabala S., Jacobsen S.E. 2014, Sensitivity of two quinoa (Chenopodium quinoa Willd.) varieties to progressive drought stress. J. Agron. Crop Sci., 200, 12–23. [Google Scholar]
- 61.Adolf V.I., Jacobsen S.-E., Shabala S. 2013, Salt tolerance mechanisms in quinoa (Chenopodium quinoa Willd.). Environ. Exp. Bot., 92, 43–54. [Google Scholar]
- 62.Hull R. 2014, In: M. Grigg, A. McBride, J.M. Quarles, et al. (eds), Current Protocols in Microbiology, John Wiley & Sons, Inc, New Jersey, Chapter 16, 16A.2.1–A2.4. [Google Scholar]
- 63.Bancroft J.B. 1972, A virus made from parts of the genomes of brome mosaic and cowpea chlorotic mottle virus. J. Gen. Virol., 14, 223–8. [Google Scholar]
- 64.Osman F., Grantham G.L., Rao A.L.N. 1997, Molecular studies on bromovirus capsid protein. IV. Coat protein exchanges between brome mosaic and cowpea chlorotic mottle viruses exhibit neutral effects in heterologous hosts. Virology, 238, 452–9. [DOI] [PubMed] [Google Scholar]
- 65.Yasui Y., Hirakawa H., Ueno M., et al. 2016, Assembly of the draft genome of buckwheat and its applications in identifying agronomically useful genes. DNA Res., 23, 215–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kajitani R., Toshimoto K., Noguchi H., et al. 2014, Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads. Genome Res., 24, 1384–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lu M., An H., Li L. 2016, Genome survey sequencing for the characterization of the genetic background of Rosa roxburghii Tratt and leaf ascorbate metabolism genes. PLoS One, 11, e0147530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sakai H., Naito K., Ogiso-Tanaka E., et al. 2015, The power of single molecule real-time sequencing technology in the de novo assembly of a eukaryotic genome. Sci. Rep., 5, 16780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sato K., Tanaka T., Shigenobu S., Motoi Y., Wu J., Itoh T. 2016, Improvement of barley genome annotations by deciphering the Haruna Nijo genome. DNA Res., 23, 21–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wendel J.F., Jackson S.A., Meyers B.C., Wing R.A. 2016, Evolution of plant genome architecture. Genome Biol., 17, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Maughan P.J., Turner T.B., Coleman C.E., et al. 2009, Characterization of Salt Overly Sensitive 1 (SOS1) gene homoeologs in quinoa (Chenopodium quinoa Willd.). Genome, 52, 647–57. [DOI] [PubMed] [Google Scholar]
- 72.Brenchley R., Spannagl M., Pfeifer M., et al. 2012, Analysis of the bread wheat genome using whole-genome shotgun sequencing. Nature, 491, 705–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Müller K., Borsch T. 2005, Phylogenetics of Amaranthaceae based on matK/trnK sequence data: evidence from parsimony, likelihood, and Bayesian analyses. Ann. Missouri Bot. Gard., 92, 66–102. [Google Scholar]
- 74.Chen L., Bush D.R. 1997, LHT1, a lysine- and histidine-specific amino acid transporter in arabidopsis. Plant Physiol., 115, 1127–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lee Y.H., Tegeder M. 2004, Selective expression of a novel high-affinity transport system for acidic and neutral amino acids in the tapetum cells of Arabidopsis flowers. Plant J., 40, 60–74. [DOI] [PubMed] [Google Scholar]
- 76.Meyer K., Cusumano J.C., Somerville C., Chapple C.C. 1996, Ferulate-5-hydroxylase from Arabidopsis thaliana defines a new family of cytochrome P450-dependent monooxygenases. Proc. Natl Acad. Sci. U. S. A., 93, 6869–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Brockington S.F., Yang Y., Gandia-Herrero F., et al. 2015, Lineage-specific gene radiations underlie the evolution of novel betalain pigmentation in Caryophyllales. New Phytol., 207, 1170–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ruiz K.B., Biondi S., Martínez E.A., Orsini F., Antognoni F., Jacobsen S.E. 2016, Quinoa – a model crop for understanding salt-tolerance mechanisms in halophytes. Plant Biosyst., 150, 357–71. [Google Scholar]
- 79.Miyakawa T., Fujita Y., Yamaguchi-Shinozaki K., Tanokura M. 2013, Structure and function of abscisic acid receptors. Trends Plant Sci., 18, 259–66. [DOI] [PubMed] [Google Scholar]
- 80.Yang S.J., Carter S.A., Cole A.B., Cheng N.H., Nelson R.S. 2004, A natural variant of a host RNA-dependent RNA polymerase is associated with increased susceptibility to viruses by Nicotiana benthamiana. Proc. Natl Acad. Sci. U. S. A., 101, 6297–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bally J., Nakasugi K., Jia F., et al. 2015, The extremophile Nicotiana benthamiana has traded viral defence for early vigour. Nat. Plants, 1, 15165. [DOI] [PubMed] [Google Scholar]
- 82.Iyer L.M., Koonin E.V., Aravind L. 2003, Evolutionary connection between the catalytic subunits of DNA-dependent RNA polymerases and eukaryotic RNA-dependent RNA polymerases and the origin of RNA polymerases. BMC Struct. Biol., 3, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tang H., Lyons E., Town C.D. 2015, Optical mapping in plant comparative genomics. Gigascience, 4, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Phytozome 11, https://phytozome.jgi.doe.gov/pz/portal.html
- 85.The Beta vulgaris Resource, http://bvseq.molgen.mpg.de/index.shtml.
- 86.Nicotiana benthamiana Genome & Transcriptome, http://benthgenome.qut.edu.au.
- 87.EMBOSS Transeq, http://www.ebi.ac.uk/Tools/st/emboss_transeq/.
- 88.Marchler-Bauer A., Derbyshire M.K., Gonzales N.R., et al. 2015, CDD: NCBI's conserved domain database. Nucleic Acids Res., 43, D222–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.